
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of active site conformations on the hydride transfer step of the thymidylate synthase reaction mechanism
Katarzyna
Świderek
ab,
Amnon
Kohen
c and
Vicent
Moliner
*a
aDepartament de Química Física i Analítica, Universitat Jaume I, 12071 Castelló, Spain. E-mail: moliner@uji.es
bInstitute of Applied Radiation Chemistry, Lodz University of Technology, 90-924 Lodz, Poland
cDepartment of Chemistry, University of Iowa, Iowa City, IA 52242, USA
First published on 2nd April 2015
Abstract
The hydride transfer from C6 of tetrahydrofolate to the reaction's exocyclic methylene–dUMP intermediate is the rate limiting step in thymidylate synthase (TSase) catalysis. This step has been studied by means of QM/MM molecular dynamics simulations to generate the corresponding free energy surfaces. The use of two different initial X-ray structures has allowed exploring different conformational spaces and the existence of chemical paths with not only different reactivities but also different reaction mechanisms. The results confirm that this chemical conversion takes place preferentially via a concerted mechanism where the hydride transfer is conjugated to thiol-elimination from the product. The findings also confirm the labile character of the substrate–enzyme covalent bond established between the C6 of the nucleotide substrate and a conserved cysteine residue. The calculations also reproduce and rationalize a normal H/T 2° kinetic isotope effect measured for that step. From a computational point of view, the results demonstrate that the use of an incomplete number of coordinates to describe the real reaction coordinate can render biased results.
Introduction
Thymidylate synthase (EC 2.1.1.45), TSase, catalyzes the reductive methylation of 2′-deoxyuridine 5′-monophosphate (dUMP) to 2′-deoxythymidine 5′-monophosphate (dTMP) using N5,N10-methylene-5,6,7,8-tetrahydrofolate (CH2H4folate) both as a reducing agent and as a methylene donor, producing 7,8-dihydrofolate (H2folate).1 The reaction mechanism involves a cascade of at least seven chemical reactions.Potential energy surfaces (PESs) of every single step of the catalysed process were studied via Quantum Mechanics/Molecular Mechanics (QM/MM) calculations.2 The findings were in good agreement with the relevant experimental data1 suggesting that the rate limiting step of the whole reaction is the reduction of an exocyclic methylene intermediate (Schemes 1 and 2). This reaction involves hydride transfer from the C6 position of the intermediate 5,6,7,8-tetrahydrofolate (H4folate), marked as the Cd atom in Scheme 1, to the C7 atom of dUMP, marked as Ca in Scheme 1. The calculations suggested several critical differences between the probable mechanism and the one originally proposed by Carreras and Santi.3 For instance, the thioether bond between the C6 of dUMP and a conserved active site cysteine residue (Cys146 in E. coli) appeared to be much more labile than previously assumed. In fact, the results showed how the breaking of this bond takes place concertedly with the hydride transfer from C6 of H4folate to the exocyclic methylene intermediate (see Scheme 2). There are experimental data suggesting that the hydride transfer is essentially irreversible3 and rate-determining on both first-order (kcat) and second-order rate constants (kcat/Km) for the overall reaction.1,4 Nevertheless, supporting evidence for the stepwise mechanism was offered by Santi and co-workers5,6 based on the close similarities between the enzyme-bound enolate intermediate and many covalent binary complexes of TSase with dUMP analogues experimentally detected.3
 | ||
| Scheme 1 Schematic representation of the active site of the E. coli TSase prior to the hydride transfer step. Atoms in the grey region are treated by QM methods and QM-MM frontiers are depicted as black thick bars. The transferred hydride is highlighted, and its donor and acceptor atoms marked as Cd and Ca, respectively. | ||
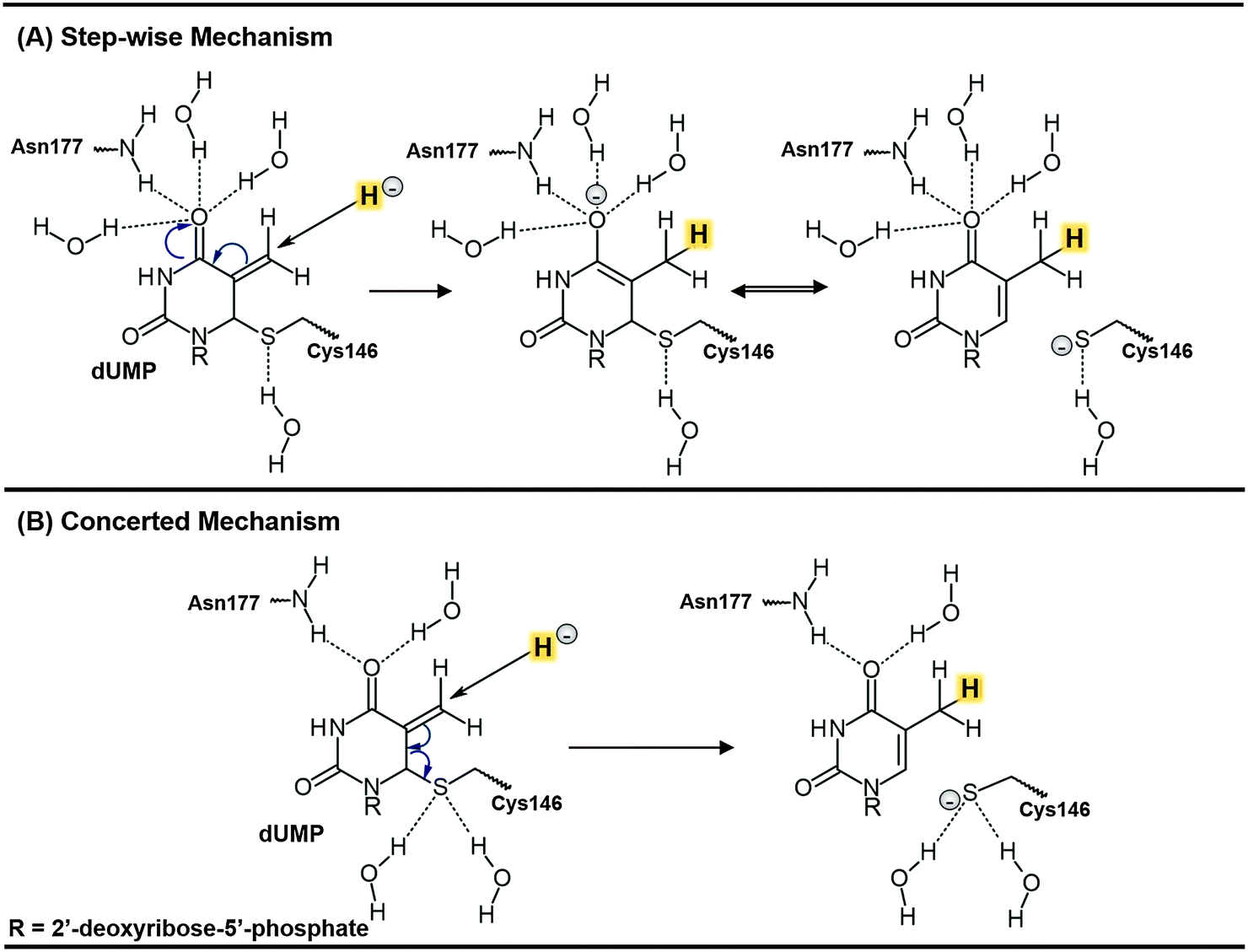 | ||
| Scheme 2 Schematic representation of the two possible mechanisms for the rate determining hydride transfer step of the TSase catalysed reaction. The transferred hydride is highlighted. | ||
Free energy surfaces, computed in terms of two dimensional potential of mean force (PMF) at four different temperatures indicated first that the hydride transfer and the scission of Cys146 take place in a concerted but asynchronous fashion, and second, a temperature independent mechanism.7 This result was in agreement with measured intrinsic Kinetic Isotope Effects (KIEs) on the same H-transfer step,1 and was completed by further computational studies carried out by means of the ensemble-averaged variational transition-state theory with multidimensional tunneling (EA-VTST/MT)8,9 combined with Grote–Hynes theory.10 KIEs, tunneling effects and quantum mechanical dynamical effects in the hydride transfer step were accomplished by computing mono-dimensional potentials of mean force (1D PMFs) along a distinguished reaction coordinate; the antisymmetric combination of distances defining the breaking and forming of bonds of the transferring hydride, d(C6FolH) − d(HC7dUMP). Multidimensional semiclassical transmission coefficients that incorporate the effects of barrier recrossing, quantized molecular vibrations and quantum mechanical tunnelling were also computed to estimate the values of rate constants. The results, calculated across the same temperature range examined experimentally, confirmed a temperature independent behavior.10
These previous computational studies showed how the 1,3-SN2 substitution appears also to be assisted by arginine 166 and several other arginine residues in the active site that polarize the carbon–sulphur bond and stabilize the charge transferred from the cofactor to the substrate. Additionally, our study of a proton abstraction step that precedes the hydride transfer also indicated that the traditionally proposed covalent bond between the protein and substrate (the C6–S bond) is very labile.11 Active site conformation not only assists cleavage of the C6–S bond to stabilize the transition state of the proton transfer step but also rearranges the H-bond network at the end of each step to prepare the active site for the subsequent chemical step.
The question of whether the rate-determining step (r.d.s) of TSase takes place by means of a concerted or a stepwise mechanism is, nevertheless, still a question of debate, with the possible implications in chemotherapeutic and antibiotic drug designs. Recently, the two proposed mechanisms for the r.d.s. were experimentally tested by H/T secondary (2°) KIE measurements on C6 of dUMP and via primary (1°) KIEs studies of the R166K mutant.12 The two possible scenarios are the stepwise mechanism, in which the hydride transfer precedes the cleavage of the covalent bond between the enzymatic cysteine and the product, and the mechanism where both happen concertedly (see Scheme 2). The normal H/T 2° KIE (1.104 ± 0.004) on kcat/KM indicated that C6 of the product dTMP is partly re-hybridized to sp2 when the hydride is transferred. This finding indicates an involvement of the C6–S cleavage in the irreversible hydride transfer step. This, together with the dramatic change in the temperature dependence of intrinsic 1° KIEs between the wild-type TSase and R166K, supported the concerted mechanism and suggested a critical role of R166 in the TS of the hydride transfer step,12 as predicted by previous QM/MM calculations.2,7,10 Nevertheless, since the measured 2° KIE was only 1.1 whereas a full rehybridization of C6–dTMP from sp3 to sp2 would lead to 2° KIE of 1.37, the level of coupling between the hydride transfer and the C6–S cleavage is still not fully understood.
In the present study, we have performed an analysis of the hydride step of the TSase catalysed reaction by exploring the effects of using a limited number of distinguished reaction coordinates on the description of such a complex chemical step. Monodimensional and bidimensional PMFs (1D PMF and 2D PMF) have been generated at different levels of theory using two initial X-ray structures (PDB IDs 2TSC and 1TLS) as starting points of our simulations. The first structure (PDB ID 2TSC) was the Escherichia coli TSase complexed with the substrate dUMP and an analogue of CH2H4folate.13–15 The second structure of the same enzyme employed in our simulations was PDB code 1TLS,16 where the enzyme is covalently bound to 5-fluoro-dUMP (FdUMP) that is covalently bound to CH2H4folate. The use of two different X-ray structures as the starting point for the simulation allows for testing the relevance of different initial protein conformations in simulations of enzyme catalyzed reactions. The use of a limited number of coordinates to study multidimensional chemical steps was also examined in the current study. The combination of such studies together with 2° KIE calculations on key atoms of the system, which are in agreement with experimental data, permit proposing a realistic picture of the catalytic activity of this important anticancer and antibiotic drug-target.
Computing methods
The model
As mentioned in the introduction section, simulations were performed based on two different starting geometries from Escherichia coli TSase: PDB codes 2TSC13–15 and 1TLS.16 The former consists of two subunits of 264 amino acids each, and an anti-folate inhibitor and a substrate in each one of the active sites. The inhibitor (10-propargyl-5,8-dideazafolate) was replaced by the cofactor in one of the active sites and the dUMP was turned into the exo-cyclice methylene intermediate. In the other starting crystal structure, 1TLS, which had both 5F-dUMP and CH2H4folate bound at the active site of each monomer, fluorine was replaced with a hydrogen atom at C5 of dUMP, the C–N bond between the methylene and N5 of THF was broken, the proton from C5 of dUMP removed, and the exo-cyclic methylene established. The backbones of both structures closely overlap. The main difference is in slightly different conformations of some side chains, the location of water molecules, and of course the different bindings of the ligands in the active site.The coordinates of the hydrogen atoms were added to both structures using the fDYNAMO17 library after computing the pKa values of ionisable amino acids with the empirical PROPKA318,19 program in order to determine their proper protonation state in the protein environment.
A total of 24 and 22 counterions (Na+) (for 2TSC and 1TLS, respectively) were placed in optimal electrostatic positions around the enzyme (further than 10.5 Å from any atom of the system and 5 Å from any other counterion, using a regular grid of 0.5 Å) because the total charge of the system was not neutral. Finally, the systems were solvated using boxes of water molecules of 100 × 80 × 80 Å3 dimensions, and the water molecules with an oxygen atom lying within 2.8 Å of any heavy atom were removed.
In both cases, the whole system was divided into a QM part and a MM part to perform combined QM/MM calculations. The QM part considers 25 atoms of the folate, 21 atoms of the dUMP and the side chain of Cys146, which gives a total of 54 QM atoms, and three hydrogen link atoms that were added at the boundary between QM and MM regions to satisfy the valence of the QM/MM frontier atoms (see Scheme 1).20 The calculations only modeled one active site in the QM region, leaving the second active site (ligands removed) in the MM region. The MM part comprises the rest of the folate and dUMP, the enzyme, the crystallization and solvation water molecules and the sodium counterions, which makes a total of 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 181 and 60826 atoms for 2TSC and 1TLS, respectively.
181 and 60826 atoms for 2TSC and 1TLS, respectively.
During the QM/MM energy optimizations, the atoms of the QM region were treated by the semiempirical Hamiltonians AM1,21 and by the M06-2X22 hybrid density functional theory (DFT) methods. The standard 6-31G+(d,p) basis set was used in the DFT calculations. The rest of the system, protein and water molecules, are described using the OPLS-AA23 and TIP3P24 force fields, respectively, as implemented in the fDYNAMO library.17 Cut-offs for the non-bonding interactions are applied using a force switching scheme within a range radius from 14.5 to 16 Å. After thermalization, QM/MM MD simulations of the system in the NVT ensemble (with the QM region treated at the AM1 level) were ran during 500 ps at a temperature of 300 K using the Langevin–Verlet algorithm using a time step of 1 fs. The molecular dynamics (MD) simulations were performed using an integration step size of 1 fs and the velocity Verlet algorithm.33 In both cases, the resulting structures had an energy fluctuation lower than 0.1%, a kinetic energy fluctuation lower than 1% and a change in temperature lower than 2.5 K over the last 2 ps of the MD. Moreover, according to the time-dependent evolution of the RMSD of those atoms belonging to the protein backbone, the systems can be considered equilibrated.
Potential energy surface, PES
After setting up the model, the full system was minimized using the Adopted Basis Newton Raphson (ABNR)23 method, keeping the backbone of the enzyme frozen. Due to the huge dimensions of the system, all the residues further than 20 Å from the QM part were kept frozen. The main interactions of the active site of these structures with the amino acids and crystallization water molecules were representative of the one proposed by Stroud and Finer-Moore,25 which can be considered as additional proof of the proper setting up of the molecular models. The structures were then used to generate a mono-dimensional (1D) and bi-dimensional (2D) QM/MM PESs for the r.d.s. of the TSase catalyzed reaction (Scheme 2). The refined TSs were used as the starting point to generate the 1D PMFs, while the geometries from the 2D PESs were used to generate the 2D PMFs.Potential of mean force, PMF
Mono-dimensional PMFs, 1D PMF, were computed using the antisymmetric combination of distances describing the hydride transfer, d(Cd–H) − d(H–Ca), as the distinguished reaction coordinate, as in the PES calculations. The weighted histogram analysis method (WHAM), combined with the umbrella sampling approach,35,36 was employed to scan the reaction coordinate in a range from −2.5 to 2.5 Å, with a window width of 0.05 Å (the total amount of windows was 101). The two-dimensional PMFs, 2D PMF, were obtained with the anti-symmetric combination of the distances describing the breaking and forming of bonds on the hydride transfer step, d(Cd–H) − d(H–Ca), and the distance between the C6 of the dUMP and the sulfur atom of the Cys146 (dC6–S). A total of 61 simulations were performed at different values of d(Cd–H) − d(H–Ca) with an umbrella force constant of 2500 kJ mol−1 A−1 for each particular value of the distance dC6–S (23 simulations with a force constant of 2500 kJ mol−1 A−1, from 1.8 to 4.0 Å).The values of the variables sampled during the simulations were then pieced together to construct a full distribution function from which the 1D-PMF and 2D-PMF were obtained. On each window, 5 ps of relaxation was followed by 10 ps of production with a time step of 0.5 ps due to the nature of the chemical step involving a hydrogen transfer. The velocity Verlet algorithm33 was used to update the velocities in each window.
All the PMFs were performed at 303 K, using as starting points of each window structures from the previously obtained 1D and 2D PESs.
Because of the large number of structures that must be evaluated during free energy calculations, QM/MM MD calculations were done with the AM1 semiempirical Hamiltonian. In order to improve the quality of our MD simulations, following the work of Truhlar et al.,26–28 a spline under tension29 has been used to interpolate this correction term at any value of the reaction coordinates ξ1 and ξ2 selected to generate the 2D PMFs. In this way we obtain a continuous function to obtain corrected PMFs:30–32
| E = EAM1/MM + S[ΔEHLLL(ξ1, ξ2)] | (1) |
Kinetic isotope effects
KIEs have been computed for isotopic substitutions of key atoms from the transition states and the reactant complex localized at the two levels of theory described above. The free energy of a state, Gi, can be expressed as a function of the molecular potential energy Ei, the total partition function Qi, and the zero point vibrational energy, ZPEi,| Gi = Ei − RTlnQi + ZPEi | (2) |
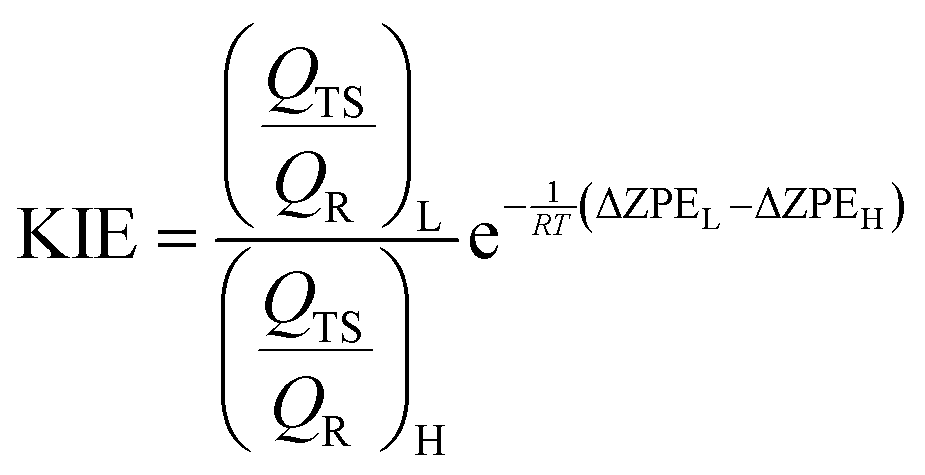 | (3) |
Results and discussion
As explained in the Computing Methods section, the free energy profile for the r.d.s. of the TSase catalyzed reaction (Scheme 2) has been first computed in terms of 1D-PMFs using as the distinguished reaction coordinate the anti-symmetric combination of distances defining the hydride transfer from the donor atom C6 of H4folate, Cd, to the acceptor atom C7 of dUMP (Ca). The resulting free energy profiles, obtained from the two different initial X-ray structures (with 2TSC and 1TLS PDB codes) are shown in Fig. 1. As observed, both profiles describe an exothermic reaction but with different reaction energy and free energy barriers. The 1D-PMF obtained from 1TLS structure presents lower barrier and appears as a more exothermic reaction than the one obtained from 2TSC. This, together with the fact that the position of maximum on the PMF for the 1TLS appears to be slightly less advanced than in the case of the 2TSC (r.c. = 0.04 Å and 0.11 Å, respectively), is in agreement with the Hammond postulate. At this point, we could already confirm the dependence of the computed PMFs on the initial conformational configuration, as previously demonstrated in other studies.37 The fact that significant energetics are obtained, even when derived from calculations based on statistical simulations such as the present PMFs, can be due to the presence of different conformations in any of the states along the full catalytic process that can not be completely overlapped during the QM/MM MD simulations carried out in every window of the full profile. Nevertheless, the fact that different conformations can render different energy barriers would explain experimental results such as the temperature dependence of KIEs38,39 or the significant different rate constants observed in single-molecule spectroscopy.40–42 The flexibility of the enzymes would be responsible for the presence of multiple reactive conformations of substrates bounded to the enzyme, each of them forming a heterogeneous branched reaction pathway with a different rate of conversion from reactants to products. The presence of a wide distribution of rates in individual enzyme molecules have also been predicted by computer simulations on different enzymatic systems.10,43–47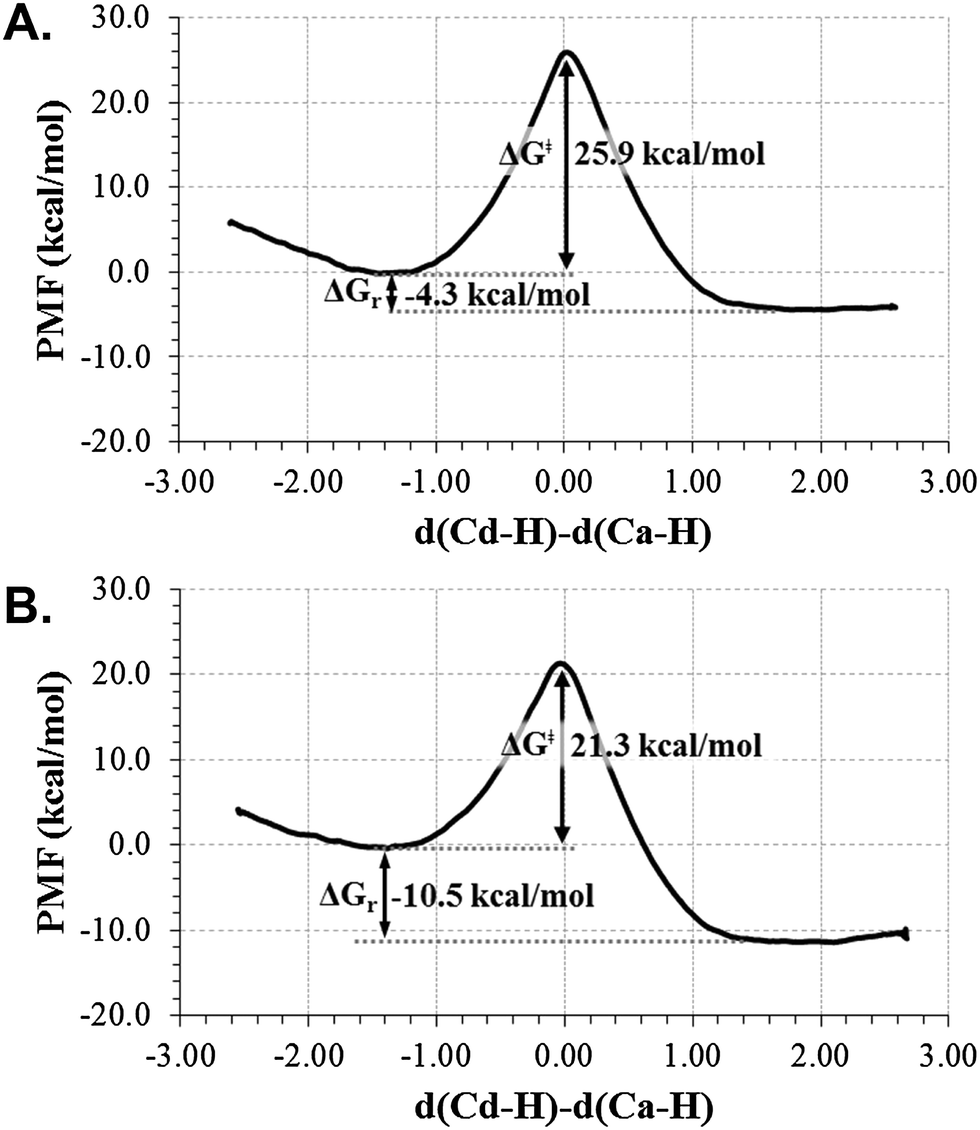 | ||
| Fig. 1 Free energy profiles for the hydride transfer step of the TSase catalyzed reaction, obtained as mono-dimensional AM1/MM potential of mean force (1D-PMF) using two different initial X-ray structure 2TSC (top) and 1TLS (bottom), at 303 K. The distinguished reaction coordinate (in Å) was the antisimmetric combination of distances definning the hydride transfer (H) from C6 (Cd) of H4folate to C7 (Ca) of dUMP. | ||
In order to perform a deeper analysis on the results, the evolution of key distances along the distinguished reaction coordinate, computed as averaged values over each window of the PMF, are presented in Fig. 2. A comparison of plots obtained from 2TSC and 1TLS structures, together with average distances of the reactants, TS and products listed in Table 1, allows obtaining interesting conclusions. First of all, the distance between the hydride donor and acceptor atoms is changing from reactants to products, reaching the minimum value at the TS in both cases (2.716 ± 0.005 and 2.756 ± 0.008 Å in 2TSC and 1TLS structures, respectively); a state where the hydride is almost in between these two carbon atoms. Nevertheless, an important difference is observed in the behaviour of the C–S bond. Thus, while in the profile of 2TSC this bond is not broken during the hydride transfer, despite small oscillations of the distance, once the TS is crossed the bond is clearly broken after reaching the TS on the PMF obtained from the 1TLS structure. In fact, the C–S distance in the TS of the former (1.953 ± 0.005 Å) presents a value that is very close to the one obtained in reactants (1.919 ± 0.002 Å), while in the case of the PMF generated from 1TLS structure, the C–S bond is clearly elongated in the TS (1.958 ± 0.005 Å and 2.063 ± 0.020 Å in reactants and TS, respectively). Then, the 1D-PMF obtained from 2TSC structure would be describing the r.d.s. of the TSase as a step-wise mechanism through an intermediate where the bond between C5 and the sulphur atom of Cys146 is not broken, while the results obtained from the 1D-PMF of the 1TLS structure would describe a concerted hydride transfer and a C–S breaking bond. According to the plots presented in the right side of Fig. 2 and data reported in Table 1, the conformation of the active site obtained in the TS of the 2TSC 1D-PMF, named the step-wise mechanism from now on, would stabilize a structure of the nucleotide (dTMP) where C5–Ca double bonds are transformed into a single bond, and at the same time the double bond character will be delocalized between O4–C4, C4–C5 and C5–C6. As observed in evolution of these distances from reactants to products, while the former is slightly elongated, the other two values are shortened in this step-wise mechanism (see Fig. 2 and Table 1). This process is associated with the development of a negative charge localized in O4 that appears to be stabilized by hydrogen bond interactions with three water molecules and Asn177 (see Table 1 and Fig. 3). On the contrary, with the 1D-PMF from 1TLS, named the concerted mechanism from now on, we arrive to a structure where the O4–C4 carbonyl bond and the C4–C5 bonds are unaltered but a double bond is clearly formed between C5 and C6 atoms. The formation of a C5–C6 double bond is associated with the C6–S breaking bond. In this case, O4 is interacting with Asn177 and only with one water molecule (see Table 1 and Fig. 3). Moreover, as observed in Table 1 and Fig. 3, the different behaviour of the S–C bond along the two PMFs can be also explained based on interatomic distances established around this sulphur center. In fact, the reaction profile in which the process of hydride transfer and C6–S bond breaking take place in a concerted manner is characterized by a local environment that favours polarization of this bond. In particular, it can be observed how Tyr94, and specially Arg166, interact with the sulphur atom through water molecules in both cases. In the case of the step-wise mechanisms, only one water molecule appears at the hydrogen bond distance from the C6–S bond, and neither Arg166 nor Tyr94 directly activate it. At this point, we could indicate that the different conformations that can be present in large systems such as the enzymes not only can render significant different kinetics, but even different mechanisms.
 | ||
| Fig. 2 Evolution of key distances along the distinguished reaction coordinate (in Å) computed as averages over the different windows generated along the (A) step-wise mechanisms (1D PMFs generated from 2TSC) and (B) concerted mechanisms (1D PMFs generated from 1TLS). | ||
| Concerted mechanism (from 1TLS) | Step-wise mechanism (from 2TSC) | |||||
|---|---|---|---|---|---|---|
| RC | TS | PC | RC | TS | PC | |
| d(Cd–H) | 1.133 ± 0.001 | 1.387 ± 0.011 | 2.812 ± 0.049 | 1.134 ± 0.000 | 1.436 ± 0.009 | 2.995 ± 0.319 |
| d(Ca–H) | 2.405 ± 0.029 | 1.344 ± 0.011 | 1.120 ± 0.000 | 2.407 ± 0.023 | 1.324 ± 0.004 | 1.120 ± 0.002 |
| d(C–S) | 1.958 ± 0.005 | 2.063 ± 0.020 | 2.996 ± 0.084 | 1.919 ± 0.002 | 1.953 ± 0.005 | 1.996 ± 0.010 |
| d(Cd–Ca) | 3.352 ± 0.025 | 2.716 ± 0.005 | 3.510 ± 0.038 | 3.262 ± 0.026 | 2.756 ± 0.008 | 3.694 ± 0.065 |
| d(O4–C4) | 1.241 ± 0.001 | 1.250 ± 0.001 | 1.248 ± 0.001 | 1.251 ± 0.001 | 1.261 ± 0.001 | 1.275 ± 0.001 |
| d(C4–C5) | 1.479 ± 0.001 | 1.448 ± 0.002 | 1.473 ± 0.001 | 1.481 ± 0.001 | 1.437 ± 0.001 | 1.404 ± 0.001 |
| d(C5–C6) | 1.483 ± 0.000 | 1.445 ± 0.003 | 1.361 ± 0.000 | 1.494 ± 0.000 | 1.469 ± 0.001 | 1.447 ± 0.002 |
| d(C5–C7) | 1.343 ± 0.000 | 1.411 ± 0.001 | 1.476 ± 0.000 | 1.342 ± 0.000 | 1.410 ± 0.001 | 1.469 ± 0.001 |
| d(C7–H′) | 1.098 ± 0.001 | 1.109 ± 0.001 | 1.118 ± 0.001 | 1.103 ± 0.000 | 1.112 ± 0.000 | 1.117 ± 0.002 |
| d(C7–H′′) | 1.101 ± 0.000 | 1.111 ± 0.001 | 1.118 ± 0.001 | 1.101 ± 0.000 | 1.112 ± 0.000 | 1.116 ± 0.002 |
| d(C6–H6) | 1.128 ± 0.001 | 1.120 ± 0.001 | 1.107 ± 0.000 | 1.128 ± 0.000 | 1.124 ± 0.001 | 1.122 ± 0.000 |
| d(O4–HAsn177) | 2.044 ± 0.048 | 1.925 ± 0.021 | 1.983 ± 0.037 | 2.120 ± 0.035 | 1.961 ± 0.019 | 1.908 ± 0.029 |
| d(O4–HWAT1) | 2.155 ± 0.029 | 2.401 ± 0.140 | 2.201 ± 0.144 | 1.906 ± 0.031 | 1.920 ± 0.031 | 1.842 ± 0.009 |
| d(O4–HWAT2) | — | — | — | 3.595 ± 0.070 | 3.653 ± 0.037 | 2.098 ± 0.051 |
| d(O4–HWAT3) | — | — | — | 2.911 ± 0.102 | 2.699 ± 0.134 | 1.978 ± 0.023 |
| d(HWAT1–OGlu58) | 1.582 ± 0.014 | 1.590 ± 0.009 | 1.705 ± 0.037 | |||
| d(S–HWAT5) | 1.789 ± 0.023 | 1.780 ± 0.007 | 1.675 ± 0.011 | 1.927 ± 0.019 | 1.904 ± 0.019 | 1.987 ± 0.030 |
| d(S–HWAT6) | 1.925 ± 0.060 | 1.950 ± 0.068 | 1.890 ± 0.034 | — | — | — |
| d(S–H1Arg166) | 3.998 ± 0.030 | 4.075 ± 0.033 | 4.130 ± 0.043 | 3.718 ± 0.024 | 3.681 ± 0.033 | 3.749 ± 0.077 |
| d(S–H2Arg166) | 5.429 ± 0.043 | 5.531 ± 0.055 | 5.737 ± 0.043 | 3.784 ± 0.081 | 3.931 ± 0.034 | 3.878 ± 0.061 |
| d(OWAT4–H1Arg166) | 1.503 ± 0.005 | 1.543 ± 0.004 | 1.615 ± 0.012 | |||
| d(OWAT5–HTyr94) | 1.642 ± 0.026 | 1.708 ± 0.014 | 1.725 ± 0.013 | |||
| d(O4–H′) | 2.680 ± 0.015 | 2.692 ± 0.017 | 3.147 ± 0.085 | 2.606 ± 0.016 | 2.649 ± 0.019 | 2.777 ± 0.255 |
 | ||
| Fig. 3 Representative snapshots of the averaged structures of reactants state obtained from the 1D PMFs generated from the (A) step-wise mechanisms (1D PMFs generated from 2TSC) and (B) concerted mechanisms (1D PMFs generated from 1TLS). | ||
Two-dimensional potential of mean force (2D-PMF)
In order to check the robustness of our results and in an attempt to elucidate whether both mechanisms can be kinetically relevant, free energy surfaces were computed starting from the two X-ray structures, as explained in the Computational Methods section. The 2D-PMFs, obtained with the antisymmetric combination of distances describing the transfer of the hydride d(Cd–H) − d(Ca–H), and the C6–S distance as the two distinguished reaction coordinates, are shown in Fig. 4. | ||
| Fig. 4 Free energy surfaces computed as AM1/MM 2D PMF performed at 303 K using two different initial X-ray structure 2TSC (A) and 1TLS (B). The distinguished reaction coordinates (in Å) are the C6(dUMP)–S(Cys146) distance and the antisymmetric combination of the breaking and forming of bonds of the transferred hydrogen atom, d(Cd–H) − d(Ca–H). Averaged values of the structures obtained along the 1D PMFs displayed in Fig. 2 are shown as squares. Values on isoenergetic lines are reported in kcal mol−1. | ||
According to the 2D PMFs on Fig. 4, when the C6–S bond is explicitly controlled during the generation of the free energy surface of the reaction, the hydride transfer and the C6–S breaking bond take place in a concerted way, not depending on the initial X-ray structure selected to perform the calculations. In any of the two surfaces shown in Fig. 4, no single minimum can be located corresponding to an intermediate where the hydride was transferred and the C–S bond was still bounded. Nevertheless, a shoulder can be identified in the lower-right corner of the surface generated from the 2TSC, which would be in agreement with the results obtained in the 1D PMF, when controlling only the coordinates associated to the hydride transfer. The average values of both coordinates obtained along the 1D PMFs shown in Fig. 2 are indicated to compare the behaviour observed when just the coordinates associated to the hydride transfer are explicitly controlled. As observed, the 1D PMF generated from the 2TSC X-ray structure did not arrive to the minimum obtained in the 2D PMF. On the contrary, in the case of the 1D PMF from 1TLS, once the TS was crossed, structures representing the products valley of the 2D PMF are also explored with the 1D PMF. Interestingly, the TS structures obtained from all methods are almost coincident regarding the advancement of the hydride transfer, and very close with respect to the C6–S interatomic distance. These geometrical coincidences are in agreement with similar free energy barriers (the two 2D PMF render almost the same value of ca. 26 kcal mol−1). Keeping in mind the possible limitations of the AM1 semiempirical Hamiltonian to report accurate energetic values of TSs, the AM1/MM 2D PMFs were corrected at the M06-2X/MM level in order to confirm whether slightly geometrical differences can have any energetic influence on the barriers. The results are shown in Fig. 5.
 | ||
| Fig. 5 M06-2X/MM 2D PMF performed at 303 K using two different initial X-ray structure 2TSC (A) and 1TLS (B). The distinguished reaction coordinates (in Å) are the C6(dUMP)–S(Cys146) distance and the antisymmetric combination of the breaking and forming bonds of the transferred hydrogen atom, d(Cd–H) − d(Ca–H). Values on isoenergetic lines are reported in kcal mol−1. | ||
According to the free energy surfaces displayed in Fig. 5, and despite the topology of the surfaces having not changed at the qualitative level with respect to the AM1/MM ones shown on Fig. 4, the concerted mechanism obtained when using the 1TLS X-ray structure as the starting point structure for the calculations appears to be clearly more favourable than the one generated from 2TSC structure. The free energy barriers that can be deduced from the M06-2X/MM surfaces are 12.9 and 26.3 kcal mol−1, respectively. Thus, the results suggest that the initial protein conformation that appeared to be better organized for the hydride transfer step describes the r.d.s. of the TSase catalysed reaction as a concerted process; a result that had been obtained even when a reduced number of coordinates are used as distinguished reaction coordinates.
Secondary kinetic isotope effects (2° KIEs)
In order to check whether our predictions derived from the exploration of free energy surfaces can be considered realistic, H/T 2° KIEs have been computed by substituting the protium atoms of C7 and C6 of dUMP by tritium. Calculations have been performed at the AM1/MM and M06-2X/MM level from the geometries of reactants and TSs generated on the exploration of the 1D PMFs in an attempt to explore differences that can be expected if two different mechanisms were possible. It must be kept in mind that when exploring the reaction in terms of 2D PMFs, essentially the same mechanism is obtaining whatever structure is employed as the starting point. The results, shown in Table 2, allow obtaining interesting conclusions that are in agreement with the evaluation of average geometries performed above. The change of the hybridization state of the hydride donor and acceptor carbon atoms (Cd and Ca) and the carbon atom involved in the C6(dUMP)–S(Cys146) breaking bond (C6) along the reaction coordinate, computed as previously proposed by Pu et al.,48 are plotted in Fig. 6 to better understand this analysis.| Mechanism | Method | H/T C6-2° KIE | H/T C7-2° KIE | 18O/16O 2° KIE | 13C/12C 1° KIE |
|---|---|---|---|---|---|
| Concerted | AM1 | 1.011 ± 0.011 | H′: 1.038 ± 0.014 | 1.005 ± 0.0029 | 1.0011 ± 0.0003 |
| H′′: 0.925 ± 0.005 | |||||
| M06-2X | 1.029 | H′: 1.044 | 1.0035 | 1.0108 | |
| H′′: 0.947 | |||||
| Step-wise | AM1 | 0.930 ± 0.008 | H′: 0.977 ± 0.006 | 1.0051 ± 0.0024 | 0.9999 ± 0.0005 |
| H′′: 0.996 ± 0.019 | |||||
| M06-2X | 0.986 | H′: 1.091 | 1.0095 | 1.0021 | |
| H′′: 1.100 |
 | ||
| Fig. 6 Evolution of the hybridization states of the hydride donor carbon (Cd) and hydride acceptor carbon (Ca), in dashed lines, and the carbon atom involved in the C6(dUMP)–S(Cys146) breaking bond (solid lines) along the reaction coordinate (in Å). Results from stepwise (A) and concerted (B) mechanisms derived from the corresponding 1D PMFs performed at 303 K. The hybridization value of 2 on the carbon atom corresponds to a sp2 molecule, while a value of 3 represents a sp3 hybridization. Positions of the TSs are indicated as a dotted vertical blue lines. | ||
Keeping in mind that hybridization of C6 from reactants to products goes from sp3 to sp2, a normal 2° H/T KIE should be expected for substitution of protium by tritium on the H atom of C6, if the C6–S bond was indeed broken during the chemical step. The 2° H/T KIE results on C6, calculated from the structures of the concerted mechanism (1D PMF from 1TLS structure), render no effects at the AM1/MM level, within the standard deviations uncertainty (1.011 ± 0.011). When computed at M06-2X/MM, a higher effect is obtained (1.029). The limit that we could obtain if the C–S bond was completely broken, computed as equilibrium isotope effects (EIE) at the AM1/MM level, is a small normal effect (1.095 ± 0.011). As shown in Fig. 6B, the change in the hybridization state of C6 between reactants and TS is very small (from 3.0 to 2.7). Thus, the computed KIE is in agreement with the other results indicating an early TS on the C–S cleavage coordinate. When the calculations are done with the structures derived from the step-wise mechanism (from 2TSC structure), the result is a small inverse effect (0.930 ± 0.008 and 0.986 at AM1/MM and M06-2X levels, respectively). In this case, the change in the hybridization state of C6 from reactants to TS is even smaller than in a previous case (from 3.0 to 2.8), as shown in Fig. 6A. Then, arguments based on evolution of the hybridization of the C6 atom in the step-wise mechanism, which would contribute to obtaining a value larger than unity, are not enough to rationalize the observed 2° H/T KIEs. The inverse effect could be explained keeping in mind the character of the C5–C6 bond between double and single, as indicated above, and confirmed when computing the EIE (0.951 ± 0.010 and 0.963 at AM1/MM and M06-2X levels, respectively) taking the structures of reactants and the intermediate (the product of the 1D PMF generated from the 2TSC structure). The H atom on C6 would be tighter in the TS than in the reactant state. Comparison of the 2° H/T KIE values with the experimentally measured normal 2° KIE (1.104 ± 0.004) on kcat/KM suggest that the concerted mechanism would be likely the one preferentially taking place in the active site of TSases, as also suggested from the lower barrier calculated for the concerted mechanism (Fig. 5). Any contribution of a stepwise mechanism would render inverse effects.
The a priori expected values of H/T 2° KIEs on the C7 position, whatever mechanism was considered, should be inverse, i.e., lower than unity, keeping in mind that now hybridization on C7 is changing from sp2 to sp3 as the hydride is transferred from Cd of folate to Ca of dUMP. Interestingly, the results reported in Table 2 suggest that, in the case of the concerted mechanism, this is only true for one of the hydrogens at C7 (H′′), but not the other one (H′) for which a small but normal effect is obtained (1.038 ± 0.014 and 1.044 from AM1/MM and M06-2X/MM calculations, respectively). Consequently, rationalization of the H/T 2° KIE values based just on the reaction center rehybridization is, again, not sufficient to explain the different behavior of the two H atoms of the C7. Analysis of the average structures (see Fig. 3 and Table 1) suggests a possible interaction between the H′ atom of C7 and O4 in reactant states that would be slightly weaker in the TS, then justifying a normal KIE. A similar behaviour is observed in both mechanisms. On the contrary, the distance between the H′ atom and the oxygen atom of a water molecule (WAT1 in Fig. 3) increases from the reactant state to TS in the concerted mechanism, a behaviour that is not detected in the step-wise mechanism. Moreover, WAT1 appears to be more activated in the concerted mechanism than in the stepwise mechanisms, due to the direct interaction with Glu58 in the former (see Fig. 3). Then, we can conclude that the two hydrogen atoms do not have the same environment, one of them (the one rendering normal 2° KIE) being more constraining in reactants than in TS. Nevertheless, the average between the KIEs measured for H′ and H′′ of C7 renders an overall inverse value in both mechanisms (0.982 and 0.987 for concerted and step-wise mechanisms, respectively) at the AM1/MM level. The average of KIEs computed at the M06-2X level are slightly inverse for the concerted mechanism (0.995) and normal for the stepwise mechanism (1.095). It is important to note that a better quantitative estimation of 2° KIEs could be obtained if quantum tunnelling corrections where included in our calculations. The values calculated here without tunnelling correction underestimate the KIEs, although they do predict their trend. This was demonstrated by Garcia-Viloca et al., in a study of hydride transfer reaction catalysed by Escherichia coli dihydrofolate reductase, EcDHFR, where 2° H/D KIEs of 1.13 and 1.03 were obtained with and without multidimensional tunnelling contributions, respectively.49 Nevertheless, as stated by Pu et al., a quasiclassical estimation of 2° KIEs, excluding tunnelling corrections, can be more informative for reflecting structural changes, such as rehybridization, at the TS.48
Heavy atom KIEs have been computed at O4 and C6 positions. The results show no measurable difference between the two mechanisms when substituting 16O4 oxygen by its 18O4 isotope, but different trends are obtained on KIEs for 12C6 to 13C6 substitution when computed between reactants and TS in a concerted and step-wise mechanism: a normal effect in the concerted mechanism and no effect in the step-wise one. As observed in Fig. 6, a larger change in hybridization on C6 from reactants to TS is observed for the concerted mechanism (Fig. 6B) than in the step-wise mechanism (Fig. 6A) since the C6–S bond is elongated in the former and no change is detected in the step-wise TS.
Finally, the analysis of the hybridization states of the hydride donor and acceptor (see Fig. 6) shows how the donor carbon atom presents a smaller change from reactants to TS than the acceptor carbon atom in both of the explored mechanisms. Then, as previously computed by Pu et al. in the EcDHFR hydride transfer catalysed reaction, the donor carbon at TS resembles the reactant state more than the product state.48 In the current study the hybridization states of the acceptor carbon would be in between the reactant and product values, especially when analysing the concerted mechanism. Anyhow, a non-perfect synchronization of the two reaction centers of the hydride transfer has been obtained in TSase, as in the EcDHFR.
Conclusions
The reduction of an exocyclic methylene intermediate by hydride transfer from the C6 position of H4folate to form dTMP and H2folate is the rate determining step (r.d.s.) of the TSase catalytic cycle.1 One of the main questions still debated in the literature is whether the breaking of the thioether bond between the C6 of dUMP and Cys146 (in E. coli) takes place concertedly with the hydride transfer or not. Based on the generation of free energy surfaces computed in terms of QM/MM 1D-PMF and 2D-PMFs, the present paper reports a theoretical study focused on addressing this question. The use of two different initial X-ray structures (with PDB codes 2TSC and 1TLS) in our simulations has allowed exploring different conformational spaces and the existence of chemical paths with different reactivities. The results derived from the 1D PMFs, computed as a function of coordinates associated exclusively to the hydride transfer, showed that different scenarios, not only with different reactivity but also with different timing of the C–S breaking bond and the hydride transfer, can be obtained. Thus, while the TSs on both 1D PMFs described the advanced hydride transfer in an almost equivalent stage, the behaviour of the C–S bond along the profile is significantly different. The C–S bond is slightly elongated in the TS of the 1D PMF obtained from 1TLS structure, and completely broken in products. This is a concerted and asynchronous mechanism. On the contrary, the 1D PMF obtained from 2TSC structure arrived to product structures where the hydride transfer leads to an enolate without cleavage of the C–S bond, i.e., a step-wise mechanism. Further analysis of the results suggests that this intermediate is stabilized by hydrogen bond interactions of the carbonyl O4 atom of dUMP with three water molecules and Asn177. Then, this bond is polarized and the transfer of the hydride to the methylene group does not significantly alter the C5–C6 bond and the C6–S bond is not broken. In the case of the concerted mechanism, the carbonyl bond on C4 and the C4–C5 bonds are unaltered but a double bond is clearly formed between C5 and C6 atoms. Moreover, the different behaviour of the S–C bond along the two PMFs is also explained based on interatomic distances established around this sulphur center: the reaction profile in which the hydride transfer and the C6–S breaking bond take place in a concerted manner presents an environment that favours polarization of this bond. In particular, the sulphur atom of the cysteine interacts with Arg166 and Tyr94 through two water molecules. In the case of the step-wise mechanisms, both Arg166 and Tyr94 are moved away and only one water molecule appears at the hydrogen bond distance from the C6–S bond. A better polarization of the C6–S bond appears to contribute to the breaking of the bond between the substrate and enzyme. To test whether the outcome of the 1D PMF is meaningful we generated 2D PMFs, using not only the antisymmetric combination of distances describing the advancement of the hydride but the C–S distance. The intermediate located in one of the 1D PMFs is just a shoulder in its corresponding 2D PMFs with a low statistical relevance in the kinetics of this enzymatic step. The two free energy surfaces (generated from the two mentioned initial X-ray structures) describe an asynchronous but a concerted process and the fact that a step-wise mechanism was obtained in one of the 1D PMF was due to the use of an incomplete number of coordinates required to reproduce the process. Nevertheless, we must keep in mind that certain conformations of the active site could provoke different behaviours in the C–S labile bond. Corrections of the 2D PMF at M06-2X/MM show that the protein conformations that favour the concerted mechanism presented a significantly lower free energy barrier.H/T 2° KIEs were computed by substituting the hydrogen atom of C6 by tritium from the geometries of reactants and TSs generated on the exploration of the 1D PMFs. The results derived from the structures of the concerted mechanism render a small normal effect on the 2° KIE at the C6 position (1.011 ± 0.011 at AM1/MM and 1.029 at M06-2X/MM). It is critical to note that an inverse effect (0.930 ± 0.008) is computed from structures of the step-wise mechanism. Keeping in mind that the limit that we could obtain if the C–S bond was completely broken, computed as equilibrium isotope effects (EIE), is a small normal effect (1.095 ± 0.011 at AM1/MM level), our results would be in agreement with a very asynchronous but concerted mechanism. Our results appear to be slightly underestimated by comparison with the published experimental measurements (Exp 2° KIE = 1.104 ± 0.004),12 although showing the same trend. The inverse effect obtained from structures of the step-wise mechanism can be explained keeping in mind the character of the C5–C6 bond, between double and single that is developed in the TS. The computed values of H/T 2° KIEs on the C7 position for both mechanisms were, a priori, unexpected. The effect is inverse for both hydrogen atoms in the stepwise mechanisms but substitution on one of the C7 hydrogen atoms renders normal 2° KIEs in the concerted mechanism. This result can be explained using not only arguments based on hybridization of the carbon atom but also on the interactions that are established between this H atom of C7 and a conserved water molecule of the active site in reactants and TS. Glu58 appears to be responsible for keeping the orientation of the water molecule in the reactant state that would be the origin of a tighter vibrational mode associated to this H atom in reactants and, consequently, rendering a normal 2° KIE. Nevertheless, the other hydrogen at C7 presented inverse 2° KIEs and, when calculating the average of the 2° KIE on the two H atoms, the result is an inverse 2° KIE. It is important to mention that without tunneling correction the predicted KIEs are probably deflated (as in ref. 48), but their trend is likely to be correct. Finally, heavy atom KIEs, computed for 12C to 13C substitution at the C6 position predict a normal effect (KIE = 1.011 at AM1/MM and 1.0108 at M06-2X/MM) in the concerted mechanism and no effect (KIE = 1) in the step-wise one. This result is not surprising as a change in hybridization has been observed from reactants to the concerted TS (the C6–S is slightly elongated in the TS), while no change has affected the C6 atom in the step-wise TS.
The above findings confirm that the r.d.s. hydride transfer in the TSase catalysed reaction takes place mainly by means of a concerted mechanism, and further demonstrate the labile character of the substrate–enzyme covalent bond between the C6 of the nucleotide and the sulphur atom of a conserved cysteine residue. From a computational point of view, our calculations stress the need of performing enough conformational sampling in the study of systems with a large amount of degrees of freedom. The use of a reduced number of coordinates to describe the reaction coordinate can render biased results that would not explain the full variety of possible reaction paths in enzyme-catalysed and other complex reactions. Single-molecule enzymology, among other tools, provides means for understanding the dynamic disorder of enzymes.50 Such examination is a requisite when defining the mechanism of action of natural biocatalysts and the implementation of this knowledge into the development of new biocatalysts or, as in the present study, the finding of possible reaction paths. In this case, our results may have implications in chemotherapeutic and antibiotic drugs designs. Theoretically predicted mechanisms can be supported by the estimation of 2° KIEs that can render information of the rate limiting transition state and, when computed as average values, they can be directly compared with experimental data.
Acknowledgements
This work was supported by the Spanish Ministerio de Economía y Competitividad for project CTQ2012-36253-C03, Universitat Jaume I (project P1 1B2014-26), Generalitat Valenciana (PROMETEOII/2014/022 and ACOMP/2014/277 projects), Polish National Center for Science (NCN) (grant 2011/02/A/ ST4/00246, 2012–2017), the Polish Ministry of Science and Higher Education (“Iuventus Plus” program project no. 0478/IP3/2015/73, 2015–2016) and the USA National Institute of Health (ref. NIH R01 GM065368). Authors acknowledge computational resources from the Servei d'Informàtica of Universitat Jaume I.References
- N. Agrawal, B. Y. Hong, C. Mihai and A. Kohen, Biochemistry, 2004, 43, 1998–2006 CrossRef CAS PubMed.
- N. Kanaan, S. Martí and V. Moliner, Biochemistry, 2007, 46, 3704–3713 CrossRef CAS PubMed.
- C. W. Carreras and S. V. Santi, Annu. Rev. Biochem., 1995, 64, 721–762 CrossRef CAS PubMed.
- H. T. Spencer, J. E. Villafranca and J. R. Appleman, Biochemistry, 1997, 36, 4212–4222 CrossRef CAS PubMed.
- W. Huang and D. V. Santi, Biochemistry, 1997, 36, 1869–1973 CrossRef CAS PubMed.
- T. W. Bruice and D. V. Santi, in Enzyme Mechanism from Isotope Effects, CRC Press, Boca Raton, 1991; pp. 457–479 Search PubMed.
- N. Kanaan, S. Martí, V. Moliner and A. Kohen, J. Phys. Chem. A, 2009, 113, 2176–2182 CrossRef CAS PubMed.
- H. Eyring and A. E. Stearn, Chem. Rev., 1939, 24, 253–270 CrossRef CAS.
- D. G. Truhlar, W. L. Hase and J. T. Hynes, J. Phys. Chem., 1983, 87, 2664–2682 CrossRef CAS Erratum: 1983, 87, 5523.
- N. Kanaan, S. Ferrer, S. Martí, M. Garcia-Viloca, A. Kohen and V. Moliner, J. Am. Chem. Soc., 2011, 133, 6692–6702 CrossRef CAS PubMed.
- Z. Wang, S. Ferrer, V. Moliner and A. Kohen, Biochemistry, 2013, 52, 2348–2358 CrossRef CAS PubMed.
- Z. Islam, T. S. Strutzenberg, I. Gurevic and A. Kohen, J. Am. Chem. Soc., 2014, 136, 9850–9853 CrossRef CAS PubMed.
- J. S. Finer-Moore, W. R. Montfort and R. M. Stroud, Biochemistry, 1990, 29, 6977–6986 CrossRef CAS.
- W. R. Montfort, K. M. Perry, E. B. Fauman, J. S. Finer-Moore, G. F. Maley, L. Hardy, F. Maley and R. M. Stroud, Biochemistry, 1990, 29, 6964–6977 CrossRef CAS.
- K. M. Perry, E. B. Fauman, J. S. Finer-Moore, W. R. Montfort, G. F. Maley, F. Maley and R. M. Stroud, Proteins: Struct., Funct., Genet., 1990, 8, 315–333 CrossRef CAS PubMed.
- D. C. Hyatt, F. Maley and W. R. Monfort, Biochemistry, 1997, 36, 4585–4594 CrossRef CAS PubMed.
- M. J. Field, M. Albe, C. Bret, F. Proust-De Martin and A. Thomas, J. Comput. Chem., 2000, 21, 1088–1100 CrossRef CAS.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins, 2005, 61, 704–721 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins, 2008, 73, 765–783 CrossRef CAS PubMed.
- M. J. Field, P. A. Bash and M. Karplus, J. Comput. Chem., 1990, 11, 700–733 CrossRef CAS PubMed.
- M. J. S. Dewar, E. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- J. S. Finer-Moore, D. V. Santi and R. M. Stroud, Biochemistry, 2003, 42, 248–256 CrossRef CAS.
- J. C. Corchado, E. L. Coitiño, Y. Chuang, P. L. Fast and D. G. Truhlar, J. Phys. Chem. A, 1998, 102, 2424–2438 CrossRef CAS.
- K. A. Nguyen, I. Rossi and D. G. Truhlar, J. Chem. Phys., 1995, 103, 5522–5530 CrossRef PubMed.
- Y. Y. Chuang, J. C. Corchado and D. G. Truhlar, J. Phys. Chem. A, 1999, 103, 1140–1149 CrossRef CAS.
- (a) R. J. Renka, SIAM J. Sci. Stat. Comput., 1987, 8, 393–415 CrossRef; (b) R. J. Renka, ACM T. Math. Software, 1993, 19, 81–94 CrossRef.
- J. J. Ruiz-Pernía, E. Silla, I. Tuñón, S. Martí and V. Moliner, J. Phys. Chem. B, 2004, 108, 8427–8433 CrossRef.
- M. Roca, V. Moliner, J. J. Ruiz-Pernía, E. Silla and I. Tuñón, J. Phys. Chem. A, 2006, 110, 503–509 CrossRef CAS PubMed.
- J. J. Ruiz-Pernía, E. Silla, E. I. Tuñón and S. Martí, J. Phys. Chem. B, 2006, 110, 17663–17670 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- B. J. Lynch, Y. Zhao and D. G. Truhlar, Effectiveness of Diffuse Basis Functions for Calculating Relative Energies by Density Functional Theory, J. Phys. Chem. A, 2003, 107, 1384–1388 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- G. D. Ruggiero, S. J. Guy, S. Martí, V. Moliner and I. H. Williams, J. Phys. Org. Chem., 2004, 17, 592–601 CrossRef CAS PubMed.
- (a) M. Garcia-Viloca, T. D. Poulsen, D. G. Truhlar and J. Gao, Protein Sci., 2004, 13, 2341–2354 CrossRef CAS PubMed; (b) G. Henkelman, M. X. LaBute, C. S. Tung, P. W. Fenimore and B. H. McMahon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15347–15351 CrossRef CAS PubMed; (c) Y. Cheng, Y. Zhang and J. A. McCammon, J. Am. Chem. Soc., 2005, 127, 1553–1562 CrossRef CAS PubMed; (d) M. Montenegro, M. Garcia-Viloca, J. M. Lluch and A. González-Lafont, Phys. Chem. Chem. Phys., 2011, 13, 530–539 RSC.
- D. R. Glowacki, J. N. Harvey and A. J. Mulholland, Nat. Chem., 2012, 4, 169–176 CrossRef CAS PubMed.
- K. Francis and A. Kohen, Curr. Opin. Chem. Biol., 2014, 21, 19–24 CrossRef CAS PubMed.
- H. P. Lu, Chem. Soc. Rev., 2014, 43, 1118–1143 RSC.
- Q. Xue and E. S. Yeung, Nature, 1995, 373, 681–683 CrossRef CAS PubMed.
- M. J. Reddish, H. L. Peng, H. Deng, K. S. Panwar, R. Callender and R. B. Dyer, J. Phys. Chem. B, 2014, 118, 10854–10862 CrossRef CAS PubMed.
- J. Pu, J. Gao and D. G. Truhlar, Chem. Rev., 2006, 106, 3140–3169 CrossRef CAS PubMed.
- A. V. Pisliakov, J. Cao, S. C. L. Kamerlin and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17359–17364 CrossRef CAS PubMed.
- A. Turner, V. Moliner and I. H. Williams, Phys. Chem. Chem. Phys., 1999, 1, 1323–1331 RSC.
- S. Ferrer, I. Tuñón, S. Martí, V. Moliner, M. García-Viloca, A. González-Lafont and J. M. Lluch, J. Am. Chem. Soc., 2006, 128, 16851–16863 CrossRef CAS PubMed.
- K. Świderek, I. Tuñón, S. Martí and V. Moliner, ACS Catal., 2015, 5, 1172–1185 CrossRef PubMed.
- J. Pu, S. Ma, M. Garcia-Viloca, J. Gao, D. G. Truhlar and A. Kohen, J. Am. Chem. Soc., 2005, 127, 14879–14886 CrossRef CAS PubMed.
- M. Garcia-Viloca, D. G. Truhlar and J. Gao, Biochemistry, 2003, 42, 13558–13575 CrossRef CAS PubMed.
- S. J. Klippenstein, V. S. Pande and D. G. Truhlar, J. Am. Chem. Soc., 2014, 136, 528–546 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |