
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–O cleavage of aromatic oxygenates over ruthenium catalysts. A computational study of reactions at step sites†
Cheng-chau
Chiu
abc,
Alexander
Genest
a,
Armando
Borgna
c and
Notker
Rösch
*ab
aInstitute of High Performance Computing, Agency for Science, Technology and Research, 1 Fusionopolis Way, #16-16 Connexis, Singapore 138632, Singapore
bDepartment Chemie and Catalysis Research Center, Technische Universität München, 85747 Garching, Germany. E-mail: roesch@mytum.de; Fax: +49 89 2891 3468; Tel: +49 89 2891 3620
cInstitute of Chemical & Engineering Sciences, Agency for Science, Technology and Research, 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
First published on 21st May 2015
Abstract
We studied the C–O cleavage of phenolate and catecholate at step sites of a Ru catalyst using periodic DFT methods at the GGA level. Both C–O scission steps are associated with activation barriers of about 75 kJ mol−1, hence are significantly more facile than the analogous reactions on Ru terraces. With these computational results, we offer an interpretation of recent experiments on the hydrodeoxygenation of guaiacol (2-methoxyphenol) over Ru/C. We hypothesize that the experimentally observed dependency of the product selectivity on the H2 pressure is related to the availability of step sites on a Ru catalyst.
1. Introduction
An important process in the sustainable production of fuels is the pyrolysis of biomass to yield a “pyrolysis oil”,1i.e., a mixture of various organic oxygenates.2,3 This pyrolysis oil has to be upgraded in a hydrodeoxygenation (HDO) step, to reduce its oxygen content.4 This is necessary as a low heating value and other undesirable properties of the pyrolysis oil are related to its high oxygen content.5,6 Experiments addressing the catalytic HDO of aromatic oxygenates have shown that Ru particles of up to 4 nm, supported on activated carbon, can act as catalyst for the conversion of the model feedstock guaiacol [2-methoxyphenol, C6H4(OH)(OCH3)] to deoxygenated aromatic products.7–10 The main product reported for reactions in the temperature range 523–673 K varied with the applied H2 pressure. Benzene was reported as main product for reactions under H2 pressure of approximately 40 bar,7 while phenol was obtained when working at H2 pressures of less than 1 bar.8–10 Experiments using Ru particles on other support materials detect a different product distribution.8,11 Here, we solely address processes on the metal catalyst. Therefore we will only refer to experiments with carbon as support which is catalytically inactive.Theoretical studies on the HDO of guaiacol on Ru indicate that the removal of oxo or hydroxyl groups from an aromatic C6 ring is kinetically challenging,12,13 consistent with the experimental observation of partially oxygenated species like catechol and phenol as intermediates or products.7–10 In these earlier calculations, the Ru catalyst was modeled as an ideal, flat Ru(0001) surface.12,13 The computational strategy of (first) addressing models of ideal close-packed surfaces is quite common and was also applied in recent studies on the HDO of aromatics or on the scission of aromatic C–O bonds over Fe,14 Ni,15 Pd,14 and Pt catalysts,16 despite of the limitation of such models. After all, they neglect certain features of a real catalyst, e.g., the effect of step and kink sites which have been shown to play important roles in various Ru catalyzed processes.17–19 Recently, we proposed a mechanism for the HDO of guaiacol on the Ru(0001) surface that is consistent with the experimental observation of phenol as main product at low H2 pressure.12 Removing the first O of guaiacol, via C–O cleavage of catecholate, is kinetically accessible whereas the removal of the second O, via C–O scission of phenolate, was calculated to be hindered by a barrier of almost 200 kJ mol−1.12 However, the mechanism for the formation of benzene, as observed in experiments under elevated H2 pressure,7 remained unclear. This open question could not be resolved either by the recently discussed HDO pathway proceeding via a decarbonylation step.13 To examine this problem, we studied the crucial C–O cleavage reactions with more elaborate catalyst models that contain step sites.
2. Computational methods and models
The results of this study were obtained from plane-wave based DFT slab model calculations at the PBE-GGA level20,21 using the software VASP (version 5.2.12).22–25 Note that standard GGA functionals are reported to yield similar energetics for reactions of aromatics on metal surfaces as calculations using the DFT-D3 approach13 or the optB88-vdW functional14 do. In other words, the known inability of GGA functionals to describe van der Waals forces properly26 is not expected to bias the results of the present work. The detailed computational methodology as well as the Ru(0001) slab models used for studying processes on terraces have previously been described.12 To model reactions at step sites, we used slab models which were constructed by cutting the Ru bulk geometry perpendicular to the crystallographic [10![[1 with combining overline]](https://www.rsc.org/images/entities/char_0031_0305.gif) 5] direction. The resulting step sites are locally of 3-fold (S3) and 4-fold (S4) symmetry (Fig. 1); see the ESI† (Section S1) for details.
5] direction. The resulting step sites are locally of 3-fold (S3) and 4-fold (S4) symmetry (Fig. 1); see the ESI† (Section S1) for details.
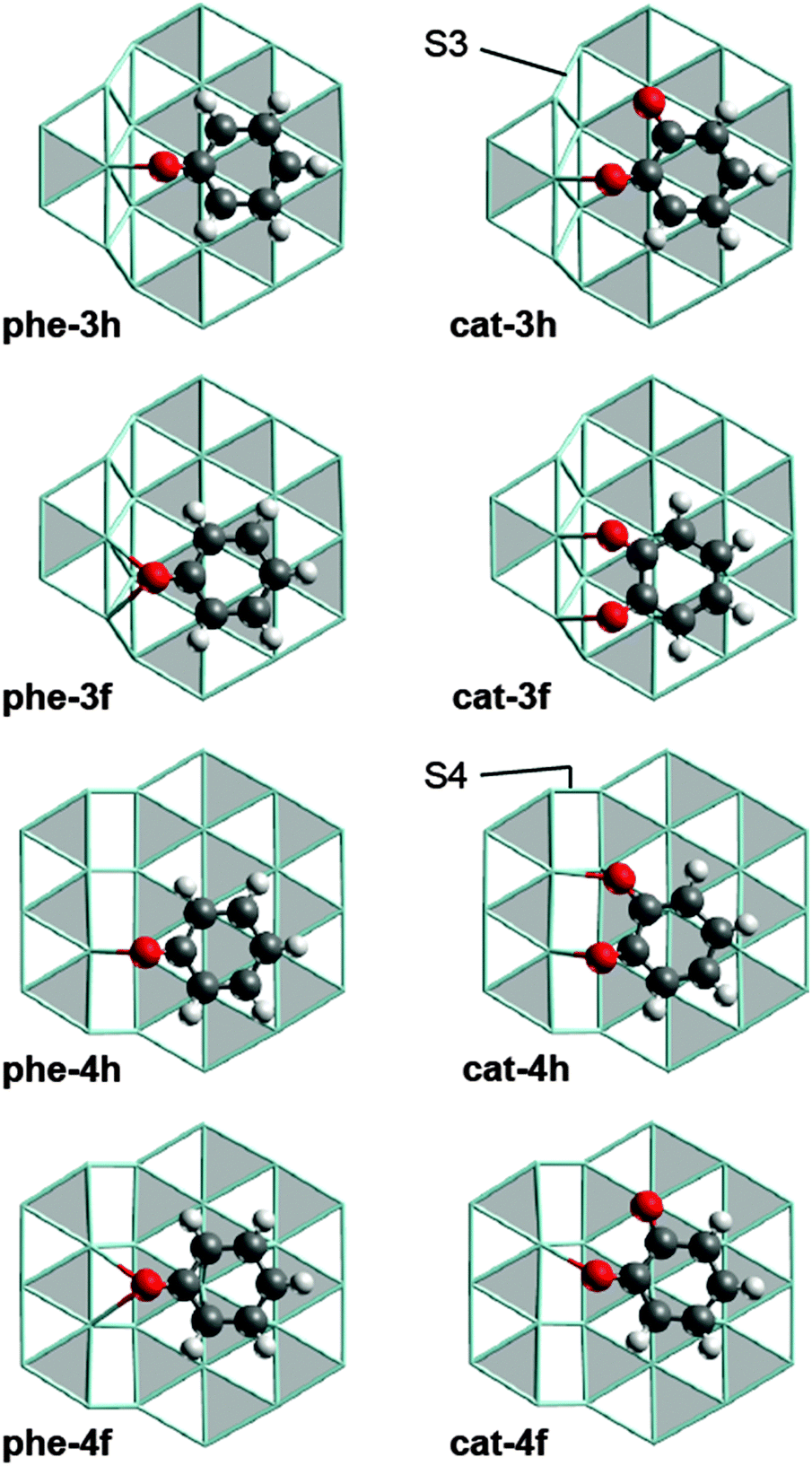 | ||
| Fig. 1 Optimized structures of phenolate and catecholate adsorption complexes at step sites. Color coding: O – red, C – gray, H – white, Ru – cyan sticks. hcp sites of the Ru surfaces are marked in gray, fcc in white. S3 and S4 type steps are discriminated in the structures of cat-3h and cat-4h. | ||
We refer to the phenolate and catecholate adsorption complexes under study by labels of the form x-yz with x = “phe”, “cat”, respectively. y can be 3, 4, or t, indicating adsorption of the substrate at a S3 step, a S4 step, or on a terrace site. z = h, f indicates whether the center of at the aromatic ring is over an hcp or an fcc site, respectively (Fig. 1). As we shall address only C–O cleavage reactions, we denote the corresponding transition state (TS) structures, derived from the initial state (IS) x-yz, by the label x-yz-ts and the structure of the co-adsorbed cleavage products as x-yz-p. Fig. S2 of the ESI† provides all stationary structures involved; their Cartesian coordinates can also be found in the ESI.†
We consider C–O cleavage at step sites as a process of three phases: (i) diffusion of the reactant from its most stable adsorption site on a terrace to a step site forming structure x-yz (y = 3, 4), (ii) the actual C–O cleavage yielding the structure x-yz-p with the cleavage products co-adsorbed at the step, and (iii) diffusion of the cleavage products to their most stable adsorption sites at formally infinite separation on a Ru(0001) surface. The latter step implies an overall low-coverage situation in our model. The changes ΔG in the Gibbs free energy associated with the three phases are denoted as ΔGt–s, ΔGC–O, and ΔGs–t, respectively, where negative values indicate exothermic processes. The labels “t–s” and “s–t” refer to the diffusion from the terrace to a step site and vice versa. For the C–O cleavage, we will present two types of activation barriers. The direct barrier ΔGa,s = ΔG(x-yz-ts) − ΔG(x-yz) is the difference between the Gibbs free energy of a TS and of the immediately preceding local minimum structure at the step site, i.e., y = 3, 4. The barrier ΔGa,t = ΔG(x-yz-ts) − ΔG(x-th) = ΔGt–s + ΔGa,s is calculated relative to the corresponding IS x-th on a terrace. Thus, all ΔGa,t values for the reactions of a given reactant are calculated relative to the same IS x-th. In other words, differences in ΔGa,t directly reflect the stability difference of the corresponding isomeric TS structures. All Gibbs free energies of this study were evaluated for 673 K, the reaction temperature of the experiments reported in ref. 7 and 8.
3. Results and discussions
First we will briefly comment on the structures of the various adsorption complexes of phenolate and catecholate and their thermodynamic stability. Then, we will turn to the reaction energetics of the C–O cleavage steps, and finally we will discuss how these results shape the interpretation of the experiments on guaiacol HDO over Ru.3.1. Adsorption of phenolate and catecholate at step sites
We identified four adsorption complexes each for phenolate and catecholate at Ru step sites (Fig. 1) where the C6 ring is adsorbed on the lower terrace and at least one C–O bond is pointing to the edge of the ascending step. As the scission of carbon monoxide and its hydrogenated derivatives at Ru step sites was reported to yield product structures in which the O center is located at the upper and the C center at the lower terrace,27–30 we assume that the C–O cleavage under study proceeds in a similar fashion. Therefore we focused on the structures shown in Fig. 1, which feature the adsorbate at the ascending steps. However, the adsorbates may also be located close to the edge of descending steps, as reported for phenol on Rh(221).31 We will start with the discussion of the structurally less complex phenolate adsorption complexes and turn to the catecholate complexes later on.In all adsorption complexes of phenolate (Fig. 1), the O atom is bonded to Ru centers at the (upward) step edge. The carbon center of the C–O group in the more stable complexes phe-4h and phe-3f is located over a hollow site, similar to the structure of the most stable phenolate complex on a Ru terrace, phe-th (Fig. S3 of ESI†).12 The ΔGt–s values for phe-4h, −9 kJ mol−1, and phe-3f, 16 kJ mol−1, indicate that these two complexes are more stable than the isomer species phe-3h, ΔGt–s = 30 kJ mol−1, and phe-4f, ΔGt–s = 57 kJ mol−1 (Table 1). The latter two structures also have their O center bonded to the step edge, but the C atom of the C–O group is located at a top site. This latter type of coordination is rather uncommon for aromatic C–O groups on close-packed Ru surfaces.12
| ΔGt–s | ΔGC–O | ΔGs–t | ΔGa,t | ΔGa,s | Δ(C–O) | γ | |
|---|---|---|---|---|---|---|---|
| a See Section 2 for the various definitions. b Calculated distances of species in the gas phase: 123 pm for both C–O bonds of catecholate; 127 pm for the CO bond of phenolate. | |||||||
| cat (gas phase) | — | 0, 0 | |||||
| cat-th | −30 | 106 | 13, 8 | 2, 4 | |||
| cat-3h | −11 | 35 | −54 | 135 | 147 | 11, 8 | 27, 1 |
| cat-3f | −24 | 64 | −70 | 52 | 76 | 9, 9 | 19, 19 |
| cat-4h | −27 | 2 | −5 | 63 | 90 | 9, 13 | 31, 5 |
| cat-4f | 27 | −55 | −2 | 132 | 105 | 11, 8 | 19, 1 |
| phe (gas phase) | — | 0 | |||||
| phe-th | −7 | 185 | 4 | 2 | |||
| phe-3h | 30 | 51 | −88 | 176 | 146 | 7 | 27 |
| phe-3f | 16 | 47 | −70 | 93 | 77 | 6 | 16 |
| phe-4h | −9 | 83 | −81 | 95 | 104 | 4 | 18 |
| phe-4f | 57 | −15 | −49 | 171 | 114 | 7 | 19 |
For a quantitative comparison of the adsorption structures, we examined the distortion of the adsorbed phenolate species relative to phenolate in the gas phase. Table 1 shows the elongation of the C–O bond and the angle γ between the C–O bond vector and the C6 plane; in gas phase γ = 0°. The C6 plane of the adsorption complexes is defined by a least-squares fit to the positions of the C centers of the ring. The two more stable step-site structures phe-4h and phe-3f have slightly shorter C–O bonds and slightly smaller angles γ than the less stable structures phe-4f and phe-3h. In other words, the phenolate moiety is less strongly distorted in the more stable adsorption complexes. This correlation between the distortion from the gas phase structure of phenolate and the stability of the complexes is only very rough; the chosen geometric parameters should not be considered as the only factors determining the stability of a phenolate complex. For instance, phe-3h is less stable than phe-4f, but features the largest angle γ of all phenolate complexes studied.
Similar to the situation of phenolate, the catecholate complexes cat-4h, ΔGt–s = −27 kJ mol−1, and cat-3f, ΔGt–s = −24 kJ mol−1, are more stable than cat-3h and cat-4f (Table 1). However, the overall topology of the more stable structures is different because both O centers of catecholate interact with Ru centers at the step (Fig. 1). This interaction induces a slight rotation of the C6 ring compared to the phenolate complexes on the analogous adsorption sites. In the less stable catecholate complexes cat-3h and cat-4f, only one O center interacts with the step edge, while the other O center binds to a top site on the terrace. The C atom bound to latter O center adsorbs over a hollow site, in a similar fashion as in phe-th (Fig. S3 of ESI†). We also tried to relate the stability of the catecholate complexes to their adsorbate structure, but were unable to identify a suitable correlation.
3.2. C–O scission at step sites
Next we will address the C–O cleavage steps, focusing mainly on the activation barriers of the reactions under study. We will start with the C–O cleavage of catecholate, the first cleavage of an aromatic C–O bond in the previously proposed HDO pathway of guaiacol on Ru.12 In the second part of this section, we will deal with the scission of the phenolate C–O bond, the second aromatic C–O cleavage step in the guaiacol HDO mechanism.12 Where appropriate, we will indicate the context of the C–O bond cleavage reactions in the HDO of guaiacol. Table 1 provides an overview of the energetics of the C–O cleavage steps.We start by comparing the values ΔGa,t for the C–O cleavage of catecholate (Table 1), thus evaluating the stability of the associated TSs. C–O cleavage via the intermediates cat-3f or cat-4h is kinetically accessible as indicated by their ΔGa,t values of 52 kJ mol−1 and 63 kJ mol−1, respectively. The associated TSs (Fig. 2) are by 54 kJ mol−1 (cat-3f-ts) and 43 kJ mol−1, (cat-4h-ts) more stable than the TS of the corresponding reaction on a terrace for which ΔGa,t = 106 kJ mol−1 (Fig. 3).12 By the same criterion, reactions via the intermediates cat-3h and cat-4f are even less likely than the reaction on a terrace (Table 1), thus we will not consider the corresponding reaction pathways in the following.
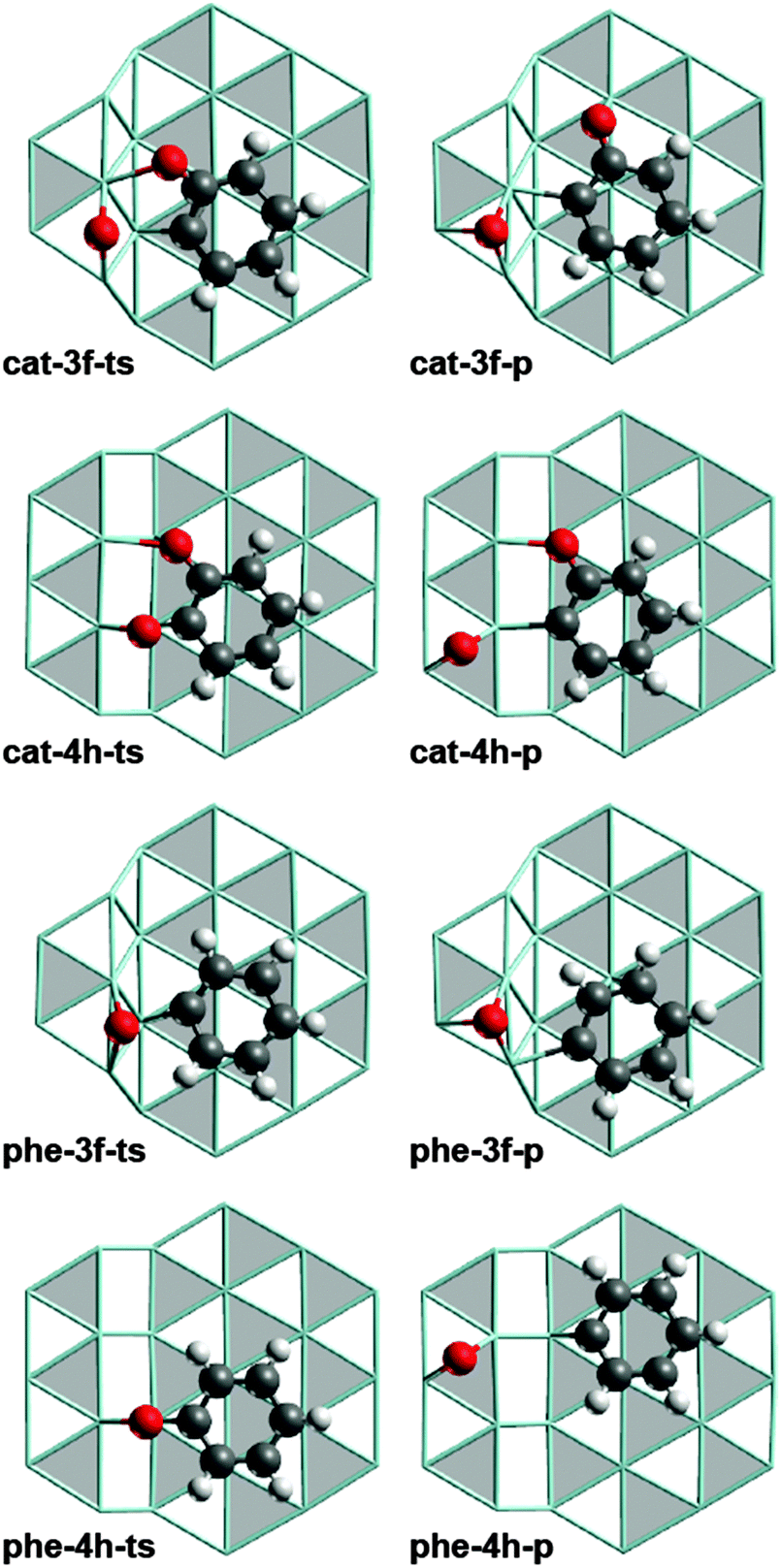 | ||
| Fig. 2 Optimized structures of TS (left-hand column) and product structures (right-hand column) of the most favored C–O cleavage pathways of catecholate and phenolate at S3 and S4 type steps. Lay-out as in Fig. 1. | ||
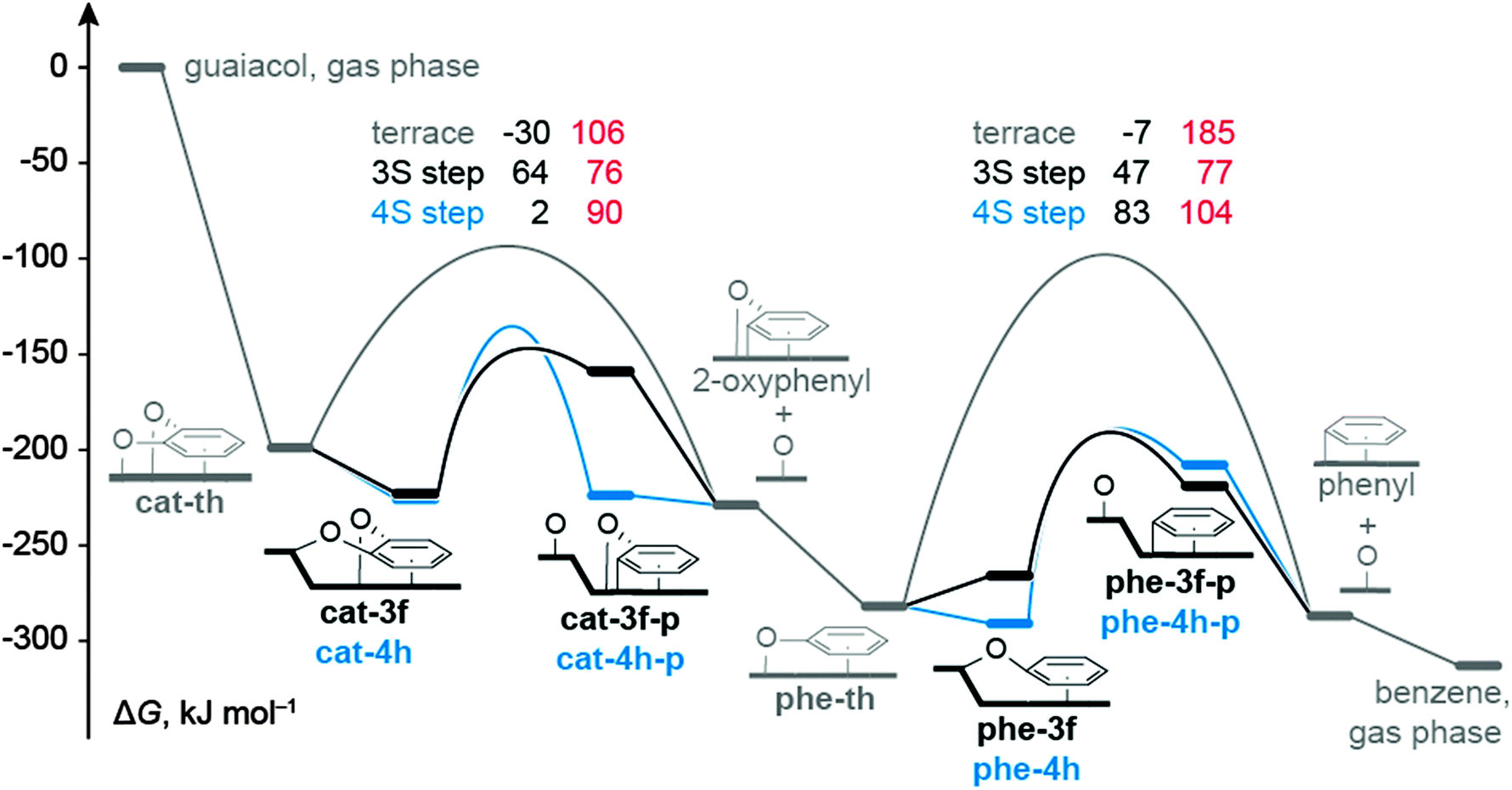 | ||
| Fig. 3 Gibbs free energy profiles for the most likely C–O bond cleavage of catecholate and phenolate reactions in the context of guaiacol HDO reported in ref. 12: profile for the pathway on a terrace in gray (data from ref. 12), at a S3 type step in black, and a S4 type step in blue. Intermediate states are represented by bars, TSs by arches. Values in black denote results for ΔGC–O, in red direct barriers, i.e., ΔGa,s for reactions at steps and ΔGa,t for reactions on terraces. All ΔG values are calculated for 673 K and 1 bar, and are referred to guaiacol in the gas phase and bare Ru surfaces. | ||
The accessible C−O cleavage pathways viacat-3f or cat-4h start with the exothermic formation of the complexes at the step site, both of similar stability, ΔGt–s = −24 kJ mol−1 and −27 kJ mol−1. The actual scission of the C–O bond is associated with direct barriers ΔGa,s of 76 kJ mol−1 and 90 kJ mol−1, thus the reaction at the S3 type step viacat-3f is kinetically slightly preferred. However, that C−O cleavage step is endothermic, ΔGC–O = 64 kJ mol−1, while the alternative C–O scission at the S4 type step viacat-4h is thermo-neutral. The difference in ΔGC–O can be related to the interaction between the O center of the “spectator” C–O bond and the Ru atoms of the step edge. In the reaction viacat-4h, both catecholate O atoms remain bound to the step edge during the entire C–O cleavage process (Fig. 1 and 2). At variance, in the reaction viacat-3f, both O centers are bonded to the step edge only up to the TS cat-3f-ts (Fig. 1 and 2), but not in the product cat-3f-p (Fig. 2). In the latter structure, the O of the spectator C–O bond is bound to a top site at the terrace. The missing interaction in cat-3f-p between the O center and the step edge results in a destabilization compared to cat-4h-p, thus rationalizing the endothermic nature of the reaction viacat-3f. The product structures cat-3f-p and cat-4h-p in Fig. 2 reflect local energy minima, reached directly from the TSs. Thus, the co-adsorbed moieties 2-oxophenyl and O do not necessarily represent their most preferred adsorption geometries.32 Rather, diffusion of these fragments to their most stable adsorption sites on a terrace in part entails a notable stabilization, ΔGt–s(phe-3f-p) = −70 kJ mol−1, ΔGt−s(phe-4h-p) = −5 kJ mol−1 (Table 1, Fig. 3).
In the HDO path for guaiacol on the ideal surface Ru(0001),12 the resulting 2-oxophenyl is easily hydrogenated in an exothermic step to yield phenolate which directly leads to the second C−O scission reaction under study. The C−O cleavage reactions viaphe-3f and phe-4h are associated with lower barriers ΔGa,s than the reaction on a terrace viaphe-th (Table 1, Fig. 3). Recall that the C–O scission of catecholate preferentially proceeds via analogous surface complexes, cat-3f and cat-4h. C–O cleavage of phenolate via the less stable intermediates phe-3h and phe-4f can be ruled out due to their high barriers ΔGa,s. Diffusion of phenolate from a terrace site, phe-th, to the step sites to form phe-3f or phe-4h, is characterized by ΔGt–s values of 16 kJ mol−1 and −9 kJ mol−1, respectively. In other words, formation of phenolate complexes at step sites is thermodynamically less favorable than the corresponding process of catecholate, (Table 1) which starts form the strained structure cat-th (Fig. S3 of ESI†).12 Release of this strain renders the diffusion of catecholate thermodynamically favorable. In contrast, no such strain is affecting the phenolate complex phe-th at the terrace (Fig. S3 of ESI†).12 The C–O cleavage steps of phenolate have direct barriers ΔGa,t of 77 kJ mol−1 (phe-3f) and 104 kJ mol−1(phe-4h), comparable to catecholate, and are endothermic processes with ΔGC–O = 47 kJ mol−1 and 83 kJ mol−1, respectively. Also here, the product species, phenyl and O, are considerably stabilized by diffusion to their preferred terrace sites. The resulting phenyl moiety can easily be hydrogenated to yield the experimentally observed benzene;7 see our earlier study.12
Having discussed the energetics for the C–O cleavage of catecholate and phenolate, we comment on some conspicuous differences between the reactions of these two adsorbates. One aspect is the relative stability of pertinent IS structures at step sites. While cat-3f and cat-4h are of similar stability, phe-4h is by 25 kJ mol−1 more stable than phe-3f. The difference between catecholate and phenolate may be related to the orientation of the aromatic substrates (Fig. 1). The two catecholate complexes differ somewhat in the orientation of the C6 rings, but the C6 moiety of both phenolate complexes are oriented in the same way, with alternating C centers at top sites.
The second topic concerns the Gibbs free energies ΔGC–O of the direct C–O cleavage. Both kinetically accessible C–O scissions of phenolate, of phe-4h and phe-3f, are significantly endothermic. In contrast, only the reaction of catecholate viacat-3f is endothermic whereas the alternative viacat-4h is thermo-neutral. As discussed above, the difference in energetics of the catecholate reactions is related to the effect of the spectator C–O group. As there is no such secondary structure component in phenolate, the C–O scission products phe-3f-p and phe-3h-p are almost degenerated (Fig. 3). The difference between the corresponding ΔGC–O values is mainly related to the stability difference of the IS structures phe-3f and phe-3h.
As central result of this computational work on Ru catalysts we note that the barriers calculated for C–O cleavage of aromatic oxygenates are significantly lower at step sites than at terrace sites. This finding can be rationalized by the larger number of surface atoms interacting with the substrate in the TS structures at steps as will be demonstrated on the example of phenolate C–O cleavage (Fig. 4). Recall a similar situation for the dissociation of N2 on Ru catalysts.19 As the involved C and O centers become under-coordinated upon bond scission, a TS is expected to be more stable if more surface atoms are interacting with these centers. In the TS structure phe-th-ts on a Ru terrace (Fig. 4) only three Ru atoms stabilize the centers C and O at two neighboring bridge sites which share one common Ru atom. Adsorption at a common Ru atom induces a repulsive interaction that destabilizes the TS structure.33 In contrast, five Ru centers are interacting with the atoms of the C–O bond in phe-3f-ts, the most stable TS associated with the C–O cleavage of phenolate. The O atom binds at a bridge site, the C atom at a hollow site, and these sites do not share any Ru atoms (Fig. 4). By the same token, one can also rationalize why C–O bond breaking via other routes is kinetically inaccessible. For instance, both the O and the C center of the energetically high lying TS structure phe-3h-ts (Fig. 4) are adsorbed at top sites. Thus only two surface atoms participate in the stabilization of the atoms of the cleaving C–O bond.
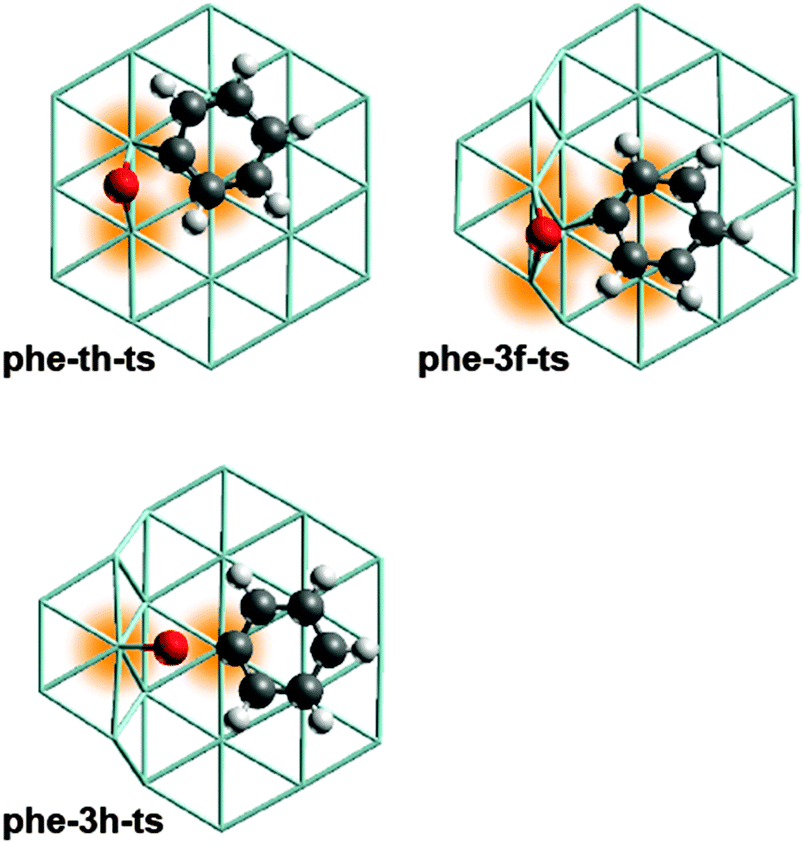 | ||
| Fig. 4 Optimized TS structures for C–O cleavage of phenolate at selected surface sites. Color coding of atoms as in Fig. 1. The Ru centers coordinating to the atoms of the cleaving C–O bond are highlighted. Structure of phe-th-ts from ref. 12. | ||
In summary, for a Ru catalyst with step sites, the C–O cleavage of catecholate and phenolate invokes similar direct barriers ΔGa,s. The lowest ΔGa,s values have been calculated for the reactions at S3 type step sites: 76 kJ mol−1 (catecholate) and 77 kJ mol−1 (phenolate). Thus, not only the C–O cleavage of catecholate, but also the corresponding reaction of phenolate is kinetically accessible (Fig. 3), consistent with the formation of benzene in guaiacol HDO over Ru observed in the experiment at approximately 40 bar H2.7 Our earlier work on guaiacol HDO over Ru(0001) revealed that the C–O bond cleavage steps determine the overall rate of the HDO process on Ru terrace sites,12 consistent with the observation of catechol and phenol as intermediates and products.7,8 This statement also holds for the HDO process over a Ru surface with step sites and close-packed terrace sites: all elementary reactions – other than Caryl–O cleavage steps – of the mechanism on Ru(0001), previously presented,12 have barriers of at most 65 kJ mol−1. Thus, even if step sites affect any of these other reactions, one does not expect these reactions to change the overall kinetics which justifies the exclusive focus of this work on C–O scission steps.
As alternative to direct C–O scission discussed here, a mechanism via the hydrogenated species phenol and catechol may seem plausible. Yet, hydrogenation of the most stable adsorption complexes of phenolate (phe-3f, phe-4h) and catecholate (cat-3f, cat-4h) at step sites is calculated highly endothermic, so that the hydrogenation products lie above the TS of “direct” C–O cleavage, ruling out C–OH cleavage via phenol or catechol.
3.3. Interpretation of experiments
Although the computational results presented allow one to rationalize the experimentally observed formation of benzene7 by the C–O bond cleavage at step sites, the dependence of the product selectivity on the H2 pressure7–10 remains to be explained. The fact that only the experimental setup with elevated H2 pressure of ∼40 bar leads to the formation of benzene as main product,7 suggests the possibility that hydrogen may play a role in the mechanism for the removal of the last O center from the aromatic substrate. The only mechanism of converting phenol to benzene on Ru, that discusses a “direct” effect of an elevated H2 pressure, invokes the partially hydrogenated intermediate C6H6OH.13 Yet, this mechanism can be ruled out for energetic reasons, due to the preceding hydrogenation of phenolate to phenol, as just discussed. Thus, hydrogen likely plays an “indirect” role for the C–O cleavage steps.Taking together the present results and those of our previous work,12 the cleavage of the phenolate C–O bond, ultimately leading to the formation of benzene, seems only possible at step sites of a Ru catalyst. However, the first C–O bond of catecholate may be cleaved on a terrace as well as at step sites of a Ru surface.12 Thus, the H2 dependent formation of benzene seems to be related to the availability of step sites. To rationalize this crucial effect of surface defects, we invoke carbon depositions at step sites which, under lower H2 pressure, may block the only sites that are catalytically active for the C–O cleavage of phenolate.
This argument is supported by DFT calculations which showed that C atoms at step sites of Ru surfaces are by ∼100 kJ mol−1 more stable than at terrace sites.28 A similar preference for the formation of carbon depositions at step sites was also determined for other metal catalysts, e.g., Pd.34,35 The formation of carbon depositions during the HDO process at elevated H2 pressure likely is reduced or even inhibited, similar to the situation reported for steam reforming and the methanation process, both over Ni.36 Recall also the experimental studies on HDO over Ru that mention coke formation as a possible reason for the deactivation of the catalyst.7,8,10 Additional experiments37 on guaiacol HDO over Ru at 40 bar H2 showed that the selectivity for benzene drops with the time on stream while the phenol selectivity increases concomitantly. This finding is in line with our assumption that the product selectivity of HDO is related to the deactivation of the catalyst. The original experiments7,8 on Ru catalyzed guaiacol HDO seem compatible with a faster catalyst deactivation under lower H2 pressure although the process conditions in those two sets of experiments are not identical. In the experiment at ∼1 bar total pressure yielding phenol,8 the guaiacol conversion rate dropped steadily from initially 100% to ∼30% after 3–4 hours on stream. In contrast, in the experiment at 40 bar yielding benzene,7 the conversion rate decreases from 50% to ∼35% within the first 2 hours of the process, but stays constant during the next 4 hours, consistent with the assumption that the C deposition is reduced.
Blocking of step sites on metal surfaces by carbonaceous species leading to the inhibition of bond breaking reactions is well known.28,35,38 For the particular case of Ru surfaces, TPD experiments demonstrated that C atoms at steps suppress the scission of carbon monoxide,28,38 which is known to occur at the step sites only.17,28,38 The concept of reduced carbon deposition at high H2 pressure is in line with the fact that transition metal catalysts based on Ni or Ru can be regenerated with H2, once they are deactivated due to coking.36,39 Not only carbonaceous species, but also oxygen may block catalytic sites, as has been discussed for the HDO process over Fe2O3 catalysts.40 However, this effect should be negligible on Ru as O atoms on Ru do not have a special preference for step sites, as shown by earlier calculations.28
4. Conclusions
In this computational study, we extended previous efforts12 to clarify mechanistic details of the Ru catalyzed hydrodeoxygenation (HDO) of aromatic oxygenates, such as guaiacol. We examined C–O cleavage of catecholate and phenolate which have previously been identified as the reaction steps with the highest barriers involved in the HDO of guaiacol.12 The present results demonstrate that pertinent adsorption complexes at step sites of a Ru surface feature notably lower barriers for the crucial C–O cleavage steps. Gibbs free energies of activation for the scission of the C–O bonds of catecholate and phenolate, adsorbed on Ru terrace sites, were calculated 106 kJ mol−1 and 185 kJ mol−1, but the corresponding reactions at step sites have barriers of only 76 kJ mol−1 and 77 kJ mol−1.Based on these results, we rationalized the experimentally observed formation of benzene in guaiacol HDO under elevated H2 pressure,7–10 which we suggest to be associated with the availability of step sites on the Ru catalyst. Carbon deposition, that preferentially occurs at the step sites, may deactivate at low H2 pressure the only sites catalytically active for the C–O cleavage of phenolate, thus preventing the formation of a fully deoxygenated product like benzene. In contrast, at elevated H2 pressure, hydrogen may reduce the carbon depositions at steps, allowing the C–O scission of phenolate to proceed with a barrier of only 77 kJ mol−1. Confirmation of the suggested scenario by experiments is highly desirable, e.g., by examining guaiacol HDO on Ru single crystals with clearly defined surface structures, the step sites of which can selectively be blocked.28,38
Acknowledgements
CCC is grateful for a fellowship by the International Graduate School of Science and Engineering at Technische Universität München and a scholarship from the A*STAR Research Attachment Program provided by the A*STAR Graduate Academy. The authors thank J. Chang, A*STAR Institute of Chemical & Engineering Sciences, for valuable discussion of the experiments. We acknowledge generous allotments of computational resources at the A*STAR Computational Resource Centre and the Gauss Centre for Supercomputing (http://www.gauss-centre.eu) provided via the SuperMUC platform of Leibniz Supercomputing Centre Garching (http://www.lrz.de).References
- R. P. Anex, A. Aden, F. K. Kazi, J. Fortman, R. M. Swanson, M. M. Wright, J. A. Satrio, R. C. Brown, D. E. Daugaard, A. Platon, G. Kothandaraman, D. D. Hsu and A. Dutta, Fuel, 2010, 89(suppl 1), S29–S35 CrossRef CAS.
- D. Mohan, C. U. Pittman and P. H. Steele, Energy Fuels, 2006, 20, 848–889 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- D. E. Resasco and S. Crossley, AIChE J., 2009, 55, 1082–1089 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- S. Boonyasuwat, T. Omotoso, D. Resasco and S. Crossley, Catal. Lett., 2013, 143, 783–791 CrossRef CAS.
- J. Sun, A. M. Karim, H. Zhang, L. Kovarik, X. S. Li, A. J. Hensley, J.-S. McEwen and Y. Wang, J. Catal., 2013, 306, 47–57 CrossRef CAS.
- D. Gao, C. Schweitzer, H. T. Hwang and A. Varma, Ind. Eng. Chem. Res., 2014, 53, 18658–18667 CrossRef CAS.
- T. Omotoso, S. Boonyasuwat and S. P. Crossley, Green Chem., 2014, 16, 645–652 RSC.
- C.-c. Chiu, A. Genest, A. Borgna and N. Rösch, ACS Catal., 2014, 4, 4178–4188 CrossRef CAS.
- J. Lu and A. Heyden, J. Catal., 2015, 321, 39–50 CrossRef CAS.
- A. Hensley, Y. Wang and J.-S. McEwen, ACS Catal., 2014, 5, 523–536 CrossRef.
- J. He, C. Zhao, D. Mei and J. A. Lercher, J. Catal., 2014, 309, 280–290 CrossRef CAS.
- K. Lee, G. H. Gu, C. A. Mullen, A. A. Boateng and D. G. Vlachos, ChemSusChem, 2015, 8, 315–322 CrossRef CAS PubMed.
- S. Shetty, A. P. J. Jansen and R. A. van Santen, J. Am. Chem. Soc., 2009, 131, 12874–12875 CrossRef CAS PubMed.
- Z.-P. Liu and P. Hu, J. Am. Chem. Soc., 2002, 124, 11568–11569 CrossRef CAS PubMed.
- S. Dahl, A. Logadottir, R. C. Egeberg, J. H. Larsen, I. Chorkendorff, E. Törnqvist and J. K. Nørskov, Phys. Rev. Lett., 1999, 83, 1814–1817 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Chem. Rev., 2011, 112, 289–320 CrossRef PubMed.
- I. M. Ciobica and R. A. van Santen, J. Phys. Chem. B, 2003, 107, 3808–3812 CrossRef CAS.
- S. B. Vendelbo, M. Johansson, D. J. Mowbray, M. P. Andersson, F. Abild-Pedersen, J. H. Nielsen, J. K. Nørskov and I. Chorkendorff, Top. Catal., 2010, 53, 357–364 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 209, 275–278 CrossRef.
- H. Li, G. Fu and X. Xu, Phys. Chem. Chem. Phys., 2012, 14, 16686–16694 RSC.
- M. L. Honkela, J. Bjork and M. Persson, Phys. Chem. Chem. Phys., 2012, 14, 5849–5854 RSC.
- C.-c. Chiu, A. Genest and N. Rösch, Top. Catal., 2013, 56, 874–884 CrossRef CAS.
- J. J. Mortensen, Y. Morikawa, B. Hammer and J. K. Nørskov, J. Catal., 1997, 169, 85–92 CrossRef CAS.
- I. V. Yudanov, A. V. Matveev, K. M. Neyman and N. Rösch, J. Am. Chem. Soc., 2008, 130, 9342–9352 CrossRef CAS PubMed.
- S. Schauermann, J. Hoffmann, V. Johánek, J. Hartmann, J. Libuda and H.-J. Freund, Angew. Chem., Int. Ed., 2002, 41, 2532–2535 CrossRef CAS.
- C. H. Bartholomew, Catal. Rev., 1982, 24, 67–112 CAS.
- J. Chang, T. Danuthai and A. Borgna, International Patent Application, WO 2013/191661 A1, 2013 Search PubMed.
- T. Zubkov, G. A. Morgan Jr., J. T. Yates Jr., O. Kühlert, M. Lisowski, R. Schillinger, D. Fick and H. J. Jänsch, Surf. Sci., 2003, 526, 57–71 CrossRef CAS.
- D. L. Trimm, Catal. Rev., 1977, 16, 155–189 CAS.
- A. J. R. Hensley, Y. Hong, R. Zhang, H. Zhang, J. Sun, Y. Wang and J.-S. McEwen, ACS Catal., 2014, 4, 3381–3392 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion of the slab models, sketches of structures and Cartesian coordinates of all stationary points, overview on electronic energies of reaction and activation. See DOI: 10.1039/c5cp01027f |
| This journal is © the Owner Societies 2015 |