
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpecific ion effects on the electrokinetic properties of iron oxide nanoparticles: experiments and simulations
Fernando
Vereda
*a,
Alberto
Martín-Molina
a,
Roque
Hidalgo-Alvarez
a and
Manuel
Quesada-Pérez
b
aGrupo de Física de Fluidos y Biocoloides, Departamento de Física Aplicada, Facultad de Ciencias, Universidad de Granada, 18071 Granada, Spain. E-mail: fvereda@ugr.es; Fax: +34-958243214; Tel: +34-958240025
bDepartamento de Física, Escuela Politécnica Superior de Linares, Universidad de Jaén, 23700, Linares, Jaén, Spain
First published on 22nd May 2015
Abstract
We report experimental and simulation studies on ion specificity in aqueous colloidal suspensions of positively charged, bare magnetite nanoparticles. Magnetite has the largest saturation magnetization among iron oxides and relatively low toxicity, which explain why it has been used in multiple biomedical applications. Bare magnetite is hydrophilic and the sign of the surface charge can be changed by adjusting the pH, its isoelectric point being in the vicinity of pH = 7. Electrophoretic mobility of our nanoparticles in the presence of increasing concentrations of different anions showed that anions regarded as kosmotropic are more efficient in decreasing, and even reversing, the mobility of the particles. If the anions were ordered according to the extent to which they reduced the particle mobility, a classical Hofmeister series was obtained with the exception of thiocyanate, whose position was altered. Monte Carlo simulations were used to predict the diffuse potential of magnetite in the presence of the same anions. The simulations took into account the ion volume, and the electrostatic and dispersion forces among the ions and between the ions and the solid surface. Even though no fitting parameters were introduced and all input data were estimated using Lifshitz theory of van der Waals forces or obtained from the literature, the predicted diffusion potentials of different anions followed the same order as the mobility curves. The results suggest that ionic polarizabilities and ion sizes are to a great extent responsible for the specific ion effects on the electrokinetic potential of iron oxide particles.
Introduction
Specific ion effects have been extensively studied since 1888, when Hofmeister discovered specific ion effects on the precipitation of purified egg white.1–6 Hofmeister showed that salts with the same cation but different anions precipitated the protein at different concentrations. This behaviour was also observed with some other colloidal suspensions.1–6 Accordingly, the effectiveness of different anions with a fixed cation, or cations with a fixed anion, can be ordered reproducibly in the so-called Hofmeister series. In colloidal science and biochemistry, there is a plethora of experiments showing that ions with the same valence can induce very different behaviours. Such experiments include protein solubility, critical micelle concentration, surface tension, forces between charged surfaces and electrophoretic mobilities, to which we will pay special attention in this work.1–6 Regardless of the huge number of studies over the years, Hofmeister phenomena are still gaining popularity nowadays. This is because, amongst other things, aqueous ion-containing interfaces are ubiquitous and play a key role in a plethora of physical, chemical, atmospheric, and biological processes.7 Hofmeister phenomena in bulk phases, surfactant-based systems and at interfaces, gels, proteins and enzymes, and real biological systems and medicine are some of the examples which have been extensively analysed in the last few decades.1–6 Among these examples, specific ion effects focused on biology are those that have mostly attracted the attention of the Hofmeister community nowadays.1,2,8 This is why there is a renewed interest in Hofmeister phenomena in colloids with biological applications.Precisely, our goal was to study the specific ion effects of iron oxide nanoparticles. Iron oxides (a general term that often includes iron oxyhydroxides) present many polymorphs with a variety of electronic and magnetic properties, and are relatively easy, and inexpensive to synthesize in the laboratory. They are currently the subject of intense research for multiple applications,9 including water splitting for the production of H2, anodes for Li-ion batteries, catalysis, and above all, for many applications in biomedical or biotechnological areas. Iron oxide phases with a relatively strong magnetic response (mainly the magnetite (Fe3O4)–maghemite (γ-Fe2O3) system) have been studied10,11 for drug and gene delivery, protein and DNA separation, tissue engineering, as contrast factors for MRI imaging, and magnetic hyperthermia. Magnetic iron oxide nanoparticles are especially attractive for biomedical uses because they can be manipulated with external magnetic fields and because of their relatively low toxicity.
Despite the technological relevance of iron oxide nanoparticles in applications that involve the use of aqueous suspensions, we have not been able to find many studies or account for ion specificity in these systems. Most of those studies are dedicated to hematite,12–14 a weak ferromagnetic iron oxide at room temperature. Regarding magnetite, we came across a study by Dixon15 on the interaction of alkaline-earth cations with the magnetite/water interface. The differences observed between different divalent cations were explained in terms of ionic radii and their degree of hydration.
From a theoretical point of view, the ionic distribution around a charged colloid immersed in an electrolytic solution is named the electric double layer (EDL). Classical models of EDL are based on the Poisson–Boltzmann (PB) equation, which do not account for ion specificity. As a consequence, the standard Derjaguin–Landau–Verwey–Overbeek (DLVO) theory of colloids is unable to explain ion specific forces between colloidal particles.16 Similarly, those electrokinetic models based on the PB equation cannot explain, a priori, why ions with identical valence cause different behaviours. In order to overcome these deficiencies, effective or fitting parameters are usually included in the classical theories of electrolytes in order to capture the specificity of ions.17 With this approach, however, the capability of prediction of these kinds of phenomenological approaches vanishes. In contrast, Ninham and coworkers pioneered the inclusion of the dispersion forces with associated hydration effects acting on ions in the colloidal formalisms (see review articles ref. 1, 2, 4 and 5, and references cited therein). The resulting theories show how specific ion effects are much more sensitive to anions than to cations, also indicating that Hofmeister effects depend on an interplay between specific surface chemistry, surface charge density, pH, buffer, and counterions with polarizabilities and ion size.5 Accordingly, mean field theories have been recently reformulated by using a modified PB equation that accounts for non-electrostatic interactions due to the size and the polarizability of each ion.18–20 Although many of these theoretical predictions qualitatively agree with experimental results, a major comprehension of different models is required to better understand the physicochemical aspects derived from ion specificity. For instance, sophisticated formalisms do not allow for a systematic analysis of the interplay between different contributions that influence the Hofmeister series.21 Under this scenario, computer simulations appear as a useful tool. On the one hand, simulations permit testing the validity of the models under hardly accessible conditions in experiments. On the other hand, systematic studies can be performed via simulations in order to assess the weight of different contributions presented in the theoretical models. Unfortunately, the use of simulations to study the specific ion effects on colloids is scarce. For instance, Tavares et al. and Boström et al. adopted the primitive model to study the effect of van der Waals interactions on forces between charged surfaces using computer simulations.22,23 These authors also used Molecular Dynamics (MD) simulations to parameterise ionic potentials in a generalized Poisson–Boltzmann equation.24 MD simulations were also used to study the ion specificity in the interaction of monovalent ions with hydrophobic and hydrophilic colloids.25
Inspired by the simulations based on the primitive model and performed by Tavares et al. and Boström et al.,22,23 we previously evaluated the effect of ionic dispersion forces on the EDL of colloids by means of Monte Carlo (MC) simulations.26,27 The results shown therein revealed that ionic van der Waals forces can sometimes contribute to the ion specificity of the electrokinetic properties of model colloids, although their relevance strongly depends on the ion size. Moreover, this subtle balance between polarizability of ions and their hydrated size was used to partially justify the specific ion effects observed in the electrophoretic mobility of latex particles by López-León et al.17
To sum up, the goal of our study was to explore the specific ion effects on iron oxide nanoparticles through both experiments and theoretical simulations. Experimentally, we synthesized magnetite nanoparticles and measured their electrophoretic mobility in the presence of several anions taken from the representative Hofmeister series given by López-León and coworkers:17
| citrate3− > SO42− > PO4H2− > F− > CH3COO− > Cl− > Br− > I− > NO3− > ClO4− > SCN− |
| (kosmotropic anions) (chaotropic anions) |
The interactions between the magnetite surface and the anions were also studied by means of MC simulations in order to complement the experimental data and deepen our understanding of the specific ion effects on the electrokinetic properties of this important colloidal system. Particularly, we wanted to find out if Lifshitz theory of van der Waals forces succeeds in justifying (and even predicting) the Hofmeister series of magnetite nanoparticles.
The work is organized as follows: (i) the experimental systems and procedures are presented in the next section; (ii) then the technical details of the model and simulations are outlined; (iii) in the third section the results are shown and discussed; (iv) finally some conclusions are drawn.
Experimental systems
Synthesis of iron oxide nanoparticles
Magnetite was chosen as the iron oxide phase due to our previous experience with this material, and because reliable information on its interfacial properties and dielectric response28–30 that was needed for the computer simulations was found in the literature.The magnetite nanoparticles were prepared in our laboratory by oxidative aging of a ferrous hydroxide precipitate with nitrate in aqueous media. This method has been thoroughly described in the past31,32 for the preparation of magnetite. It is well known that if the process is carried out in an excess of OH−, i.e. at a relatively large basic pH far from the isoelectric point of magnetite (ca. 6.5–7.0), the resulting particles are single crystals with sizes between 50 and 200 nm. We carried out the process at an excess of OH− of 0.075 M.33 Doubly distilled water was used for the preparation of the solutions: the first solution, containing KNO3 (Scharlau, reagent grade, 99%) and KOH (Panreac, chemically pure, 90%), was introduced into a 5-neck jacketed reactor and purged with N2 for an hour to get rid of dissolved atmospheric O2. At that point, a FeSO4·7H2O (Sigma Aldrich, reagent grade, 99%) solution was prepared in 0.01 M H2SO4 that had also been purged for an hour with N2. Upon mixing the two solutions, which resulted in the immediate formation of the dark green ferrous hydroxide precipitate, the circulation of hot water through the jacket of the reactor was started. This made the temperature of the reactant mixture reach 90 °C in approximately 12 minutes. The final concentrations of OH−, NO3− and Fe2+ were 0.125 M, 0.20 M and 0.025 M respectively, and the total volume of the reactant mixture was 800 mL. The system was kept at 90 °C for 4 hours. The main differences between this synthesis process and those reported in the past31,33 relied on the fact that this time moderate mechanical stirring of the reactant mixture and a N2 flow were maintained during the 4 hours of aging. The N2 flow implied that the pressure inside the reactor was practically atmospheric pressure, whereas in previous reports the process was carried out in sealed screw-cap flasks and the pressure must have been higher. Its value, however, was not reported.
After 4 hours of aging, the final black precipitate was washed several times with distilled water with the help of a permanent magnet to speed the separation of the particles. The particles were finally stored at 5 °C in ethanol.
Preparation of aqueous suspensions for electrophoretic mobility measurements
Over a period of time, the nanoparticles stored in ethanol settled down. When needed, the sediment would be redispersed in the ethanol supernatant by means of stirring and sonication, and small aliquots of this magnetite–ethanol dispersion were dried at room temperature. The resulting black powder would be dispersed in the appropriate aqueous solution for the electrophoretic measurements.Electrophoretic mobility as a function of pH
The magnetite nanoparticles in the dried powder form were dispersed in 2 mM NaNO3 solutions. The pH of these suspensions was lowered with HNO3 or raised with NaOH, and the electrophoretic mobility was measured at the resulting pH values.Electrophoretic mobility in the presence of various electrolytes
Because we wanted to study the specific behaviour of several anions in the iron oxide–water interface, all measurements reported in this manuscript were carried out at pH = 4 so that the nanoparticle surface was positively charged and the anions were counterions. The dry powder was dispersed in an aqueous HNO3 solution at pH = 4 to obtain a stock suspension of nanoparticles. Meanwhile, solutions of a variety of electrolytes (NaSCN, NaClO4, CH3COONa, Na2SO4, and Na3C6H5O7) were prepared with concentrations of 5 mM, 20 mM, 50 mM, 100 mM and 200 mM. The pH of these solutions was adjusted to 4 with HNO3 as well. Finally, a fixed volume of the stock suspension was added to each one of the electrolyte solutions. The pH of the final suspension was checked before the mobility measurements. The solid concentration of the final suspensions was approximately 0.01 mg mL−1.Electrophoretic mobility measurements
The electrophoretic mobility was measured at room temperature on a Malvern Zetasizer Nano ZS. Each reported data point is the average of 3 or 4 measurements, each one of them carried out on a different 0.8 mL aliquot of the dispersion.Model and simulation
Model
In this work, simulations were performed within the primitive model, in which ions are considered charged hard spheres and the solvent is thought of as a continuum of the dielectric constant εr. Many previous theoretical studies and simulations have proved that this representation of reality is able to encompass the essential features of real systems and overcome some limitations of the classical electric double layer (EDL) theory. In fact, this model has been particularly useful in the study of mesoscopic systems governed by electrostatic interactions as well as by the effects of the ion size.34–38A planar EDL in water (at 298 K) has been simulated here. Such an EDL is generated by a uniformly charged wall (that represents the particle surface) located at z = 0. The interaction energy between two ions, i and j, can be split into the electrostatic energy (uelec), the ionic dispersion energy (udisp) and the hard sphere term (uhs), which accounts for the ion size:
 | (1) |
The interaction energy between ions and the colloid surface can also be divided into analogous terms:
 | (2) |
Simulations
The conventional Metropolis algorithm is applied to a canonical ensemble for a collection of N ions confined in a rectangular prism (or cell) of dimensions W × W × L. More specifically, this simulation cell contains an ionic mixture corresponding to the bulk electrolyte solution together with an excess of counterions neutralizing the surface charge. The impenetrable charged wall is located at z = 0 (as mentioned previously) whereas at z = L, another impenetrable wall without charge is placed. In addition, periodic boundary conditions were used in the lateral directions (x and y). The system was always thermalized before collecting data for averaging (as usual) and the acceptance ratio was kept between 0.3 and 0.5. The L-values used in our simulations are (at least) one order of magnitude larger than the Debye length. When this is the case, a considerable portion of the solution bulk is included in the simulation box. The so-called Lekner–Sperb method is applied to account for corrections associated with the long range of the electrostatic interactions in the energy computation. More information about this method and simulations can be found elsewhere.26,39Dispersion constants
As our work focused on the effect of dispersion forces, the values of Banion/anion, Banion/cation, Bcation/cation, Banion/m and Bcation/m played a key role. According to Lifshitz theory of van der Waals forces,23,40 the constant between two ionic species (i and j) can be calculated as: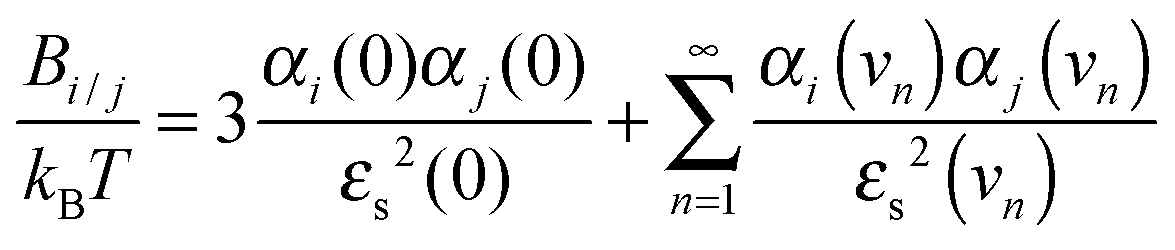 | (3) |
Here kB is the Boltzmann constant, T is the absolute temperature, αi(ν) is the effective polarizability of ion i in water and at frequency ν, εs(ν) is the dielectric constant of the solvent at the same frequency, and νn = 2πkBTn/h, where h is Planck's constant and n an integer. Analogously, the dispersion constant characterizing the interaction between a small ion and a colloidal particle (whose surface is assumed to be planar as compared to ions) is given by:
 | (4) |
 | (5) |
 | (6) |
Some authors had previously calculated some of the dispersion constants that we needed for our simulations using Lifshitz theory. Specifically, Tavares et al. computed BNa/Na as well as Banion/anion and Banion/Na for ClO4− and SO42−,23 whereas Boström et al. estimated Banion/anion and Banion/Na for SCN−.22 In this work, however, we estimated Bi/m and BNa/m for magnetite particles and all of the dispersion constants in which citrate ions are involved (Bcitrate/citrate, Bcitrate/Na, and Bcitrate/m), since these values have not been found in previous literature.
It should be mentioned that eqn (6) was not applied in the computation of dispersion constants involving magnetite. Instead, an expression published by Faure et al.30 describing the dielectric response of magnetite particles in terms of two characteristic frequencies (νIR and νUV) was employed. According to these authors:
 | (7) |
From eqn (4)–(7) and the ionization frequencies summarized by Tavares et al.,23BNa/m and Banion/m for anions ClO4− and SO42− were calculated. In the case of SCN− the value of polarizability employed in the computation of Banion/m was previously used by Boström et al.41 Unfortunately, the ionization frequency of this species was not found in studies where Lifshitz theory was applied to similar colloidal systems.2,22,23,42 In a previous work, Boström et al. faced the same problem.42 After analysing the characteristic frequencies of different ions computed by Mahan et al.,43 these authors concluded that νI typically ranges from 1.6 × 1015 to 8 × 1015 Hz and estimated the dispersion constants corresponding to these limit values.42 It can be easily shown from eqn (4) and (5) that Bi/m increases with νI, which means that the effects of van der Waals forces are stronger for ions with greater characteristic frequencies. For this reason, we preferred to compute Bi/m (for the thiocyanate–magnetite pair) corresponding to the upper limit of νI.
This νI-value was also employed in the computation of the three dispersion constants involving acetate and citrate ions (Bacetate/acetate, Bacetate/Na, Bacetate/m, Bcitrate/citrate, Bcitrate/Na, and Bcitrate/m) since the ionization frequency of this species was also unknown for us. The polarizability of acetate ions was extracted from the review by Lo Nostro et al.2 In the case of citrate, an estimate of the static effective polarizability in water was also made as follows. Citrate ions are expected to be the largest anions among those studied in this survey (see the next subsection, in which ionic diameters are estimated). In fact, their volume could be twice larger than ClO4− and SO42− anions, whose static polarizabilities are 5.4 and 6.3 Å3,23 respectively. Since the electronic polarizability is proportional to the molecular volume in the harmonic oscillator model, a static polarizability of 10 Å3 (in round numbers) was assumed for citrate anions. The values of the dispersion constants used in this work are summarized in Table 1.
| Electrolyte |
|
|
|
|
|
|---|---|---|---|---|---|
| Na2SO4 | 4.69 × 10−3 | 2.01 × 10−4 | 1.31 × 10−5 | 1.27 × 10−2 | 1.83 × 10−3 |
| NaClO4 | 1.59 × 10−3 | 8.97 × 10−5 | 1.31 × 10−5 | 4.07 × 10−3 | 1.83 × 10−3 |
| NaSCN | 4.69 × 10−3 | 2.00 × 10−4 | 1.31 × 10−5 | 6.34 × 10−2 | 1.83 × 10−3 |
| Na acetate | 3.86 × 10−4 | 11.0 × 10−6 | 1.31 × 10−5 | 6.43 × 10−2 | 1.83 × 10−3 |
| Na citrate | 1.28 × 10−3 | 2.00 × 10−5 | 1.31 × 10−5 | 11.7 × 10−2 | 1.83 × 10−3 |





Ionic diameters
Some previous studies clearly suggest that the effects of van der Waals forces are significantly influenced by the ion size. We should also bear in mind that ions between the particle surface and the outer Helmholtz plane are expected to be dehydrated. Thus we have considered a simple model of dehydration in our simulations. According to this model, ions trying to cross the outer Helmholtz plane were allowed to reduce their diameters to a bare ion value. Certainly, this is a simple model for dehydration, but it offers the advantage of requiring only one additional parameter (bare ion size). Table 2 shows the diameters of hydrated and bare ions employed in our simulations. The values corresponding to sodium were straightforwardly extracted from Israelachvili.40 The bare diameters of ClO4−, SCN−, SO42−, CH3CH2OO− and the citrate ion were assumed to be practically identical to the molecular diameters of their acids, which in turn were estimated from the mean molecular volume occupied in the liquid state.40 The hydrated ion diameters of these anions were estimated by adding up the thickness of a hydration layer similar to that of NO3−, 0.08 nm.40| Ion | Bare ion diameter (nm) | Hydrated ion diameter (nm) |
|---|---|---|
| Na+ | 0.19 | 0.72 |
| ClO4− | 0.52 | 0.68 |
| SO42− | 0.50 | 0.66 |
| SCN− | 0.40 | 0.56 |
| CH3COO− | 0.51 | 0.67 |
| Citrate | 0.65 | 0.81 |
Charge of citrate ions
Although experiments were carried out at pH = 4, we should keep in mind that the local pH near the surface of a positively charged colloidal particle is expected to be larger (since the concentration of H3O+ is smaller than in the bulk of the solution). As the pKa of the second and third protons of citric acid are 4.77 and 6.70, respectively, we have assumed that the doubly charged citrate anions are the predominant species.Computation of diffuse potential
The diffuse potential (ψd) is defined as the electric potential at the plane of the closest approach of the hydrated ions to the charged surface (the outer Helmholtz plane). This EDL property is extremely useful for comparing with experiments, since it is intimately related to the ζ-potential, which is a crucial quantity to analyse electrokinetic phenomena. From the ionic profiles provided by simulations, the diffuse potential of a planar EDL can be computed as follows: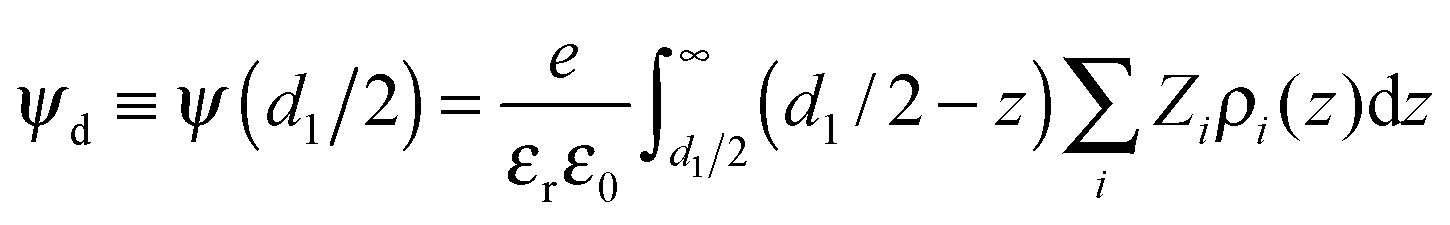 | (8) |
Results
Magnetite nanoparticles
The magnetite nanoparticles were predominantly octahedral, with a typical size of 50 nm (see Fig. 1), which is in agreement with previous synthesis processes33 also carried out in an excess of OH− of 0.075 M. It was determined in that study that these octahedral particles were single crystals, that their stoichiometry corresponded to a slightly oxidized form of magnetite, and that their saturation magnetization was ca. 82 emu g−1 at room temperature. | ||
| Fig. 1 TEM micrograph of the iron oxide nanoparticles prepared by oxidation of ferrous hydroxide with KNO3 in an excess of OH−. | ||
Electrophoretic mobility
The measurements of the electrophoretic mobility (μ) as a function of pH were carried out to ensure that our nanoparticles' electrokinetic behaviour was similar to that reported for magnetite in the literature. From the resulting curve, shown in Fig. 2, we concluded that the isoelectric point of our particles was in the vicinity of 6.7. This is in good agreement with previous observations by our group44 and with the values reported for the isoelectric point of magnetite (Fe3O4) by Regazzoni et al.28 and Vidojkovic et al.29 Note that these two articles included a collection of point of zero charge (PZC) and isoelectric point (IEP) data for magnetite taken from the literature, and that most of those data are between pH = 6.5 and pH = 7.0.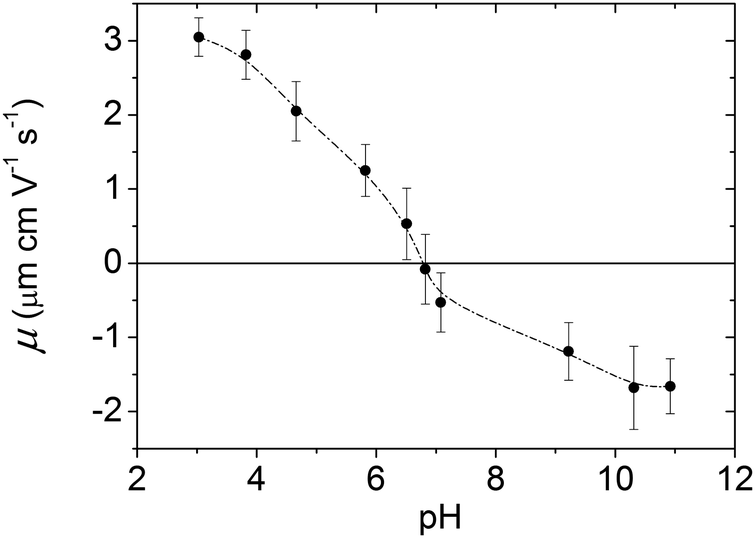 | ||
| Fig. 2 Electrophoretic mobility of the magnetite nanoparticles as a function of pH. Nanoparticles were dispersed in a 2 mM KNO3 solution. The pH was adjusted with NaOH or HNO3. The line is provided as guide to the eye. | ||
The evolution of the electrophoretic mobility (μ) of the magnetite nanoparticles, at pH = 4, and as a function of the concentration of different anions, is shown in Fig. 3. At pH 4 and in the absence of any electrolyte other than the nitrate ions and the protons from the nitric acid used to fix the pH, μ was approximately 3.0 μm cm V−1 s−1. In all cases the addition of an electrolyte lowered the mobility due to a decrease of the effective positive surface charge of the particles as the counterions (anions) approach the surface. However, we observed a very different behaviour between the chaotropic (SCN−, ClO4−, see the representative Hofmeister series given in the Introduction) and kosmotropic (SO42−, HC6H5O72−) anions. The effect of the chaotropic anions was limited to the screening of the particles' surface charge, bringing μ practically to zero, whereas the kosmotropic anions induced a mobility reversal that was especially fast (i.e. it was noticeable at remarkably low anion concentrations) in the case of the citrate ion.
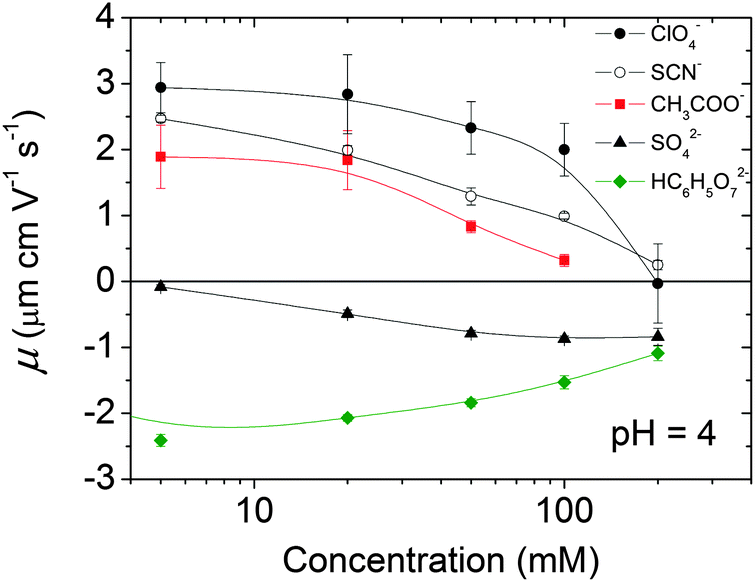 | ||
| Fig. 3 Electrophoretic mobility of the magnetite nanoparticles in the presence of different anions. The cation was always Na+ and the pH was fixed with HNO3. | ||
In general, the effectiveness of the anions at decreasing mobility of the particles follows the Hofmeister series given in the Introduction. Only the position of the anions ClO4− and SCN− seems to be exchanged regarding the original Hofmeister series. However, this anomalous behaviour has already been observed for other hydrophilic colloids by López-León et al.45 Therein, Hofmeister effects on the electrokinetics and stability of a cationic latex covered with a protein (IgG) were extensively analysed, the anomalous behaviour of SCN− being explained in terms of entropic mechanisms associated with rearrangements in the water structure. Building on this, the authors claimed that the chaotropic/kosmotropic concept can be readily extended from ions to any type of surface. This means that the behavior of hydrophilic surfaces is similar to that of kosmotropic ions, in the sense that they interact strongly with neighboring solvent molecules and promote their ordering, whereas hydrophobic surfaces behave like a chaotropic (structure-breaking) system. This entropically controlled process would explain why the adsorption of chaotropic ions is favoured in hydrophobic surfaces and precluded in hydrophilic ones.45
The mobility reversals observed in Fig. 3 deserve further discussion. Mobility reversals of colloids induced by counterions have been extensively reported in the literature.46–52 In general, they were induced by multivalent counterions, high surface charge densities and high salt concentrations. Under such conditions, the phenomenon of mobility reversal can be justified in terms of ion–ion correlations.53–55 However, the reversal of charged colloids induced by monovalent ions or very low multivalent salt concentrations is much scarcer and cannot be explained in terms of ionic correlations, thus other mechanisms are suggested.17,25,56–58 For instance, in the case of soft particles made of polyelectrolyte-grafted colloids, the mobility reversal observed in the presence of monovalent salt is governed by the properties of the grafted layer,58 whereas the reversals of anionic liposomes at low concentrations of trivalent cations were explained in terms of solvent mediated effects.59 In the case of hard particles, mobility reversal induced by monovalent ions can be dominated by the chaotropic/kosmotropic character of ions and the hydrophobic/hydrophilic character of surfaces.25,57 In our case, only the sulphate and citrate anions induced negative mobilities of the iron oxide particles. The monovalent salts did not induce this phenomenon.
The singular behavior of the mobility measured in the presence of citrate can be explained in terms of (i) a strong chemical affinity between the citrate ion and the surface of magnetite, which leads to a large charge reversal at relatively low concentrations of the ion; and by (ii) screening of the particle's charge as the ion concentration increases, which results in a decrease of the magnitude of the mobility. The affinity of the carboxyl groups for the surface of iron oxides is well known.60,61 This affinity depends on the structure of the particular organic acid, but in the case of citrate it seems to lead to the formation of metal–carboxylate surface complexes.62 In fact, citric acid has been routinely (and even commercially) used for the stabilization of iron oxide magnetic nanoparticles.10 Citrate adsorbs onto the iron oxide surface, leaving at least one carboxylic group exposed to the solution at neutral pH, which charges negatively the surface and provides stabilization. The behaviour of the mobility in the presence of sulphate follows a trend clearly different from that of the citrate curve, but in agreement with that observed on multivalent ions on other colloidal systems:50,52 there is a reversal of sign in the mobility and the reversed mobility increases in magnitude with ion concentration, until eventually it reaches a plateau. Interestingly, the affinity of the sulphate ion for the surface of the iron oxides is not so well known, but specific adsorption of sulphate was reported by Breeuwsma and Lyklema13 for hematite particles and by Andrés-Vergés et al.63 for magnetite particles that were synthesized following a method very similar to ours. These latter authors confirmed the presence of sulphate ions by means of infrared spectroscopy, and concluded that the ions were strongly attached since they were not eliminated by washing in water under sonication, or even after treatment with hydrochloric acid.
Simulation results
It is quite instructive to find out to what extent the electrokinetic behaviour described above can be justified (and even predicted) from the polarizabilities and the size of the different ions involved. To tackle this issue, the diffuse potentials of a planar magnetite surface in the presence of different concentrations of the same ions used in electrophoretic experiments were calculated by means of MC simulations (Fig. 4). | ||
| Fig. 4 Diffuse potential calculated using MC simulations, as a function of the concentration of salt for the same anions used in Fig. 3. Input data for simulations are shown in Tables 1 and 2. | ||
Assuming that tendencies for ψd should be similar to those found experimentally for μ, a qualitative comparison between both magnitudes can be done. As can be seen, there are different curves for ions with the same valence, which evidences the specificity of the interaction of these ions with magnetite. What is more, the general trends shown in Fig. 3 are captured by simulations. More specifically, the Hofmeister series for hydrophilic systems exhibited by our particles in experiments is also observed in simulation results: ψd(ClO4−) > ψd(SCN−) > ψd(CH3COO−) > ψd(SO42−) > ψd(HC6H5O72−). This means that chaotropic ions screened the charge of the particles more feebly than the kosmotropic ones. We could also mention that a diffuse potential reversal was achieved in simulations. However, this reversal was found only for HC6H5O72− at moderate and high salt concentrations, whereas mobility reversals were clearly observed for citrate and sulphate anions over a wider range of electrolyte concentrations. This qualitative difference between experiments and simulations will be discussed later in more detail.
Now let us elaborate on the simulation results shown in Fig. 4. Although the agreement between simulation predictions and experimental data is only qualitative, it is important to emphasize that the specific ion effects that characterize the electrokinetic behaviour of iron oxide particles were reproduced by simulations without requiring any fitting parameters. We should also keep in mind that input data used in simulations shown in Tables 1 and 2 were estimated using Lifshitz theory of van der Waals forces and the literature, respectively (see the Model and simulations section). Undoubtedly, the qualitative resemblance between Fig. 3 and 4 supports the capability of prediction of this theoretical approach.
Additionally, it suggests that ionic polarizabilities and sizes play a key role in justifying the ion specificity observed. In other words, these two magnitudes are to a great extent responsible for the specific effects of Hofmeister series on the electrokinetic potential of oxide iron particles. This finding agrees with the general assumptions of the most recent theories on surface hydration, which combined with hydrated nonelectrostatic potentials have been developed to predict experimental zeta potentials and hydration forces.5
Now, let us turn to the discrepancies in the charge reversal phenomena found in the experiments and simulations. It is evident that mobility reversal induced by kosmotropic anions is not properly predicted by simulations. SO42− and HC6H5O72− anions induce a clear reversal in the sign of μ whereas the diffuse potential reversal is only achieved in the presence of HC6H5O72− anions at moderate and high electrolyte concentrations, as mentioned before. At this point it is appropriate to recall that mobility reversal and potential reversal may not be necessarily equivalent. The reader should bear in mind that the diffuse potential was evaluated at the plane of the closest approach of the hydrated ions to the charged surface, also called the Outer Helmholtz Plane (OHP). On the other hand, electrophoretic mobility is associated with the ζ-potential defined in the shear plane. Although the approximation ψd ≈ ζ is acceptable for ideal surfaces, the OHP and shear planes are not strictly the same. For real colloids, the shear plane is expected to be further from the surface than the OHP.64 Accordingly the salt concentration at which mobility reversal was experimentally reached would not correspond to that required to induce a reversal in the diffuse potential In order to illustrate this feature, the electrostatic potential was calculated as a function of the distance from the magnetite surface (normalized by the ionic diameter) in the presence of SO42− anions at 100 and 200 mM concentrations (Fig. 5).
 | ||
| Fig. 5 Electrostatic potential calculated using MC simulations as a function of the distance from the surface for two concentrations of SO42−. Input data for simulations are shown in Tables 1 and 2. | ||
As can be seen in Fig. 5, the electrostatic potential at the OHP is always positive. However, for larger distances from the surface, a potential reversal can be reached, especially for the highest salt concentrations. Accordingly, better agreement with experiments would have been observed if an electrostatic potential further from the surface had been used to characterize electrokinetic phenomena. Indeed, this approach was employed in a previous work in which diffuse potentials, theoretically calculated from an integral equation theory, fit mobility reversal data for latex particles.65 Nevertheless, since the distance between the OHP and the shear plane is not clearly identified, this magnitude would become a fitting parameter in the calculations, and this procedure would reduce the predictive effectiveness of simulations and we preferred to avoid it.
In a previous paragraph, the diverse origins of the mobility reversals previously described in the literature were briefly commented. In a sense, it would be interesting to look into the origin of the reversal of the diffuse potential of magnetite nanoparticles observed in simulations in the presence of citrate anions. Given that this species was assumed to be divalent, the role of ion–ion correlations might be important. Computer simulations are extremely useful to shed light on this kind of issue since some interactions can be deliberately switched off. For instance, Fig. 6 shows the diffuse potential obtained in the presence of citrate anions if all the dispersion constants vanish and dispersion interactions are left out. The figure also includes the results previously obtained for the citrate and perchlorate anions, the latter serving as a reference of monovalent species in which the reduction of diffuse potential caused by dispersion forces is not intense. Let us focus on the results obtained in the absence of dispersion forces. As can be seen, when the valence of the anion is doubled (changing perchlorate into citrate ions), the diffuse potential considerably decreases but this curve does not exhibit a change of sign. The reversal only appears when dispersion forces are additionally switched on. This suggests that ion–ion correlations cannot justify the change of sign on its own.
 | ||
| Fig. 6 Diffuse potential calculated using MC simulations with and without dispersion forces in the presence of citrate anions as a function of the electrolyte concentration. The results obtained for perchlorate ions are also plotted for comparison. | ||
Finally, we are aware that the primitive model is excessively simple to accurately explain all the specific ion effects in colloidal systems. It leaves out many effects that sometimes might not be negligible, and more sophisticated models that include quantum dispersion forces are required. We could have also considered the affinity of the carboxyl groups and the sulphate ions for the surface of iron oxides, previously mentioned, in our simulations. The inclusion of this interaction would probably improve the agreement between experiment and simulations, but it would require the use of interaction parameters not known a priori. Actually, the fact that simulations did not take into account the chemical affinity could explain the discrepancy between the trends of the experimental and simulation citrate curves. It should be noted that the main difference between the curves is the larger mobility inversion measured at low ion concentrations, which, as mentioned above, must be related to the chemisorption of the citrate ion on the iron oxide surface.
Conclusions
We observed ion specific phenomena in colloidal suspensions of magnetite nanoparticles. Concretely, the electrophoretic mobility of the magnetite particles evolved very differently when exposed to increasing concentrations of different anions. The effect of an increasing concentration of chaotropic anions was milder and was limited to the screening of the charge until the mobility was negligible. In contrast, kosmotropic anions reduced the positive charge more effectively and caused a mobility inversion even at low concentrations. If the anions were ordered according to their effectiveness in decreasing the positive charge of the particles, they followed a Hofmeister series, with the exception of SCN− and ClO4− ions, which had their positions exchanged. This exception had also been observed by López-León and coworkers45 in some cationic hydrophilic colloids. It should be reminded that the bare surface of magnetite is known to be hydrophilic and positively charged at pH 4.Monte Carlo simulations were used to gain more insight into the origin of the behavior of different anions. Qualitatively speaking, the diffuse potential predicted for them using Lifshitz theory of van der Waals forces exhibited features very similar to those found in the electrophoretic mobility experiments. In particular, the same Hofmeister series was observed for the diffuse potential, which supports that simulations are a useful tool to qualitatively predict the specific ion effects on the electrokinetic behavior of iron oxide particles and other colloidal particles. Simulations also suggested that the polarizability of citrate ions and the associated dispersion forces play an important role in justifying the reversal observed in the presence of these anions, at least at high salt concentrations.
Acknowledgements
The authors thank the financial support from the following institutions: (i) ‘Ministerio de Economía y Competitividad, Plan Nacional de Investigación, Desarrollo e InnovaciónTecnológica (I + D + i)’, Projects MAT2013-44429-R, MAT2012-36270-C04-04 and -02. (ii) ‘Consejería de Innovación, Ciencia y Empresa de la Junta de Andalucía’, Projects P09-FQM-4698, P10-FQM-5977, and P11-FQM-7074. (iii) European Regional Development Fund (ERDF). Fernando Vereda is especially grateful to the “Programa de Reincorporación de Doctores” of the Universidad de Granada. Dr Ortega-Vinuesa is also sincerely acknowledged for helpful discussions.References
- A. Salis and B. W. Ninham, Chem. Soc. Rev., 2014, 43, 7358–7377 RSC.
- P. Lo Nostro and B. W. Ninham, Chem. Rev., 2012, 112, 2286–2322 CrossRef CAS PubMed.
- J. Lyklema, Chem. Phys. Lett., 2009, 467, 217–222 CrossRef CAS PubMed.
- W. Kunz, P. Lo Nostro and B. W. Ninham, Curr. Opin. Colloid Interface Sci., 2004, 9, 1–18 CrossRef CAS PubMed.
- D. F. Parsons, M. Boström, P. Lo Nostro and B. W. Ninham, Phys. Chem. Chem. Phys., 2011, 13, 12352–12367 RSC.
- A. Salis, F. Cugia, D. F. Parsons, B. W. Ninham and M. Monduzzi, Phys. Chem. Chem. Phys., 2012, 14, 4343–4346 RSC.
- P. Jungwirth and D. J. Tobias, Chem. Rev., 2006, 106, 1259–1281 CrossRef CAS PubMed.
- S. Bae, H. Son, Y.-G. Kim and S. Hohng, Phys. Chem. Chem. Phys., 2013, 15, 15829–15832 RSC.
- P. Tartaj, M. P. Morales, T. Gonzalez-Carreño, S. Veintemillas-Verdaguer and C. J. Serna, Adv. Mater., 2011, 23, 5243–5249 CrossRef CAS PubMed.
- K. Turcheniuk, A. V Tarasevych, V. P. Kukhar, R. Boukherroub and S. Szunerits, Nanoscale, 2013, 5, 10729–10752 RSC.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutiérrez, M. P. Morales, I. B. Böhm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306 RSC.
- F. Dumont and A. Watillon, Discuss. Faraday Soc., 1971, 52, 352–360 RSC.
- A. Breeuwsma and J. Lyklema, J. Colloid Interface Sci., 1973, 43, 437–448 CrossRef CAS.
- M.
![[C with combining breve]](https://www.rsc.org/images/entities/char_0043_0306.gif) olić, D. W. Fuerstenau, N. Kallay and E. Matijević, Colloids Surf., 1991, 59, 169–185 CrossRef.
olić, D. W. Fuerstenau, N. Kallay and E. Matijević, Colloids Surf., 1991, 59, 169–185 CrossRef. - D. R. Dixon, Colloids Surf., 1985, 13, 273–286 CrossRef CAS.
- M. Boström, D. Williams and B. Ninham, Phys. Rev. Lett., 2001, 87, 168103 CrossRef.
- T. López-León, A. B. Jódar-Reyes, D. Bastos-González and J. L. Ortega-Vinuesa, J. Phys. Chem. B, 2003, 107, 5696–5708 CrossRef.
- P. H. R. Alijó, F. W. Tavares and E. C. Biscaia, Jr, Colloids Surf., A, 2012, 412, 29–35 CrossRef PubMed.
- F. W. Tavares, M. Boström, E. R. A. Lima and E. C. Biscaia, Fluid Phase Equilib., 2010, 296, 99–105 CrossRef CAS PubMed.
- E. R. A. Lima, F. W. Tavares and E. C. Biscaia, Phys. Chem. Chem. Phys., 2007, 9, 3174–3180 RSC.
- V. Deniz, M. Boström, D. Bratko, F. W. Tavares and B. W. Ninham, Colloids Surf., A, 2008, 319, 98–102 CrossRef CAS PubMed.
- M. Boström, F. W. Tavares, B. W. Ninham and J. M. Prausnitz, J. Phys. Chem. B, 2006, 110, 24757–24760 CrossRef PubMed.
- F. W. Tavares and D. Bratko, J. Phys. Chem. B, 2004, 108, 9228–9235 CrossRef CAS.
- E. R. A. Lima, D. Horinek, R. R. Netz, E. C. Biscaia, F. W. Tavares, W. Kunz and M. Boström, J. Phys. Chem. B, 2008, 112, 1580–1585 CrossRef CAS PubMed.
- C. Calero, J. Faraudo and D. Bastos-González, J. Am. Chem. Soc., 2011, 133, 15025–15035 CrossRef CAS PubMed.
- A. Martín-Molina, J. G. Ibarra-Armenta and M. Quesada-Pérez, J. Phys. Chem. B, 2009, 113, 2414–2421 CrossRef PubMed.
- M. Quesada-Pérez, R. Hidalgo-Álvarez and A. Martín-Molina, Colloid Polym. Sci., 2010, 288, 151–158 Search PubMed.
- A. E. Regazzoni, M. A. Blesa and A. J. G. Maroto, J. Colloid Interface Sci., 1983, 91, 560–570 CrossRef CAS.
- S. Vidojkovic, V. Rodriguez-Santiago, M. V. Fedkin, D. J. Wesolowski and S. N. Lvov, Chem. Eng. Sci., 2011, 66, 4029–4035 CrossRef CAS PubMed.
- B. Faure, G. Salazar-Alvarez and L. Bergström, Langmuir, 2011, 27, 8659–8664 CrossRef CAS PubMed.
- T. Sugimoto and E. Matijevic, J. Colloid Interface Sci., 1980, 74, 227–243 CrossRef CAS.
- F. Vereda, J. de Vicente and R. Hidalgo-Alvarez, J. Colloid Interface Sci., 2013, 392, 50–56 CrossRef CAS PubMed.
- F. Vereda, J. De Vicente, M. Del Puerto Morales, F. Rull and R. Hidalgo-Álvarez, J. Phys. Chem. C, 2008, 112, 5843–5849 CAS.
- M. Dijkstra, Curr. Opin. Colloid Interface Sci., 2001, 6, 372–382 CrossRef CAS.
- P. Linse, Adv. Polym. Sci., 2005, 185, 111–162 CAS.
- A. V. Dobrynin, Curr. Opin. Colloid Interface Sci., 2008, 13, 376–388 CrossRef CAS PubMed.
- R. Kovács, M. Valiskó and D. Boda, Condens. Matter Phys., 2012, 15, 23803 CrossRef.
- M. Valiskó, D. Henderson and D. Boda, Condens. Matter Phys., 2013, 16, 43601 CrossRef.
- M. Quesada-Pérez, A. Martín-Molina and R. Hidalgo-Alvarez, J. Chem. Phys., 2004, 121, 8618–8626 CrossRef PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces/Jacob N. Israelachvili, Academic Press, London, San Diego, 1991 Search PubMed.
- M. Boström, D. Parsons and A. Salis, Langmuir, 2011, 27, 9504–9511 CrossRef PubMed.
- M. Boström, D. Williams and B. Ninham, Biophys. J., 2003, 85, 686–694 CrossRef.
- G. D. Mahan, J. Chem. Phys., 1982, 76, 493 CrossRef CAS PubMed.
- F. Vereda, J. de Vicente and R. Hidalgo-Álvarez, Colloids Surf., A, 2008, 319, 122–129 CrossRef CAS PubMed.
- T. López-León, M. J. Santander-Ortega, J. L. Ortega-Vinuesa and D. Bastos-González, J. Phys. Chem. C, 2008, 112, 16060–16069 Search PubMed.
- R. Ottewill and J. Shaw, J. Colloid Interface Sci., 1968, 26, 110–119 CrossRef CAS.
- M. Elimelech and C. R. O'Melia, Colloids Surf., 1990, 44, 165–178 CrossRef CAS.
- M. Tanaka and A. Y. Grosberg, J. Chem. Phys., 2001, 115, 567 CrossRef CAS PubMed.
- M. Quesada-Pérez, A. Martín-Molina, F. Galisteo-González and R. Hidalgo-Álvarez, Mol. Phys., 2002, 100, 3029–3039 CrossRef PubMed.
- A. Martín-Molina, M. Quesada-Pérez, F. Galisteo-González and R. Hidalgo-Álvarez, Colloids Surf., A, 2003, 222, 155–164 CrossRef.
- K. Besteman, M. A. G. Zevenbergen, H. A. Heering and S. G. Lemay, Phys. Rev. Lett., 2004, 93, 170802 CrossRef CAS.
- A. Martín-Molina, J. A. Maroto-Centeno, R. Hidalgo-Álvarez and M. Quesada-Pérez, Colloids Surf., A, 2008, 319, 103–108 CrossRef PubMed.
- A. Y. Grosberg, T. T. Nguyen and B. I. Shklovskii, Rev. Mod. Phys., 2002, 74, 329–345 CrossRef CAS.
- Y. Levin, Rep. Prog. Phys., 2002, 65, 1577–1632 CrossRef CAS.
- M. Quesada-Pérez, E. González-Tovar, A. Martín-Molina, M. Lozada-Cassou and R. Hidalgo-Alvarez, ChemPhysChem, 2003, 4, 234–248 CrossRef PubMed.
- T. López-León, a. B. Jódar-Reyes, J. L. Ortega-Vinuesa and D. Bastos-González, J. Colloid Interface Sci., 2005, 284, 139–148 CrossRef PubMed.
- A. Martín-Molina, C. Calero, J. Faraudo, M. Quesada-Pérez, A. Travesset and R. Hidalgo-Álvarez, Soft Matter, 2009, 5, 1350 RSC.
- S. Raafatnia, O. A. Hickey and C. Holm, Phys. Rev. Lett., 2014, 113, 238301 CrossRef CAS.
- A. Martín-Molina, C. Rodríguez-Beas and J. Faraudo, Phys. Rev. Lett., 2010, 104, 168103 CrossRef.
- L. Shen, P. E. Laibinis and T. A. Hatton, ACS Macro Lett., 1999, 15, 447–453 CAS.
- A. Wooding, M. Kilner and D. B. Lambrick, J. Colloid Interface Sci., 1991, 144, 236–242 CrossRef CAS.
- E. Tombácz, I. Y. Tóth, D. Nesztor, E. Illés, A. Hajdú and M. Szekeres, Colloids Surf., A, 2013, 435, 91–96 CrossRef PubMed.
- M. Andrés Vergés, R. Costo, a. G. Roca, J. F. Marco, G. F. Goya, C. J. Serna and M. P. Morales, J. Phys. D: Appl. Phys., 2008, 41, 134003 CrossRef.
- R. J. Zasoski, in Encyclopedia of Soil Science SE - 644, ed. W. Chesworth, Springer, Netherlands, 2008, pp. 841–845 Search PubMed.
- A. Martín-Molina, M. Quesada-Perez, F. Galisteo-Gonzalez and R. Hidalgo-Alvarez, Trends Colloid Interface Sci. XVI, 2004, vol. 123, pp. 114–118 Search PubMed.
| This journal is © the Owner Societies 2015 |