
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
XPS of guanidinium ionic liquids: a comparison of charge distribution in nitrogenous cations†
Ana R.
Santos
,
Rebecca K.
Blundell
and
Peter
Licence
*
School of Chemistry, The University of Nottingham, Nottingham NG7 2RD, UK. E-mail: peter.licence@nottingham.ac.uk; Tel: +44 (0)115 8466176
First published on 2nd April 2015
Abstract
Herein, we investigate the first X-ray photoelectron spectroscopy (XPS) data for a range of functionalised guanidinium based systems that are commonly employed in the dissolution of biomolecules. We define a peak fitting model which allows the direct comparison to more common cation sets including dialkyl-imidazolium, pyrrolidinium, and quaternary ammonium based systems. The measured binding energies (BEs) of the N 1s and C 1s components are presented and notable variations discussed. These data show a large difference between measured binding energies for the Ncation 1s when compared to other families of ionic liquids. These results suggest a weaker anion/cation interaction thus the anion is more able to interact with a solid matrix, i.e. keratin, silk, chitin, collagen, cellulose, and become more active in dissolution.
Introduction
In recent years, ionic liquids (ILs) have gained applications as electrolyte materials,1,2 reaction solvents,3–5 and extraction solvents6–8 attributed to their unique properties, such as excellent thermal stability, negligible vapour pressure, high conductivity stability and designable structures. ILs can be designed to be environmentally friendly, with large potential benefits for sustainable chemistry.9Although, research mainly concentrates on imidazolium, quaternary ammonium, pyrrolidinium, alkylpyridinium, and tetraalkylphosphonium ILs, guanidinium-based ILs have found many useful applications in numerous fields such as in dye sensitized solar cells, biological and molecular recognition, and as energetic materials.10–17 Moreover, the high degree of delocalisation of the cationic centre, the tuneability of the alkyl functionality of the nitrogen atoms, and other characteristic properties are making the synthesis and the application of guanidine salts increasingly popular.18,19 It is demonstrated that guanidinium ILs are environmental friendly solvents which have good designability, and more potential than traditional imidazolium ILs.20
Remarkably, guanidinium-based ILs have been showing clear signs of stabilisation and sometimes destabilisation of peptides and proteins, as well dissolution of biomolecules of interest such as cellulose. This shows the incredible potential of these molecules to be designed as task-specific solvents, which increases the need to recognise the main interactions between solute and the IL solvent system, but also how these ILs interact on a molecular level, hence the effect upon inter-ion interactions must be explored.
Binding energies obtained from X-ray photoelectron spectroscopy (XPS) from ionic liquid solutions can help identifying the main interactions occurring between the ionic liquid solvent and the solute and can be used to investigate the nature of the molecular interactions within ions, providing both elemental and chemical information. XPS is an established and commonly applied surface analysis Ultra High Vacuum (UHV) technique which is exceptionally suitable for negligible vapour pressure compounds such as ionic liquids.21 XPS also provides information on the impact of structural changes on the electronic environment and communication between ions, resulting in a shift in the measured binding energy, as well as the determination of chemical composition and purity.22,23
XPS has been a valuable technique in a wide range of different fields, providing vital information in areas such as bulk and surface composition of ILs systems,24,25in situ reaction monitoring,26 and cation–anion interactions.27 Not surprisingly, imidazolium and pyrrolidinium cation–anion interactions have been the emphasis of research, where the focus of interest has been on the effect of the anion upon the electronic environment of the cation, but interestingly there are fewer studies into the nature of the cation and its influence.22,23,27 These results were correlated to NMR spectroscopy, theoretical calculations and Kamlet–Taft parameters aiming for a better understanding of ionic liquid properties.22
It has been reported that cyclic guanidinium cations are similar, in terms of their performance and robustness, to more-traditional families of ionic liquids when exposed to energetic radiolytic sources,18 here we explore if guanidinium ILs will be stable to prolonged exposure to X-ray irradiation and/or argon etching. To the best of our knowledge, guanidinium-based ionic liquids have not yet been studied by XPS, hence a detailed investigation into how cationic structure influences cationic N 1s binding energies and the comparison with other nitrogen-based cations, can be established. The high degree of charge delocalisation of the cationic core may be used as an indicator of cation–anion interaction. This interaction could be significantly weaker when compared with other nitrogen counterparts, which could be an explanation for the ability of the anion to interact with solute molecules. XPS studies upon the cation–anion interactions can help raise some light onto this assumption. The charge on pyrrolidinium ILs is localised on the single nitrogen atom, whereas in the imidazolium ILs is distributed across the two nitrogen atoms. This results in a higher measured N 1s binding energy on the pyrrolidinium cation (when the anion is the same), evidencing a stronger interaction with the anion.23 These results could be indispensable for the development of appropriate force fields for molecular simulations of guanidinium ionic liquids, which are crucial for molecular understanding of ILs properties and interactions with other molecules.28–31 One recent theoretical publication even predicted the distilability of some guanidinium carboxylates using a combination of molecular dynamics and ab initio approaches.32 Furthermore, XPS has also been used to monitor the electronic environment of the constituent atoms for a binary IL mixture by varying the composition of the anionic components, highlighting how IL mixtures can be used to tune the electronic environment of their cation–anion system.33
Research onto ILs based on the guanidinium cation may be of particular importance, as they can offer interesting physicochemical properties, in comparison to other nitrogen based relatives, given by the high degree of delocalisation of the cationic core. The work described herein utilises XPS to probe cation–anion interactions for guanidinium-based ILs and how the cationic structure influences cationic N 1s binding energies. Furthermore, comparisons are made to other nitrogen-based ionic liquids, as a new potential system for binary IL mixtures can be acknowledged.
Experimental
Materials
All ionic liquids investigated in this study were prepared in our laboratory using known literature methods.17 The structures of the compounds investigated are shown in Table 1. Unless otherwise stated, chemical precursors were obtained from commercial suppliers and used without further purification. All compounds were dried in vacuo (p ≤ 10−2 mbar) at 50 °C and stored under argon prior to analysis by 1H (400 MHz), 13C (101 MHz), 31P (162 MHz) and 19F (377 MHz) NMR (recorded at room temperature on a Bruker AV 400); IR (recorded as liquid films on a Perkin Elmer 1600 FT spectrometer); ESI-MS (Bruker MicroTOF 61 spectrometer), XPS (Kratos Axis Ultra spectrometer). CHN microanalysis (Exeter CE-440 Elemental Analyser) and differential scanning calorimetry, DSC (TA Instruments DSC Q2000). Full data including synthetic methods and XPS spectra with deconstruction models for all materials investigated appear in the ESI.†| Abbreviation | Structure | Name |
|---|---|---|
| [(C8C1)2dmg]+ |
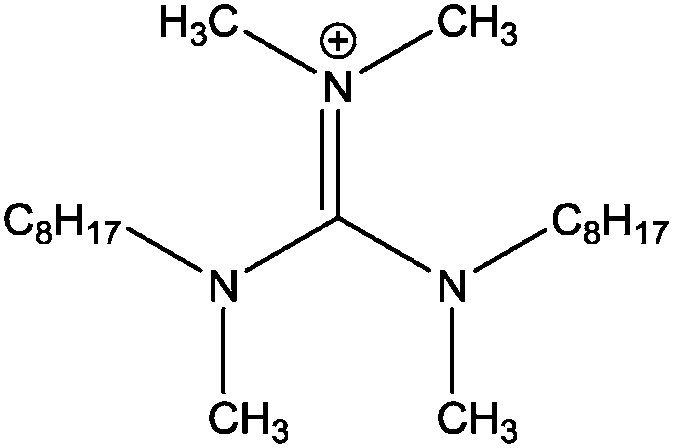
|
Bis-(methyloctylamino)-dimethylguanidinium |
| [(C4C1)2dmg]+ |
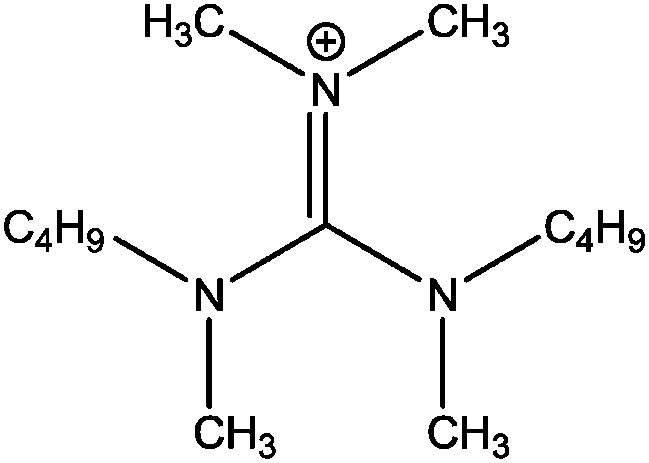
|
Bis-(butylmethylamino)-dimethylguanidinium |
| [(C4C4)2dmg]+ |
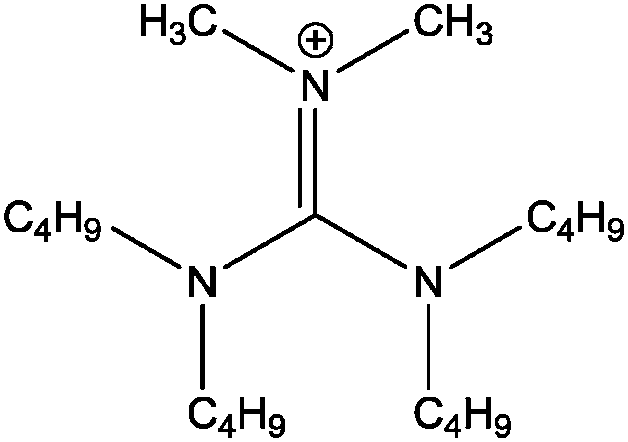
|
Bis-(dibutylamino)-dimethylguanidinium |
| [BF4]− |
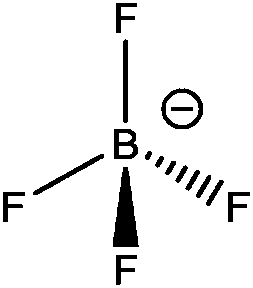
|
Tetrafluoroborate |
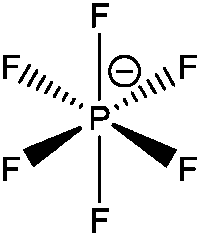
| [PF6]− |
|
Hexafluorophosphate |
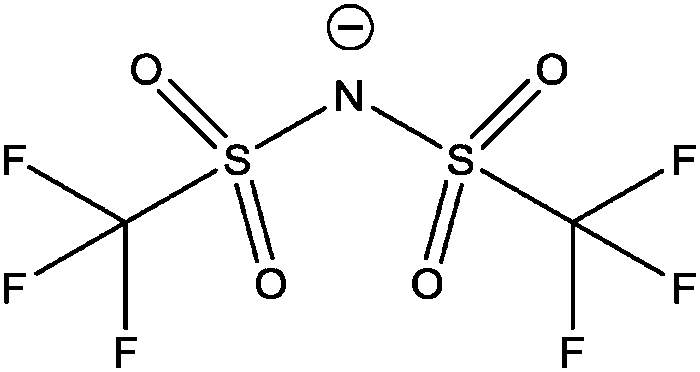
| [NTf2]− |
|
Bis(trifluoromethanesulfonyl)imide |
XPS data collection
XP spectra were recorded using a Kratos Axis Ultra spectrometer employing a focused, monochromated Al Kα source (hν = 1486.6 eV), hybrid (magnetic/electrostatic) optics, concentric hemispherical analyser, a multi-channel plate and delay line detector (DLD) with an X-ray incident angle of 30° and a collection angle of 0° (both relative to the surface normal). X-ray source was operated at 10 mA emission current and 12 kV anode potential. All spectra were recorded using an entrance aperture of 300 × 700 μm with pass energy of 80 eV for survey scans and 20 eV for high resolution scans. The instrument sensitivity was 7.5 × 105 counts s−1 when measuring the Ag 3d5/2 photoemission peak for a clean Ag sample recorded at pass energy of 20 eV and 450 W emission power. Ag 3d5/2 full width at half maximum (FWHM) was 0.55 eV for the same instrument settings. Binding energy calibration was made using Au 4f7/2 (83.96 eV), Ag 3d5/2 (368.21 eV) and Cu 2p3/2 (932.62 eV). Charge neutralisation (used for solid samples only) was applied using a standard Kratos charge neutraliser consisting of a filament, coaxial with the electrostatic and magnetic transfer lenses, and a balance plate which creates a potential gradient between the neutraliser and sample. Charge neutralisation was applied at 1.9 A filament current and 3.3 V balance plate voltage. Sample stubs were earthed via the instrument stage using a standard BNC connector.Room temperature ionic liquid samples were prepared by placing a small drop (≈10 mg) of ionic liquid onto a stainless steel multi-sample bar; solid samples were fixed to the bar using double-sided adhesive tape. All samples were pre-pumped in a preparative chamber to pressures lower than 1 × 10−6 mbar before transfer into the main analytical chamber.
When required, surface contaminants such as oxygen and silicon were removed via argon ion bombardment using a Kratos Minibeam III ion gun using a sputter energy of 4 kV for five minutes. Solid samples that required argon bombardment were prepared by placing a small amount of solid in a standard stainless steel or gold-coated powder stub, heating the sample until melted and a uniform film of material was produced, then allowing it cool and solidify, followed by argon bombardment. Surface segregated oxygen and silicon containing impurities were removed for each the compounds investigated, however irreversible damage to the sample occurred for compounds when the alkyl chain was short (i.e. n < 8).
XPS data analysis
All XPS data were analysed using the CASAXPS software. Relative sensitivity factors (RSF) were taken from the Kratos Library (RSF of F 1s = 1.000) and used to determine relative atomic percentages of the most intense photoelectron peak for each element, from high-resolution spectra. The estimated error in the relative atomic concentrations of each element was taken to be between 10 and 20%.34 Peak areas were measured after performing a two-point linear or Shirley background subtraction. Peaks were fitted using GL(30) lineshapes (70% Gaussian, 30% Lorentzian). Peak fitting areas for each element take into account spin orbit coupling and/or stoichiometry ratios. The full width at half maximum (FWHM) for all components in the C 1s region are constrained to be between 0.8 and 1.2 eV. For compounds with a long alkyl chain (n ≥ 8) charge-referencing was achieved by setting the experimentally determined binding energy of the aliphatic carbon component (Caliphatic) equal to 285.0 eV.23,34 For compounds with a short alkyl chain (n < 8) charge-referencing was achieved by setting the measured binding energy of the cationic nitrogen photoemission peak equal to that for its long chain analogue, i.e. Ncation 1s for [(C4C1)2dmg][BF4] was set equal to 400.4 eV (where 400.4 eV is the measured binding energy for Ncation 1s for [(C8C1)2dmg][BF4]). The error in these binding energies has been shown to vary by a maximum of ±0.1 eV across a range of different ionic liquid standards.23,34Results and discussion
Sample purity and argon etching studies
Attaining high purity of ionic liquids is essential for the accurate characterisation of their physical and chemical properties. XPS is a highly surface sensitive technique and can be used to establish elemental composition by, utilising survey and high resolution scans. Fig. 1 shows the survey scan for the solid [(C8C1)2dmg][BF4] which exhibits all the expected photoelectron and Auger lines. Earlier XPS studies, in the field of ionic liquids, have shown the presence of hydrocarbon and silicon-based impurities in the near-surface region, which cannot be detected by NMR or other bulk sensitive techniques.35–37 Some of the samples investigated in this work exhibited a O 1s and Si 2s/Si 2p signal (see ESI†) and this is likely to be a result of laboratory high-vacuum silicon grease during sample preparation and has no apparent effect on the recorded binding energies.34,35,38 Silicon grease, and other surface contaminants, can be removed by argon bombardment, a description of this procedure is provided within the experimental section. Additionally, there is no evidence of metal-based cations or halides that may be carried over anion metathesis chemistry involved in the preparation of these materials. Experimental stoichiometries, calculated from high resolution spectra, are presented in Table 2.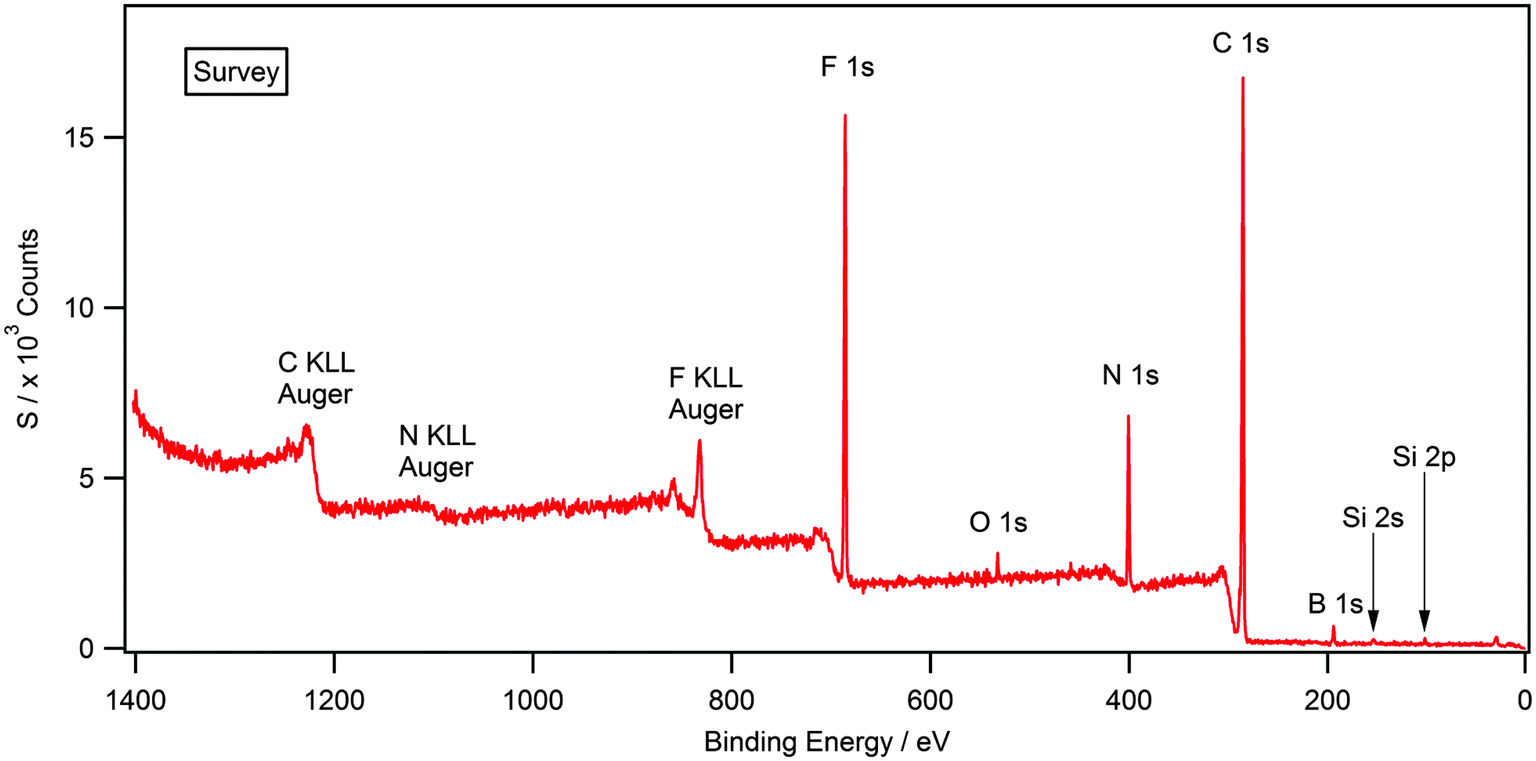 | ||
| Fig. 1 Survey XP spectrum for [(C8C1)2dmg][BF4]. O 1s and Si 2s/2p photoelectron lines are clearly visible indicating surface contamination. | ||
| Compound | Compositiona (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| C 1s | N 1s | F 1s | O 1s | S 2p | P 2p | B 1s | Cl 2p | ||
| a Measured experimental and nominal (in brackets) stoichiometries for the guanidinium compounds studied. Relative sensitivity factors (RSF) taken from the Kratos library. | |||||||||
| RSF35,39 | 0.278 | 0.477 | 1.000 | 0.780 | 0.668 | 0.486 | 0.159 | 0.891 | |
| [(C8C1)2DMG]+ | Cl− | 85.7 (84.0) | 10.8 (12.0) | — | — | — | — | — | 3.5 (4.0) |
| [(C4C1)2DMG]+ | Cl− | 78.6 (76.5) | 16.4 (17.6) | — | — | — | — | — | 5.1 (5.9) |
| [(C4C4)2DMG]+ | Cl− | 83.9 (82.6) | 12.4 (13.0) | — | — | — | — | — | 3.7 (4.3) |
| [(C8C1)2DMG]+ | [BF4]− | 74.5 (72.4) | 9.3 (10.3) | 12.0 (13.8) | — | — | — | 4.3 (3.4) | — |
| [(C4C1)2DMG]+ | [BF4]− | 68.8 (61.9) | 11.2 (14.3) | 14.3 (19.0) | — | — | — | 5.6 (4.8) | — |
| [(C4C4)2DMG]+ | [BF4]− | 72.4 (70.4) | 10.3 (11.1) | 13.5 (14.8) | — | — | — | 3.8 (3.7) | — |
| [(C8C1)2DMG]+ | [PF6]− | 68.9 (67.7) | 8.6 (9.7) | 19.4 (19.4) | — | — | 3.1 (3.2) | — | — |
| [(C4C1)2DMG]+ | [PF6]− | 62.1 (56.5) | 12.6 (13.0) | 22.2 (26.1) | — | — | 3.1 (4.3) | — | — |
| [(C4C4)2DMG]+ | [PF6]− | 69.1 (65.5) | 9.1 (10.3) | 18.6 (20.7) | — | — | 3.2 (3.4) | — | — |
| [(C8C1)2DMG]+ | [NTf2]− | 61.4 (59.0) | 10.0 (10.3) | 14.9 (15.4) | 9.0 (10.3) | 4.7 (5.1) | — | — | — |
| [(C4C1)2DMG]+ | [NTf2]− | 53.7 (48.4) | 12.3 (12.9) | 16.9 (19.4) | 11.6 (12.9) | 5.5 (6.5) | — | — | — |
| [(C4C4)2DMG]+ | [NTf2]− | 59.8 (56.8) | 10.6 (10.8) | 14.8 (16.2) | 10.2 (10.8) | 4.5 (5.4) | — | — | — |
It should be noted here that isolated photoemission lines exhibit errors that are slightly larger than anticipated (and do not lie within the acceptable ±20%), this does not appear to be systematic or linked with any component in either the sample or indeed the synthetic route to the material. Further investigations into the nature of contaminants, i.e. bulk or surface impurities, are described below.
In the particular case of [(C4C1)2dmg][BF4], whereby the experimentally determined stoichiometries for nitrogen and fluorine exceed the error, elemental microanalysis was used to determine the C, H and N content (see ESI†). The CHN microanalysis for [(C4C1)2dmg][BF4] demonstrates the high purity for this compound where C 49.66 (49.54), H 9.49 (9.59), N 13.12 (13.33) theoretical values are shown in brackets. Also, for [(C8C1)2dmg][BF4], C 59.10 (59.01), H 10.91 (10.85), N 9.81 (9.83) was determined. Hence, the results of CHN microanalysis are well within the error for this experimental technique (±0.4%) which strongly suggests that the error observed in percentage quantification by XPS is a result of a surface contamination, which could be very small in terms of mol fraction, rather than bulk contamination.
It has been previously shown that surface contaminants such as oxygen and silicon can be removed via argon ion bombardment.35 A major problem in etching samples which are granular solids is the disturbance of solid particles within the chamber by electrostatic effects. Solid samples that required argon bombardment were initially prepared by placing a small amount of solid in a stainless steel stub, heating the sample until melted, followed by argon bombardment. Fig. 2 (blue) shows the XPS spectra of [(C8C1)2dmg][BF4] after this experiment.
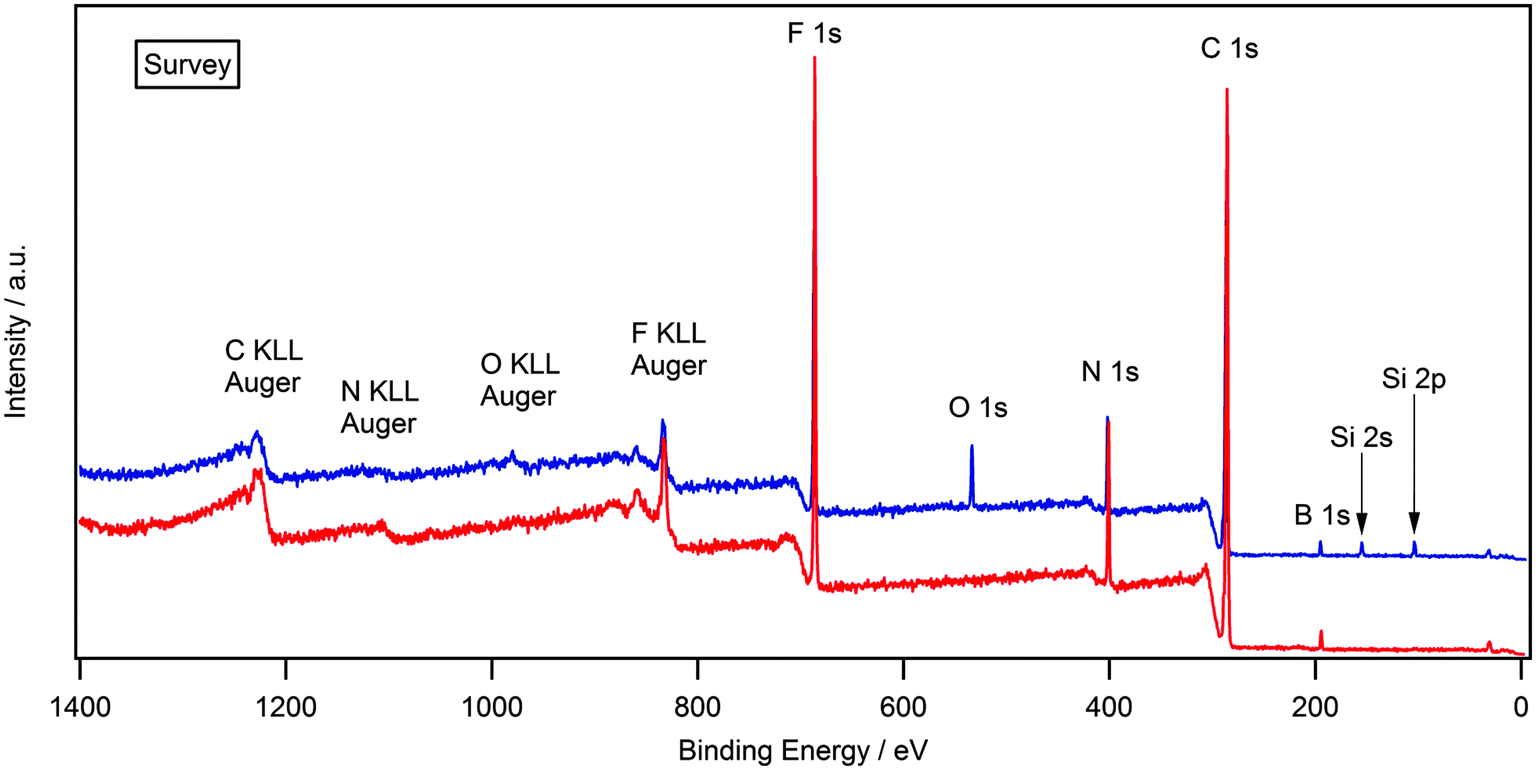 | ||
| Fig. 2 Survey XP spectrum for [(C8C1)2dmg][BF4] after melting, followed by argon bombardment (blue). Survey XP spectrum for [(C8C1)2dmg][BF4] after melting, then cooling, followed by argon bombardment (red). | ||
It is clear to see that the O 1s and Si 2s/2p photoelectron lines are still visible, which demonstrates that argon bombardment has not removed the contaminant. This result can be explained by the nature of the sample and the experiment; by heating the sample to the melt and maintaining this temperature, the mobility of the ions and contaminant is increased, consequently the rate of surface segregation of any contaminant is also increased. Therefore the removal of a surface segregated layer of impurities by argon bombardment is successful, but is quickly replaced by a new layer of segregated contaminant. To examine this possibility, the sample was prepared by placing a small amount of solid in a stainless steel powder stub, heating the sample until melted, then allowing it to cool and form a glass, followed by argon bombardment. Forming a glass instead of a granular solid allows argon etching of the surface without any disturbance of solid particles and also reduces the mobility of ions and contaminant, thus reducing surface segregation between scans. If silicon and oxygen were still present, this would suggest that the impurities were part of the bulk, however if they were removed, it would prove that these were surface contaminants and therefore not representative of the bulk. Fig. 2 (red) shows the XPS spectra of [(C8C1)2dmg][BF4] after this experiment.
Although surface etching displayed that oxygen and silicon can be removed for the compounds with a long alkyl chain (n ≥ 8), irreversible damage to the sample occurred for compounds with a short alkyl chain (n < 8). Fig. 3 illustrates a comparison of N 1s and C 1s high resolution scans for [(C4C1)2dmg][BF4] before and after argon bombardment. The full width at half maximum (FWHM) of the N 1s peak for the [(C4C1)2dmg] series is consistently equal to 1.1 eV when analysed without etching; however, after argon bombardment, a broadening effect is clear to see on the N 1s region, showing clear signs of degradation as the Gaussian–Lorentzian shape is visibly lost with an associated increase of the FWHM. The same behaviour can also be seen in the C 1s region. This damage to the sample prevents the deconstruction of the different carbon environments, which is required to charge reference the Caliphatic to 285.0 eV. In summary, ILs with short alkyl chain (n < 8) are unstable and decompose under argon bombardment, which is consistent with previous results.40
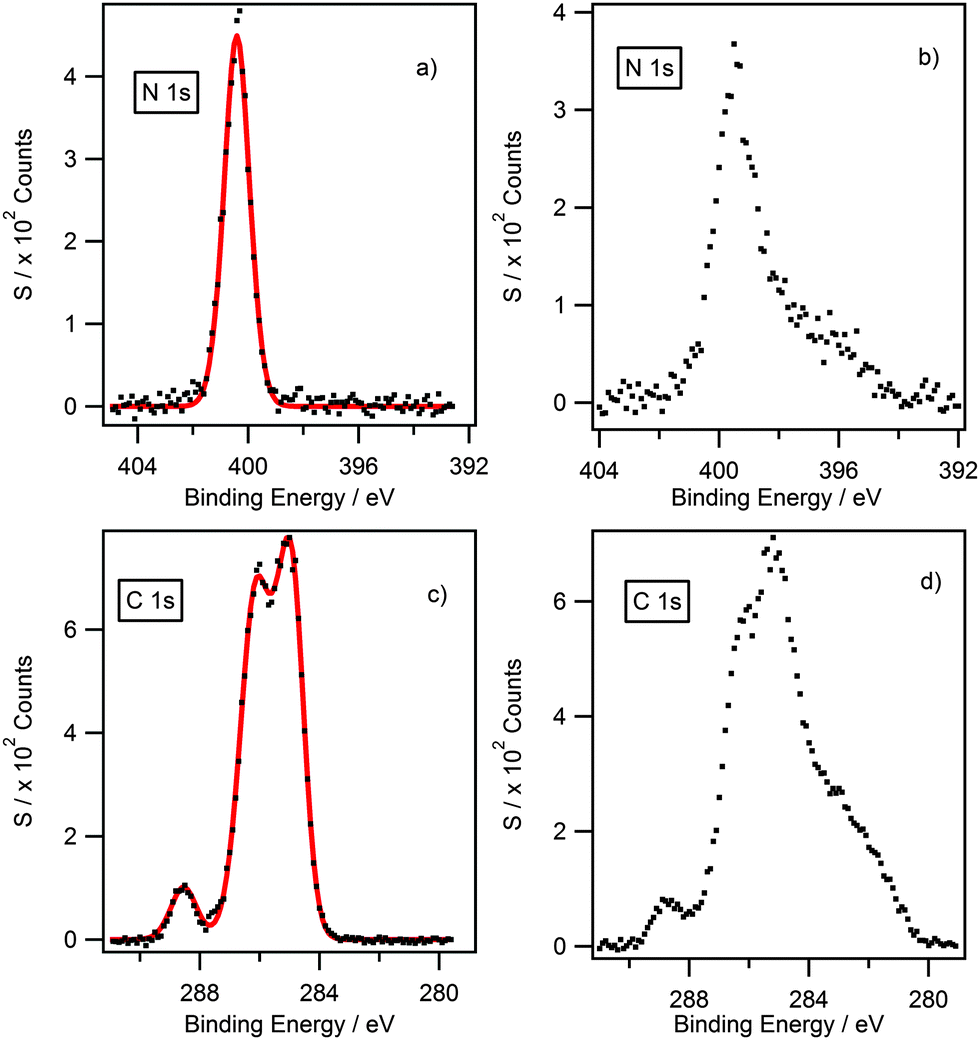 | ||
| Fig. 3 XPS high resolution spectra for the N 1s (a) and (b) and C 1s (c) and (d) of [(C4C1)2dmg][BF4] before (left) and after (right) argon bombardment. C 1s spectra are truncated to 279–290 eV to highlight changes to the lineshape. Spectra showing the full region are shown in the ESI.† | ||
Subsequently, we have shown that heating the sample to the melt, then allowing it to cool prior to argon bombardment, does indeed remove the surface contaminants for compounds with long alkyl chain (n ≥ 8). As a result, experimental values determined by XPS are now within the error of the nominal stoichiometry of ± 20% for compounds with long alkyl chain (n ≥ 8). For shorter alkyl chain (n < 8) compounds, the surface contaminants could not be removed by argon bombardment, nevertheless quantification obtained by CHN microanalysis indicates the silicon and oxygen impurities observed by XPS originate through surface-segregation rather than bulk contamination. This is further reinforced by the argon-etching studies upon the longer-chain analogues (see ESI†).
Development of C 1s fitting model for [(CRCR′)2DMG]X ILs
The binding energies measured for all compounds investigated in this study are displayed in Table 3. In order to extract reliable binding energies, a suitable charge-referencing method must be applied. In the case of ionic liquids with a long alkyl chain (n ≥ 8), charge-referencing can be accomplished by setting the binding energy of the aliphatic carbon component (Caliphatic) equal to an internal reference value, which is 285.0 eV.22,23,34 However, for ionic liquids with shorter alkyl chains, charge referencing is achieved by setting the measured binding energy of Ncation 1s equal to that of their long-chain equivalent.22 To determine that this charge referencing technique is viable a series of short alkyl chain analogues were investigated. The cation size has been reduced to [(C4C1)2dmg]+ and [(C4C4)2dmg]+ to evaluate whether the length of the alkyl chain impacts on cation–anion interactions. A comparison between the binding energies in Table 3 for the same anion, shown that a decrease in alkyl chain length does not influence the electronic environment of the anion. For example, Ncation 1s for [(C4C1)2dmg][BF4] is set to 400.4 eV (where 400.4 eV is the measured binding energy of Ncation 1s for [(C8C1)2dmg][BF4]), which has been charge referenced by setting Caliphatic to 285.0 eV. It must be stressed, however, that the nature of the anion does effect the binding energy of Ncation 1s.22| Compound | Binding energy/eV | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cation | Anion | |||||||||||||
| Cation | Anion | Caliphatic 1s | Cinter 1s | Chetero 1s | Ccore 1s | Ncation 1s | CCF3 1s | Nanion 1s | O 1s | F 1s | S 2p3/2 | P 2p3/2 | B 1s | Cl 2p3/2 |
| [(C8C1)2DMG]+ | Cl− | 285.0 | 285.5 | 286.0 | 288.1 | 400.1 | — | — | — | — | — | — | — | 196.4 |
| [(C4C1)2DMG]+ | Cl− | 284.7 | 285.3 | 285.8 | 288.2 | 400.1 | — | — | — | — | — | — | — | 196.4 |
| [(C4C4)2DMG]+ | Cl− | 284.9 | 285.4 | 286.0 | 288.2 | 400.1 | — | — | — | — | — | — | — | 196.4 |
| [(C8C1)2DMG]+ | [BF4]− | 285.0 | 285.8 | 286.2 | 288.5 | 400.4 | — | — | — | 685.7 | — | — | 194.0 | — |
| [(C4C1)2DMG]+ | [BF4]− | 284.9 | 285.3 | 286.2 | 288.5 | 400.4 | — | — | — | 685.6 | — | — | 193.8 | — |
| [(C4C4)2DMG]+ | [BF4]− | 284.9 | 285.4 | 286.2 | 288.6 | 400.4 | — | — | — | 685.5 | — | — | 193.8 | — |
| [(C8C1)2DMG]+ | [PF6]− | 285.0 | 285.6 | 286.4 | 288.8 | 400.7 | — | — | — | 686.5 | — | 136.3 | — | — |
| [(C4C1)2DMG]+ | [PF6]− | 285.1 | 285.5 | 286.4 | 288.8 | 400.7 | — | — | — | 686.5 | — | 136.5 | — | — |
| [(C4C4)2DMG]+ | [PF6]− | 285.2 | 285.8 | 286.5 | 288.8 | 400.7 | — | — | — | 686.5 | — | 136.4 | — | — |
| [(C8C1)2DMG]+ | [NTf2]− | 285.0 | 285.5 | 286.5 | 288.9 | 400.7 | 292.9 | 399.3 | 532.5 | 688.8 | 168.9 | — | — | — |
| [(C4C1)2DMG]+ | [NTf2]− | 285.1 | 285.5 | 286.5 | 288.9 | 400.7 | 293.0 | 399.5 | 532.7 | 688.9 | 169.0 | — | — | — |
| [(C4C4)2DMG]+ | [NTf2]− | 285.2 | 285.5 | 286.5 | 288.8 | 400.7 | 293.0 | 399.4 | 532.6 | 688.9 | 168.9 | — | — | — |
Different electronic environments of carbon often lead to unresolved peaks in the C 1s high-resolution XP spectrum. Therefore, it is essential that an appropriate fitting model is developed to describe these environments. Fig. 4 shows some examples of the C 1s region for [(C8C1)2dmg]X (X = Cl−, [BF4]−, [NTf2]−). In the case of [(C8C1)2dmg][NTf2], there are four clear peaks two of which are unresolved. The peak at ≈293 eV is assigned to the CF3 group of the [NTf2] anion as seen previously for other [NTf2]-based ionic liquids.22,23,27,34 For all the ILs investigated a small, well-defined peak is also observed, at a lower binding energy of 288–290 eV, alongside two unresolved peaks between 283–288 eV. Consequently, a suitable multicomponent model must be developed and applied in the deconstruction of the measured photoemission envelopes. A series of multi-component models has been developed previously to describe the C 1s XP spectra for a range of ionic liquid families including dialkyl-imidazolium, alkyl-pyridinium, pyrrolidinium, quaternary ammonium and phosphonium based cations.23,27,34 Herein we present first XPS data and appropriate fitting models for a range of guanidinium based cations.
 | ||
| Fig. 4 High resolution XP spectra of the C 1s region with component fittings for (a) [(C8C1)2dmg][NTf2], (b) [(C8C1)2dmg][BF4] and (c) [(C8C1)2dmg][Cl]. Intensities are normalised to the Ncation 1s photoemission peak for [(C8C1)2dmg][BF4]. All XP spectra charge corrected to Caliphatic = 285.0 eV. | ||
A four-component model was applied to describe the guanidinium cation. Upon looking at the structure of [(C8C1)2dmg]+ (Fig. 4) we would expect four different chemical environments of carbon. The carbon bound exclusively to nitrogen (Ccore) is expected to have the highest binding energy as it is directly bound to three electron-withdrawing nitrogen atoms. The label Chetero is given to the first carbon of the alkyl-chain which is bound to a single nitrogen atom, and is expected to be at lower binding energy to Ccore. This is followed by (Cinter), designated as an “intermediate” carbon environment, where the electron-withdrawing impact of the nitrogen centre is less noticeable, resulting in an even lower binding energy. The remaining carbons are outside of the reach of the inductive pull of the cationic core and form the aliphatic component (Caliphatic).
It is important that parameter constraints are utilised to ensure that the calculated model accurately describes the chemical environments of the compound. Component areas are constrained to their nominal stoichiometries, for example Caliphatic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Chetero:Cinter:Ccore for [(C8C1)2dmg][BF4] equates to 12:6:2:1. However, approximately 20% of all photoelectrons originating from carbon atoms involved in unsaturated bonding will be affected by shake up/off phenomena and the corresponding C 1s peaks should be fitted to account for this, i.e. by reducing the total component area for Ccore by 20%.35 As the peak areas for the signals characteristic of aliphatic carbons are not affected by shake up/off phenomenon, the relative peak area ratios for the four cationic components can now be set to 12:6:2:0.8, correspondingly. Binding energy constraints were not applied as the binding energy shift between Chetero, Cinter and Ccore is expected to be dependent upon the nature of the anion.22,23 The fitting envelopes (red curve), shown in Fig. 4a–c, correspond well to the experimental spectra (black dots). C 1s fittings for [(C8C1)2dmg][PF6] and [(C4C1)2dmg][X] (X = Cl−, [BF4]−, [PF6]−, [NTf2]−) are included in the ESI† and are also in agreement with the experimental spectra.
:Chetero:Cinter:Ccore for [(C8C1)2dmg][BF4] equates to 12:6:2:1. However, approximately 20% of all photoelectrons originating from carbon atoms involved in unsaturated bonding will be affected by shake up/off phenomena and the corresponding C 1s peaks should be fitted to account for this, i.e. by reducing the total component area for Ccore by 20%.35 As the peak areas for the signals characteristic of aliphatic carbons are not affected by shake up/off phenomenon, the relative peak area ratios for the four cationic components can now be set to 12:6:2:0.8, correspondingly. Binding energy constraints were not applied as the binding energy shift between Chetero, Cinter and Ccore is expected to be dependent upon the nature of the anion.22,23 The fitting envelopes (red curve), shown in Fig. 4a–c, correspond well to the experimental spectra (black dots). C 1s fittings for [(C8C1)2dmg][PF6] and [(C4C1)2dmg][X] (X = Cl−, [BF4]−, [PF6]−, [NTf2]−) are included in the ESI† and are also in agreement with the experimental spectra.
Anionic effects on cation–anion interactions
XPS has been successfully used to probe cation–anion interactions in ionic liquids.22,23,27,41 It is been previously acknowledged that anion basicity influences the electronic environment of the cation, and is thought to occur through a transfer of charge from anion to cation. This is reflected in the binding energy of the Ncation 1s and Chet 1s where binding energies follows the trend [NTf2]− > [BF4]− > Cl−.22,27,34 Unsurprisingly, the same trend is also observed for guanidinium-based ionic liquids which could be seen in Fig. 5.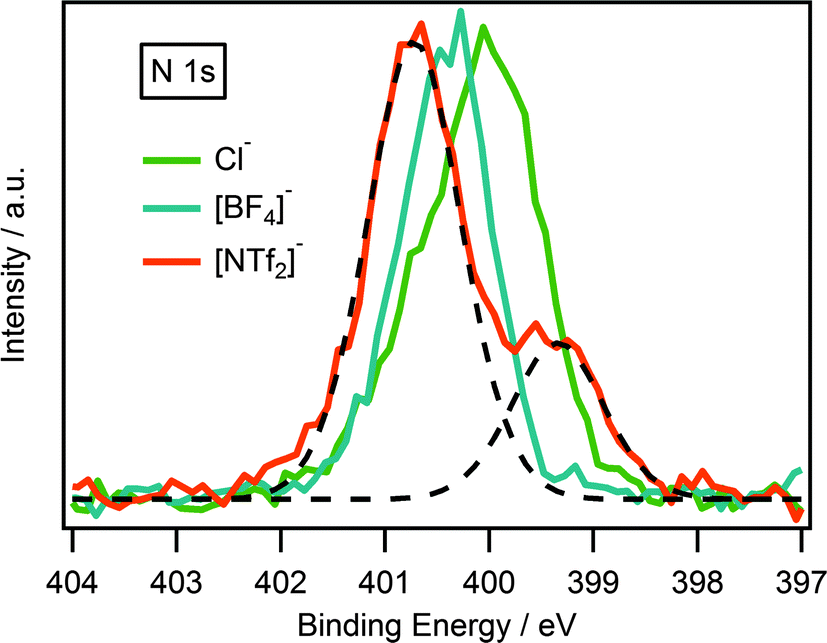 | ||
| Fig. 5 N 1s XP spectra of [(C8C1)2dmg][NTf2], [(C8C1)2dmg][BF4] and [(C8C1)2dmg][Cl]. Intensities are normalised to the Ncation 1s peak for [(C8C1)2dmg][BF4]. XP spectra were charge corrected by referencing the aliphatic C 1s photoemission peak to 285.0 eV. Deconstruction of the N 1s XP photoemission peaks (dotted line) for the [(C8C1)2dmg][NTf2] highlighting the two nitrogen components, Ncation at higher binding energy and Nanion at lower binding energy. | ||
Comparison of the cationic N (Ncation): [(C8C1)2dmg]+, [C8C1Im]+, [C8C1Pyrr]+ and [N6,6,6,14]+
Having obtained reliable and reproducible binding energies, comparisons can be made between N 1s binding energies of [NTf2] guanidinium-based ILs with other nitrogen-based cations such as pyrrolidinium,23 imidazolium22 and ammonium.27 Normalisation was accomplished using the F 1s photoelectron peak for the NTf2 anion for [C8C1Pyrr][NTf2].As shown in Fig. 6a, there are two nitrogen peaks which correspond to two nitrogen electronic environments, i.e. the three nitrogen atoms of the cation and the single nitrogen atom of the NTf2 anion. Considering the stoichiometry of the molecule the ratio of Ncation:Nanion is expected to be 3:1. The N 1s high resolution scan shows that these results are in good agreement with the expected ratio. Fig. 6b, illustrates two nitrogen peaks which correspond to two nitrogen electronic environments in the imidazolium ionic liquid. However, the ratio of Ncation to Nanion is expected to be 2:1 from the stoichiometry of the molecule, and the N 1s high resolution scan is in good agreement with this. The analogous is also true for the pyrrolidinium and ammonium ionic liquid, Fig. 6c and d, where the ratio of Ncation to Nanion is expected to be 1:1 and these results are also in good agreement with the expected ratio.
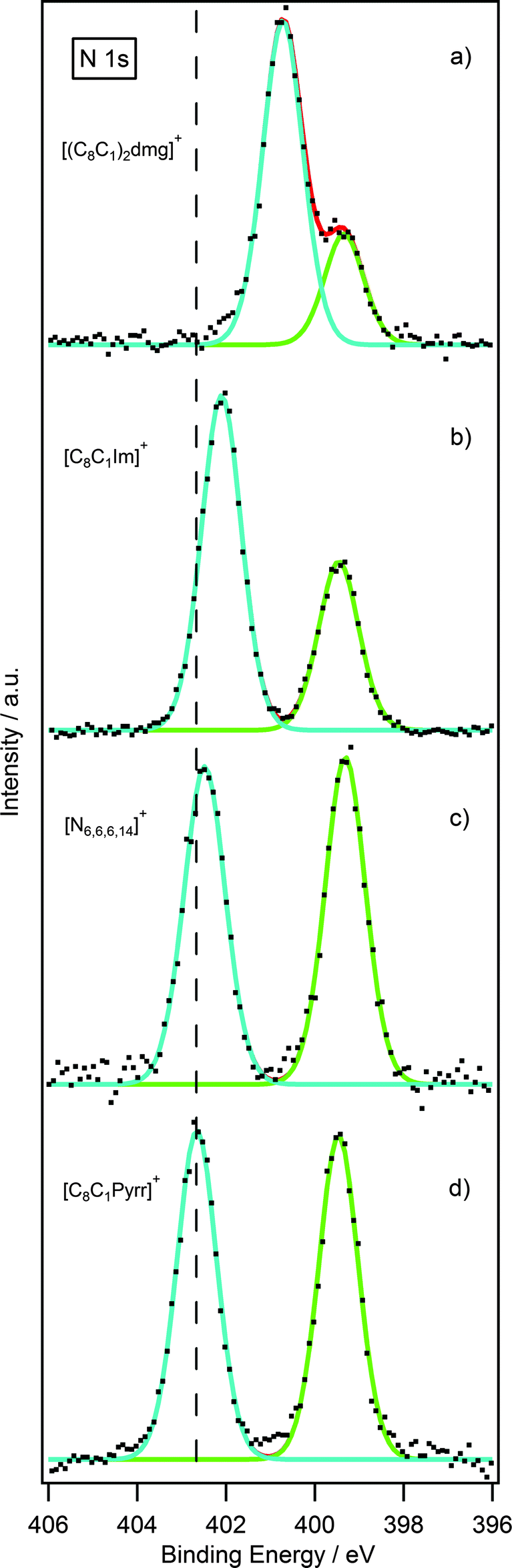 | ||
| Fig. 6 N 1s XP spectra of (a) [(C8C1)2dmg][NTf2], (b) [C8C1Im][NTf2], (c) [N6,6,6,14][NTf2] and (d) [C8C1Pyrr][NTf2]. Intensities are normalised to the F 1s peak for [C8C1Pyrr][NTf2]. XP spectra were charge corrected by referencing the aliphatic C 1s photoemission peak to 285 eV. | ||
Interestingly, there is a notable difference between the binding energies of the Ncation 1s peak for the four ionic liquids, Table 4. Looking firstly at the pyrrolidinium cation, it noticeably possesses the highest binding energy for the Ncation 1s peak. The reason for this may be explained by the charge being localised entirely on the single nitrogen atom of the pyrrolidinium ring. The ammonium cation shows a binding energy very similar to the pyrrolidinium, due to the charge being localised on a single nitrogen atom. For the imidazolium, however, the positive charge is distributed across the π-ring system resulting in a slightly lower binding energy by 0.6 eV. The Ncation 1s peak for the guanidinium cation occurs at an even lower binding energy with a striking difference of 2.0 eV from that of the pyrrolidinium cation. This can be explained by considering that the positive charge of the guanidinium cation is distributed across three nitrogen atoms as opposed to just one in the pyrrolidinium cation.
| [(C8C1)2dmg][NTf2] | [C8C1Im][NTf2] | [N6,6,6,14][NTf2] | [C8C1Pyrr][NTf2] | |
|---|---|---|---|---|
| Ncation 1s B.E. (eV) | 400.7 | 402.1 | 402.5 | 402.7 |
Conclusions
The successful measurement of XP spectra for a range of guanidinium-based ionic liquids, varying both cation aliphatic chain length, n, and the anion, demonstrates that this class of ILs are stable to measure X-ray irradiation with no observable decomposition across the timescale of our programmed scan cycle (≈30 min per cycle). Experimentally, we have applied an effective method of etching solid samples by heating to the melt, followed by cooling to form a glassy state solid, to avoid the scattering of powder during argon bombardment, surface contaminants were successfully removed via this method. However, ILs with shorter alkyl chains (n < 8) are observed to be less stable and have been shown to decompose under argon bombardment.40,42 The electronic environments of all elements were identified and an appropriate fitting model for the C 1s region was developed, the binding energy of the aliphatic carbon (Caliphatic 1s) was shown to be reliable and robust as an internal charge reference. Variation of the cation bound alkyl chain length, with a common anion, was shown to have minimal impact on the electronic environment of the anion. Comparison of binding energies for different nitrogen-based cationic structures revealed significant differences. This could be explained by considering that the positive charge of the guanidinium cation is well distributed across three nitrogen atoms, as opposed to just one in the pyrrolidinium cation, reflecting the weak bond between the guanidinium cation and the anion through the delocalisation of the positive charge around the three nitrogen centre.These data will inform design principles to deliver a material with a very specific series of physical and chemical properties. XPS binding energy data can be used to probe the magnitude of cation–anion based interactions which will allow us to investigate the dissolution behaviour of small molecular weight biomolecules. Therefore, future studies on ionic liquids mixtures will be further investigated.
Acknowledgements
We gratefully acknowledge the University of Nottingham and EPSRC (EP/K005138/1) for financial support and Dr Emily F. Smith for helpful discussions and critical advice.References
- A. Rehman and X. Zeng, Acc. Chem. Res., 2012, 45, 1667–1677 CrossRef CAS PubMed.
- Q. Li, Q. Tang, N. Du, Y. Qin, J. Xiao, B. He, H. Chen and L. Chu, J. Power Sources, 2014, 248, 816–821 CrossRef CAS PubMed.
- H. Weingärtner, C. Cabrele and C. Herrmann, Phys. Chem. Chem. Phys., 2012, 14, 415–426 RSC.
- Y. Gu, Green Chem., 2012, 14, 2091–2128 RSC.
- Q. Zhang, K. D. O. Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- J. W. Lee, J. Y. Shin, Y. S. Chun, H. Bin Jang, C. E. Song and S.-G. Lee, Acc. Chem. Res., 2010, 43, 985–994 CrossRef CAS PubMed.
- A. B. Pereiro, J. M. M. Araújo, J. M. S. S. Esperança, I. M. Marrucho and L. P. N. Rebelo, J. Chem. Thermodyn., 2012, 46, 2–28 CrossRef CAS PubMed.
- P. R. V. Rao, K. A. Venkatesan, A. Rout, T. G. Srinivasan and K. Nagarajan, Sep. Sci. Technol., 2012, 47, 204–222 CrossRef.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- Y. Gao, S. W. Arritt, B. Twamley and J. M. Shreeve, Inorg. Chem., 2005, 44, 1704–1712 CrossRef CAS PubMed.
- H. Xie, S. Zhang and H. Duan, Tetrahedron Lett., 2004, 45, 2013–2015 CrossRef CAS PubMed.
- S. Li, Y. Lin, H. Xie, S. Zhang and J. Yu, Org. Lett., 2006, 8, 391–394 CrossRef CAS PubMed.
- S. Li, Y. Lin, J. Cao and S. Zhang, J. Org. Chem., 2007, 72, 4067–4072 CrossRef CAS PubMed.
- T. Jiang, H. Gao, B. Han, G. Zhao, Y. Chang, W. Wu, L. Gao and G. Yang, Tetrahedron Lett., 2004, 45, 2699–2701 CrossRef CAS PubMed.
- Y. Peng, G. Song and F. Huang, Monatsh. Chem., 2005, 136, 727–731 CrossRef CAS.
- P. Blondeau, M. Segura, R. Pérez-Fernández and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC.
- N. M. M. Mateus, L. C. Branco, N. M. T. Lourenço and C. A. M. Afonso, Green Chem., 2003, 5, 347–352 RSC.
- I. A. Shkrob, T. W. Marin, J. R. Bell, H. Luo and S. Dai, J. Phys. Chem. B, 2013, 117, 14400–14407 CrossRef CAS PubMed.
- K. A. Schug and W. Lindner, Chem. Rev., 2005, 105, 67–113 CrossRef CAS PubMed.
- R. F. M. Frade, A. A. Rosatella, C. S. Marques, L. C. Branco, P. S. Kulkarni, N. M. M. Mateus, C. A. M. Afonso and C. M. M. Duarte, Green Chem., 2009, 11, 1660–1665 RSC.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H.-P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158–5190 CrossRef CAS PubMed.
- T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, R. Wöelfel, P. S. Schulz, P. Wasserscheid, H. Weber, J. Thar, B. Kirchner, F. Maier and H.-P. Steinrück, Chem. – Eur. J., 2010, 16, 9018–9033 CrossRef CAS PubMed.
- S. Men, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 15244–15255 RSC.
- K. R. J. Lovelock, C. Kolbeck, T. Cremer, N. Paape, P. S. Schulz, P. Wasserscheid, F. Maier and H.-P. Steinrück, J. Phys. Chem. B, 2009, 113, 2854–2864 CrossRef CAS PubMed.
- F. Maier, T. Cremer, C. Kolbeck, K. R. J. Lovelock, N. Paape, P. S. Schulz, P. Wasserscheid and H.-P. Steinrück, Phys. Chem. Chem. Phys., 2010, 12, 1905–1915 RSC.
- I. Niedermaier, C. Kolbeck, N. Taccardi, P. S. Schulz, J. Li, T. Drewello, P. Wasserscheid, H.-P. Steinrück and F. Maier, ChemPhysChem, 2012, 13, 1725–1735 CrossRef CAS PubMed.
- R. K. Blundell and P. Licence, Phys. Chem. Chem. Phys., 2014, 16, 15278–15288 RSC.
- X. M. Liu, S. J. Zhang, G. H. Zhou, G. W. Wu, X. L. Yuan and X. Q. Yao, J. Phys. Chem. B, 2006, 110, 12062–12071 CrossRef CAS PubMed.
- X. Liu, G. Zhou, S. Zhang, G. Wu and G. Yu, J. Phys. Chem. B, 2007, 111, 5658–5668 CrossRef CAS PubMed.
- X. Liu, G. Zhou and S. Zhang, Fluid Phase Equilib., 2008, 272, 1–7 CrossRef CAS PubMed.
- X. Liu, X. Zhang, G. Zhou, X. Yao and S. Zhang, Sci. China: Chem., 2012, 55, 1573–1579 CrossRef CAS PubMed.
- G. Yu, X. Chen, C. Asumana, S. Zhang, X. Liu and G. Zhou, AIChE J., 2011, 57, 507–516 CrossRef CAS PubMed.
- I. J. Villar-Garcia, K. R. J. Lovelock, S. Men and P. Licence, Chem. Sci., 2014, 5, 2573–2579 RSC.
- I. J. Villar-Garcia, E. F. Smith, A. W. Taylor, F. Qiu, K. R. J. Lovelock, R. G. Jones and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 2797–2808 RSC.
- E. F. Smith, F. J. M. Rutten, I. J. Villar-Garcia, D. Briggs and P. Licence, Langmuir, 2006, 22, 9386–9392 CrossRef CAS PubMed.
- E. F. Smith, I. J. Villar-Garcia, D. Briggs and P. Licence, Chem. Commun., 2005, 5633–5635 RSC.
- K. R. J. Lovelock, E. F. Smith, A. Deyko, I. J. Villar-Garcia, P. Licence and R. G. Jones, Chem. Commun., 2007, 4866–4868 RSC.
- A. W. Taylor, S. Men, C. J. Clarke and P. Licence, RSC Adv., 2013, 3, 9436–9445 RSC.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- A. W. Taylor and P. Licence, ChemPhysChem, 2012, 13, 1917–1926 CrossRef CAS PubMed.
- B. B. Hurisso, K. R. J. Lovelock and P. Licence, Phys. Chem. Chem. Phys., 2011, 13, 17737–17748 RSC.
- K. R. J. Lovelock, J. P. Armstrong, P. Licence and R. G. Jones, Phys. Chem. Chem. Phys., 2014, 16, 1339–1353 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp01069a |
| This journal is © the Owner Societies 2015 |