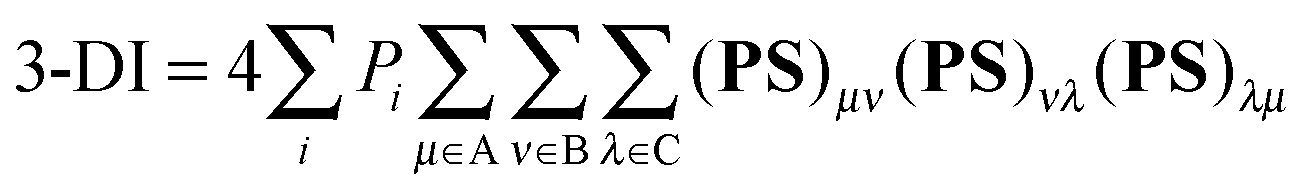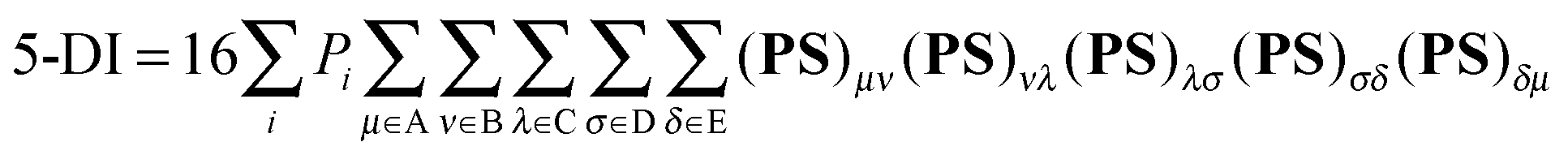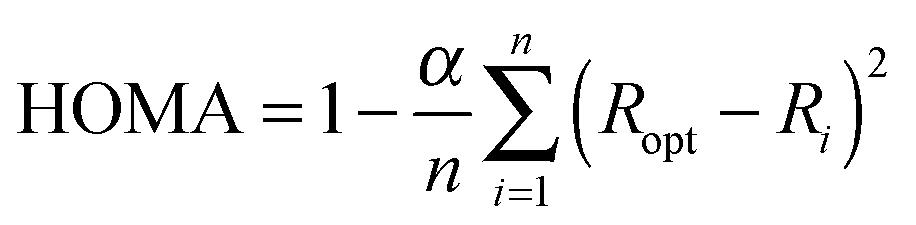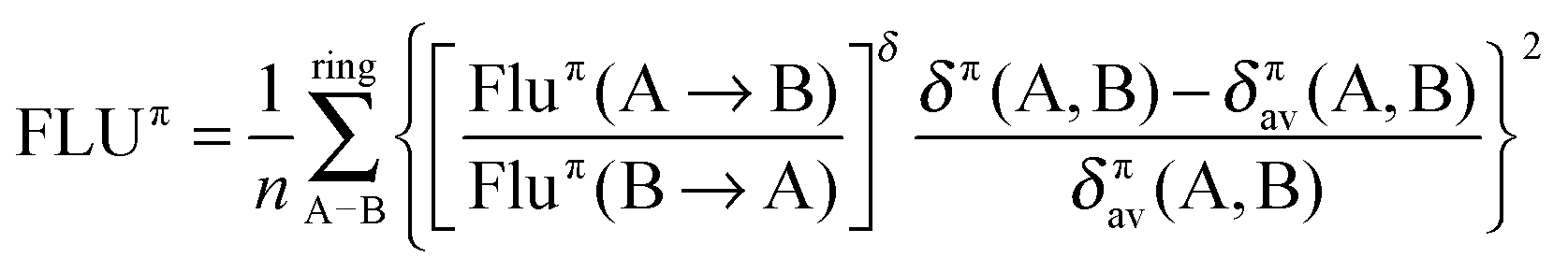Tunable aromaticity in bicalicenes†
Received
16th February 2015
, Accepted 2nd June 2015
First published on 2nd June 2015
Abstract
The unusual aromatic stability of cyclic bicalicene has been suggested to come from a tetraionic structure, where positive and negative charges are located on the cyclopropene and cyclopentadiene rings, respectively. Energetic, magnetic, geometric and electron delocalization analysis performed on a series of bicalicene derivatives, incorporating different electron donating and withdrawing groups, and electrically perturbed bicalicene structures provide additional proof of the role played by this tetraionic structure in the aromatic stability of bicalicene. In this work the aromatic stabilization is chemically and electrically tuned, enhancing or disrupting the electron delocalization and aromatic stability of the cyclopropene and cyclopentadiene rings by increasing or decreasing their corresponding charges. It is shown how the electron delocalization within these rings is similar to that of cyclopropene cation and cyclopentadiene anion for a perfect polarization of one electron.
Introduction
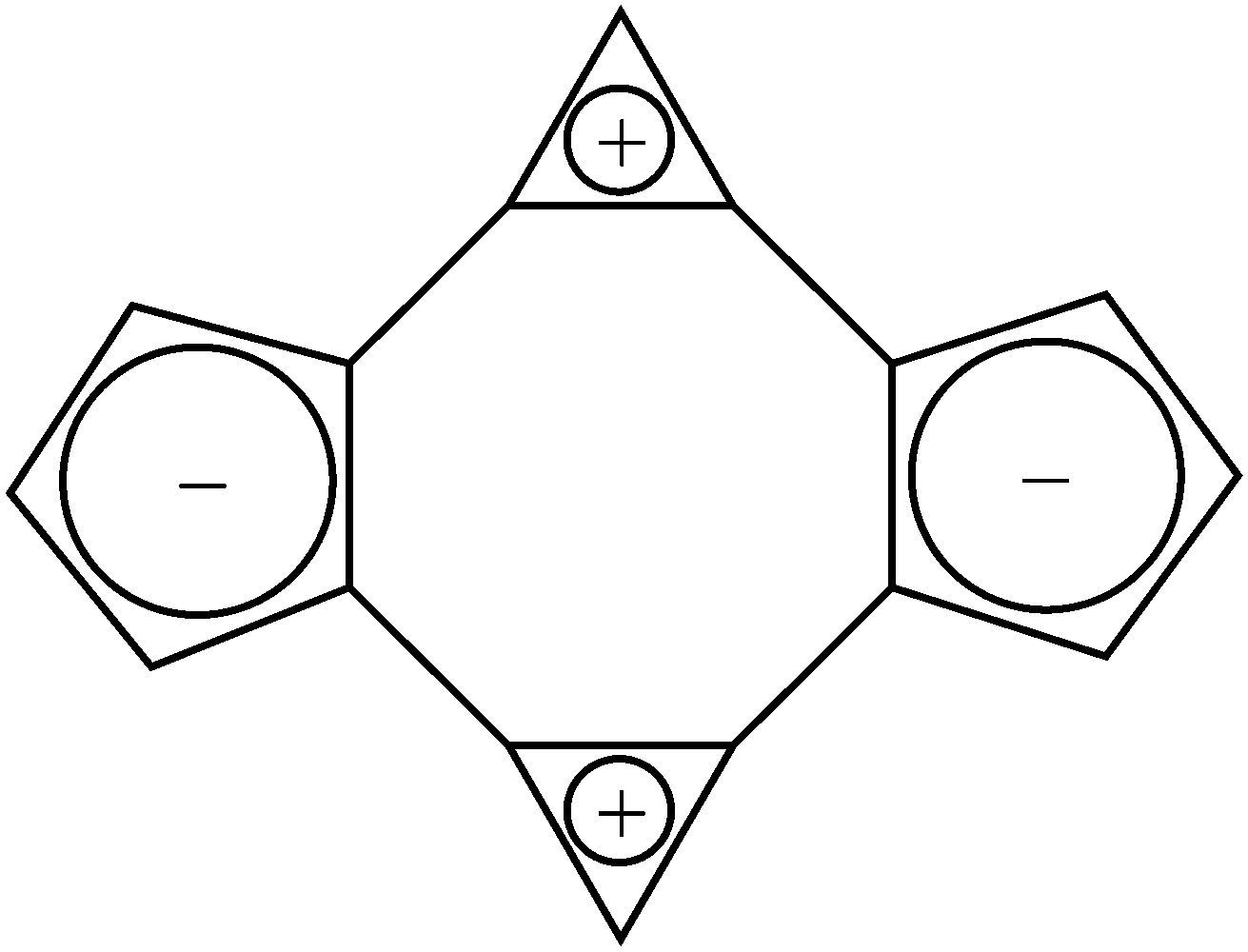
Cyclic bicalicene represents an anomalous case of a planar 16π system stabilized by aromaticity.1 Several experimental and theoretical proofs of its aromatic stabilization2–8 and also that of other calicene systems9–11 have been reported since its first synthesis in 1982.1 Thus, cyclic bicalicene displays a planar geometry with non-alternant carbon–carbon bonds (D2h symmetry), significant stability at room temperature and absence of upfield chemical shifts in its 1H-NMR spectrum.2 A few theoretical studies have been carried out in order to characterize the aromaticity of this compound as well as to identify the origin of such. Thus, the global aromatic character of bicalicene, confirmed by measures of homodesmotic reaction energies,8 topological and bond resonance energies5–7 and ring current maps,8 was ascribed to the cyclopropene and cyclopentadiene rings. The aromaticity arisen from these rings would overcome the antiaromatic character of the peripheral 16π and internal 8π rings. In order to become aromatic, the cyclopropene and cyclopentadiene rings in bicalicene would adopt positive and negative charges, respectively, approaching their behaviour to the 2π cyclopropene cation and 6π cyclopentadiene anion aromatic rings (see Scheme 1).
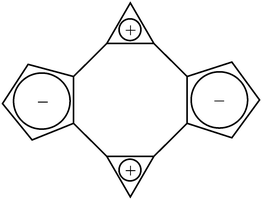 |
| | Scheme 1 | |
The role played by the tetraionic form of Scheme 1 can be definitely revealed by analysing the changes exerted by different electron donating and electron withdrawing groups (EDGs and EWGs) as well as external electric perturbations on the global and local aromaticity of bicalicene. Thus, EDGs and EWGs will exert an opposite effect on the ring electron charges that must be reflected on the electron delocalization and aromatic stability. As long as the aromaticity of bicalicene has its origin in this tetraionic form, the changes experienced by the ring electron delocalization and aromatic stability must be ruled by the orientation of these groups. Additionally, the ring electron charge can be also ‘modulated’ by means of external electric perturbations. Thus, electric perturbations that enhance the polarization represented by Scheme 1 must reinforce also the electron delocalization within the cyclopropene and cyclopentadiene rings and the aromaticity of bicalicene. On the contrary, electric perturbations which tend to reverse this polarization must reflect just the opposite. In this work we show these arguments are indeed correct, demonstrating the tetraionic structure of Scheme 1 is the origin of the aromatic stabilization of bicalicene through local aromatic rings of cyclopropene and cyclopentadiene.
Computational details
All the geometries and wavefunction calculations were performed at the HF/6-311G(d,p) level since multicenter delocalization indices can be calculated exactly only at HF level.12 Differences between, for instance, HF and DFT(B3LYP) ring currents were found to be negligible,8 which indicates electron correlation plays a minor role in the aromaticity of bicalicene. However, we have reoptimized the structures using the B3LYP level and compared the energetic parameters obtained with the HF ones.
Multicenter indices and ring charges were calculated using the Mulliken atomic partitioning scheme through home-made Fortran codes. The density matrices required for the calculation of these magnitudes were obtained from Gaussian 09 program.13 The program GaussView 5.014 was employed for visualization of the electron deformation orbitals and associated electron densities, which were previously calculated with a home-made program. The reader is referred to ref. 12, 15–19 and 20 for a full description and theoretical backgrounds of multicenter delocalization indices and electron deformation orbitals, respectively. In this work, we have employed the renormalized version of multicenter delocalization indices defined in ref. 16. For the case of 3-center and 5-center delocalization indices (3-DI and 5-DI) using Mulliken atomic partition, they adopt the following forms;
| | 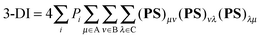 | (1) |
| | 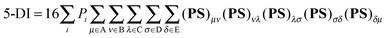 | (2) |
where A, B, C,… are atomic labels,
Pi is a permutation operator, which includes all the possible permutations of these atomic labels in
eqn (1) and (2),
PS represents the product matrix of the density,
P, and overlap,
S, matrices, and
μ,
ν,
λ,… are atomic basis functions.
In order to compare with aromaticity indices based on different criteria, we have also calculated the HOMA index (Harmonic Oscillator Model of Aromaticity),21 the NICS index (Nucleus Independent Chemical Shift)22 and the FLU index (Aromatic Fluctuation).23 In particular, we have employed here the zz component of the magnetic shielding tensor calculated 1 Å over the ring center, NICSzz(1), and also the π component of this index, NICSπzz(1), after classification of molecular orbitals (MOs) in σ and π symmetry. The orbital decomposition of the magnetic shielding tensor was performed with the help of the NBO program package.24 These indices were characterized as the best NICS indicators of aromaticity in planar polycyclic compounds with a hydrocarbonated frame.25 For the calculation of the HOMA indices we have employed the following expression,
| | 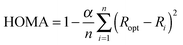 | (3) |
where
n represents de number of bonds forming the ring,
α is constant adjusted to give HOMA = 0 for a model nonaromatic ring and HOMA = 1 for a ring with all bonds equal to and optimal value
Ropt.
Ri is the bond distance of the
i bond. In this work we have obtained
α(81.03) and
Ropt (1.387 Å) from the calculated values of the C–C bond distances in ethane and ethene following the procedure described in
ref. 21.
For the FLU index, we have opted for the π version (FLUπ), which does not require of a reference system and is the best choice for π aromatic compounds where the σ–π partitioning of molecular orbitals is possible. FLUπ is obtained from the following expression,
| | 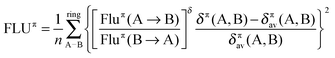 | (4) |
where Flu
π(A → B) is a function that measures the importance of the π electron fluctuation between atoms A and B with respect to the total π electron fluctuation in atom A and depends on the π 2-center delocalization indices (see
ref. 23 for details) and
δπ(A,B) and
δπav(A,B) are the π 2-center delocalization index between A and B and the average value of
δπ, respectively.
Energies for homodesmotic reactions in Fig. 3 represent only the change in the electronic energy and do not include the zero point vibrational energy.
Results and discussion
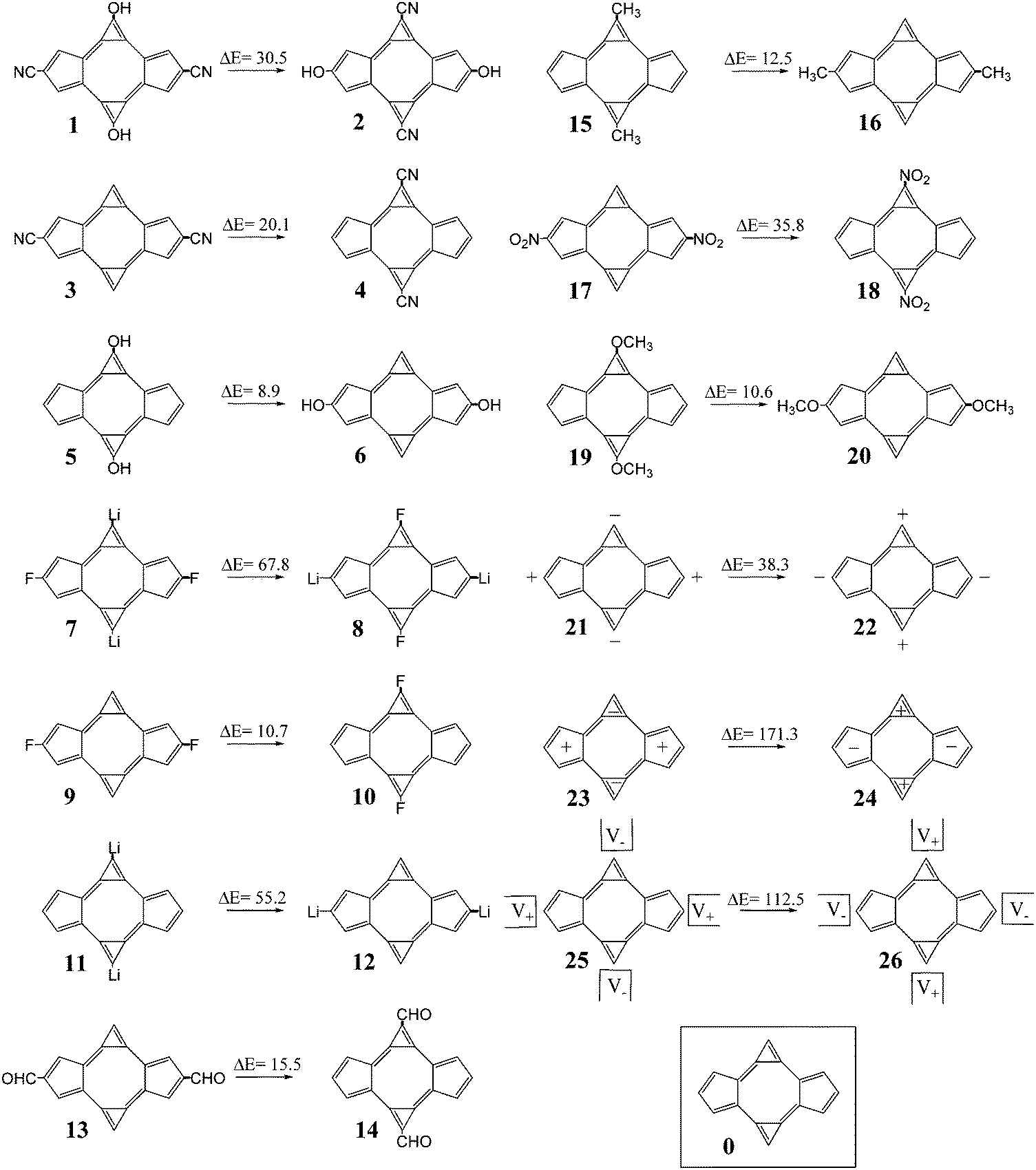
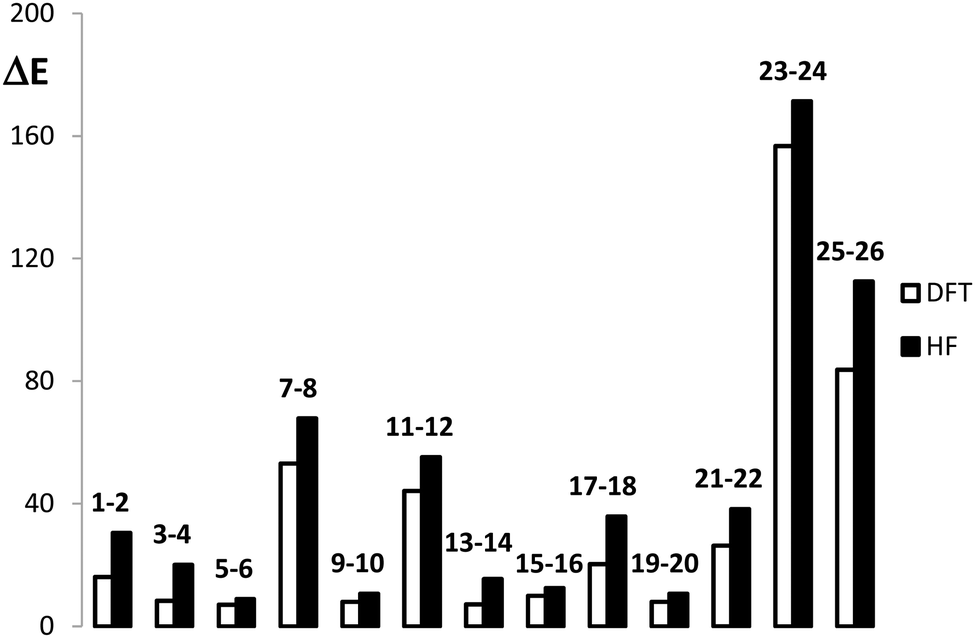
The structures considered for this work comprise different derivatives and electrically perturbed bicalicene systems which are collected in Fig. 1. All the optimized structures are planar and the atomic Cartesian coordinates obtained at the HF and B3LYP levels are included in the ESI† (Tables S1 and S2). As EDGs and EWGs, for the bicalicene derivatives, we have considered three groups with positive resonance effects (+R) and negative inductive effects (−I), –OH, –OCH3 and –F, two groups with negative resonance effects (−R), –CN and –CHO, a group with negative resonance effects (−R) and negative inductive effects (−I), –NO2, and two groups with positive inductive effects (+I), –CH3 and –Li. Overall, –OH, –OCH3, –CH3 and –Li, are EDGs whereas –CN, –CHO, –NO2 and –F are EWGs. We have incorporated these groups and some combinations of them either into the cyclopropene or the cyclopentadiene rings, so that forming different positional isomers. The energies of these isomers have been compared and the corresponding isomerization energies calculated and collected in Fig. 1. As one can see, the most stable isomers are those incorporating EDGs and EWGs in the cyclopropene and cyclopentadiene rings, respectively. This result confirms the stability of the different isomers is controlled by the orientation of the groups. The B3LYP isomerization energies reflect exactly the same trend, the comparison with the HF ones (Fig. 2) indicate that electron correlation does not change the relative stability of the isomers but also reflect a slight overestimation of the isomerization energy by HF.
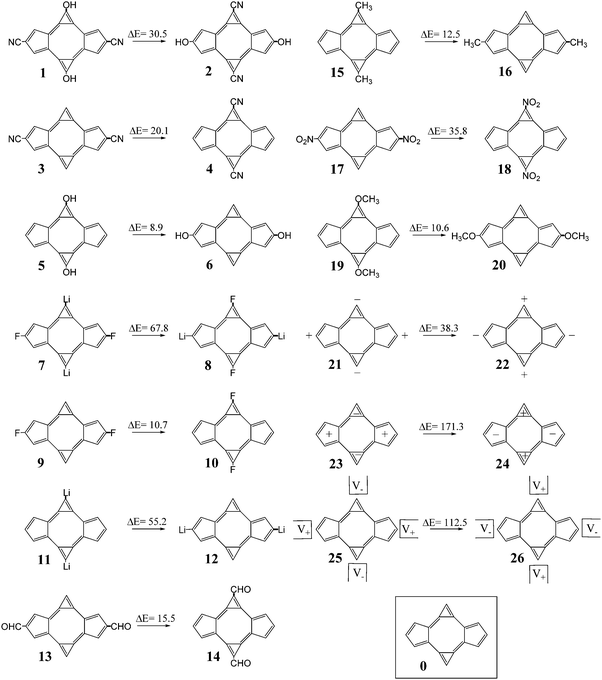 |
| | Fig. 1 Structures and nomenclature of the different bicalicene derivatives and bicalicene structures subjected to different electric perturbations. The outside charges in 21 and 22 correspond to 1 au whereas inside charges in 23 and 24 correspond to 0.1 au. The quadrupole field intensity in 25 and 26 is 8 × 10−3 au. In the figure are reflected, in kcal mol−1, the values of the corresponding isomerization energies calculated at HF level. | |
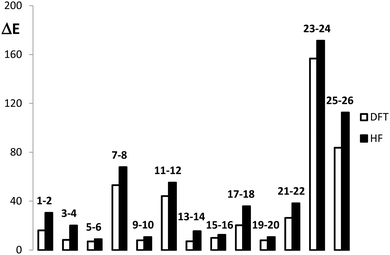 |
| | Fig. 2 Comparison of the B3LYP and HF energy differences (in kcal mol−1) calculated for the processes reported in Fig. 1. | |
On the other hand, the effect of different external electric perturbations on the energetic stability of bicalicene is also shown in Fig. 1. The electric perturbations comprise positive and negative point charges placed outside and inside of the rings (structures 21–24) and external quadrupole electric fields (structures 25, 26). In all cases when the perturbation tends to concentrate positive and negative charges on the cyclopropene and cyclopentadiene rings, respectively, the stability increases significantly with respect to those that tend to polarize the charge in the opposite direction. Both HF and B3LYP energy differences reflect this trend with a small overestimation for the HF case (see Fig. 2). However, the total energy differences given in Fig. 1 depend on two terms (eqn (5));
the difference in the electrostatic interaction between the external electric field and the charge distribution in the polarized molecule (Δ
Eelec); and the change in the intramolecular deformation energy (Δ
Edef). The first term is purely electrostatic and depends on the electron and nuclear charge distribution and the strength of the external electric perturbation, whereas the second one accounts for the energetic cost of polarizing the ground state electron density against the internal electron–nuclei forces. Thus, we have calculated the electrostatic and deformation terms for the pairs
21–22,
23–24 and
25–26. The results are collected in
Table 1, both electrostatic and deformation energies decrease in structures
21,
23 and
25 with respect to structures
22,
24 and
26. The most relevant result for this study is related to the values of the deformation energy. External charge and quadrupole field orientations in structures
21,
23 and
25 reinforces the charge polarization represented by the
Scheme 1 whereas this charge polarization is reduced in structures
22,
24 and
26. This means the energetic cost of increasing the positive and negative charges of cyclopropene and cyclopentadiene rings, respectively, in bicalicene is less than the energetic cost of reducing them. This result is remarkable taking into account that the molecule already supports significant positive and negative charges on these rings
8 (0.20 au for cyclopropene and −0.57 au for cyclopentadiene for the total Mulliken charges, and 0.46 au and −0.44 au for the π Mulliken charges calculated in this work at the HF level). Respect to all of mentioned above, the most outstanding case is the pair
23–24 with a large change in the deformation energy (22.8 kcal mol
−1). In this pair the electric perturbation is located at the center of the rings, producing the most ‘symmetric’ polarization of the π charge within the rings,
i.e., all the carbon atoms within a given ring increases or decreases its π charge depending on the external field orientation. On the contrary, in the structures
21,
22,
25 and
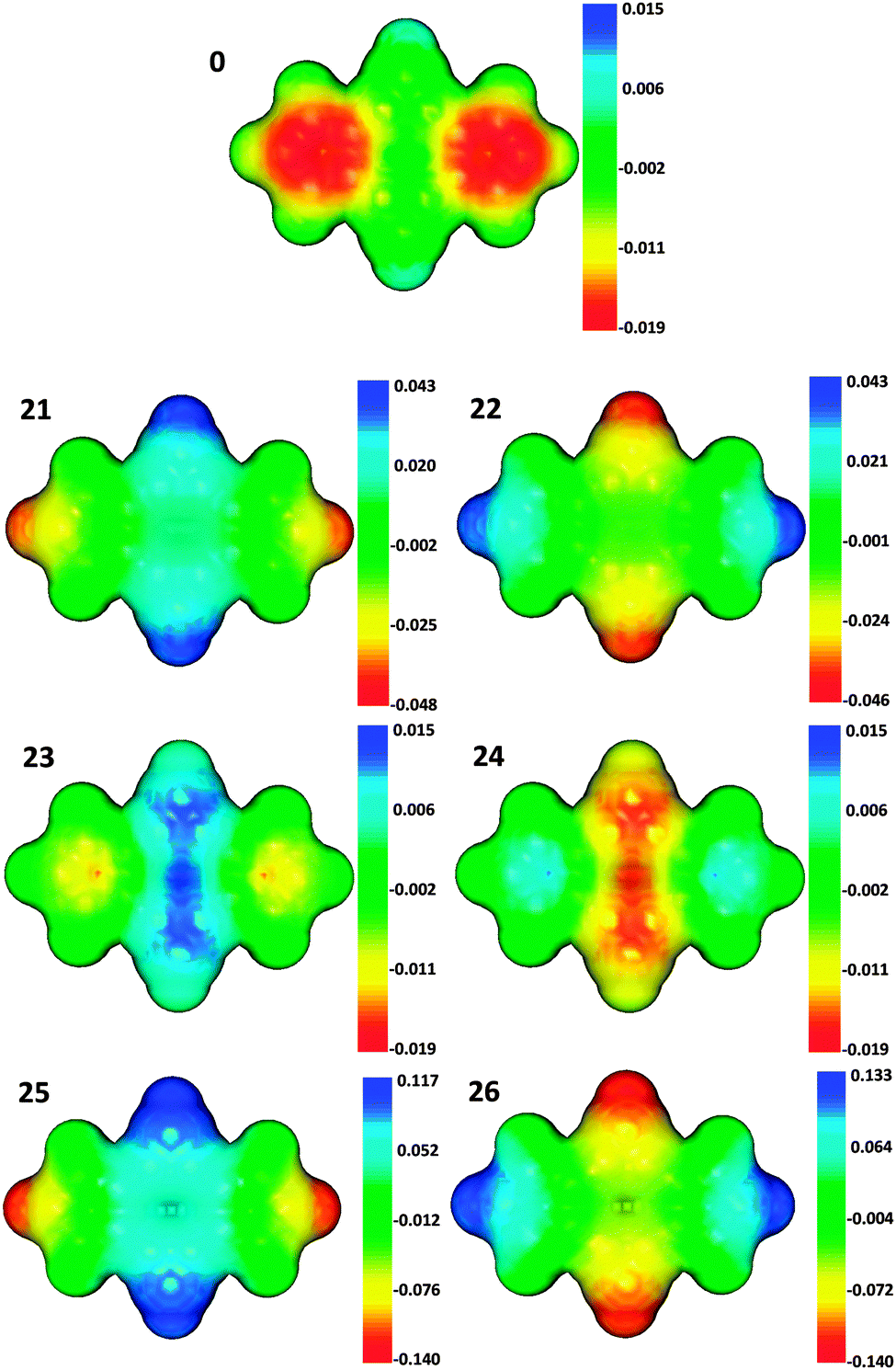
26, the electric perturbation produce opposite changes in the π charge of carbon atoms located in the same ring, even though the change of the total π ring charge follows the expected trend. In these structures the largest charge accumulation/depletion takes place at the most external atom of the ring, which is the nearest atom to the electric perturbation. These differences in the atomic charge polarization are well-reflected in the molecular electrostatic potentials (MEPs) of these structures relative to unperturbed bicalicene (
0) (
Fig. 3). The plots shown in
Fig. 3 for the structures
21–26 represent the difference in the MEP of each structure minus the MEP in
0, so that the changes introduced by the electric perturbation can be appreciated more clearly. Thus,
Fig. 3 reflects perfectly the positive and negative MEP over the cyclopropene and cyclopentadiene rings in
0 and the reinforcement of this picture in structures
21,
23 and
25 and the reverse trend followed by structures
22,
24 and
26. In addition, the symmetric changes in the ring charge polarization in the pair
23–24 and the asymmetric changes in the pairs
21–22 and
25–26 are also noticeable in the plots of
Fig. 3. This ‘asymmetric’ polarization is not fully in agreement with
Scheme 1 and could be in part the cause of the smaller energy differences displayed by the pairs
21–22 and
25–26 with respect to
23–24. This claim is reinforced by the fact that the π ring charges (3-Q
π and 5-Q
π data in
Table 2) experience changes with respect to bicalicene larger in these pairs than in pair
23–24, so that the relative stability is not only related to the magnitude of the total ring charge but also to the distribution of this charge along the atoms of the rings. Unfortunately, the information provided by the MEP in bicalicene derivatives in the cyclopropene and cyclopentadiene regions is contaminated by the part of the electrostatic potential associated to the nuclei and electron charges of the substituent groups. Since this contribution to the MEP is quite different among the groups, a relation between electron donating and electron withdrawing effects and MEPs cannot be unequivocally established.
Table 1 Electrostatic and deformation components of the energy differences given in Fig. 1 between the electrically perturbed bicalicene structures. All data are in kcal mol−1
| |
ΔE |
ΔEelec |
ΔEdef |
|
21–22
|
38.3 |
38.1 |
0.2 |
|
23–24
|
171.3 |
144.5 |
26.8 |
|
25–26
|
112.5 |
108.7 |
3.8 |
Table 2 Total and π 3-center and 5-center delocalization indices, 3-DI and 5-DI, together with the corresponding ring charges, 3-Q and 5-Q. All values are in au and relative to the bicalicene molecule (0)
| |
3-Q |
5-Q |
3-Qπ |
5-Qπ |
3-DI |
5-DI |
3-DIπ |
5-DIπ |
|
0
|
0.199 |
−0.568 |
0.465 |
−0.442 |
0.658 |
0.882 |
1.136 |
0.836 |
|
1
|
0.282 |
0.145 |
−0.052 |
−0.056 |
−0.536 |
0.076 |
0.113 |
0.034 |
|
2
|
0.164 |
0.407 |
−0.129 |
0.028 |
−0.212 |
−0.203 |
−0.229 |
−0.193 |
|
3
|
0.032 |
0.159 |
0.052 |
−0.004 |
0.082 |
0.011 |
0.070 |
−0.027 |
|
4
|
0.175 |
0.108 |
−0.106 |
0.111 |
−0.235 |
−0.142 |
−0.201 |
−0.138 |
|
5
|
0.256 |
−0.016 |
−0.091 |
−0.059 |
−0.571 |
0.075 |
0.067 |
0.072 |
|
6
|
−0.003 |
0.292 |
−0.016 |
−0.084 |
−0.001 |
−0.049 |
−0.022 |
−0.043 |
|
7
|
−0.263 |
0.191 |
0.147 |
−0.202 |
0.645 |
0.148 |
0.180 |
0.170 |
|
8
|
0.327 |
−0.344 |
−0.156 |
0.126 |
−0.702 |
−0.092 |
−0.057 |
−0.122 |
|
9
|
0.012 |
0.353 |
0.009 |
−0.084 |
0.033 |
−0.007 |
0.015 |
0.009 |
|
10
|
0.328 |
−0.017 |
−0.100 |
−0.004 |
−0.602 |
0.002 |
0.020 |
−0.001 |
|
11
|
−0.274 |
−0.156 |
0.140 |
−0.123 |
0.618 |
0.176 |
0.169 |
0.173 |
|
12
|
−0.006 |
−0.317 |
−0.063 |
0.136 |
−0.089 |
−0.096 |
−0.087 |
−0.126 |
|
13
|
−0.030 |
−0.005 |
0.040 |
0.046 |
0.060 |
−0.044 |
0.053 |
−0.076 |
|
14
|
0.012 |
−0.067 |
−0.058 |
0.092 |
−0.369 |
−0.113 |
−0.178 |
−0.109 |
|
15
|
0.059 |
−0.004 |
0.003 |
−0.036 |
−0.054 |
0.043 |
0.046 |
0.043 |
|
16
|
0.002 |
0.018 |
−0.010 |
0.000 |
−0.007 |
0.018 |
−0.014 |
−0.014 |
|
17
|
−0.052 |
−0.355 |
0.066 |
0.001 |
0.104 |
−0.047 |
0.088 |
−0.048 |
|
18
|
−0.323 |
−0.093 |
−0.131 |
0.167 |
−0.992 |
−0.207 |
−0.275 |
−0.200 |
|
19
|
−0.264 |
0.022 |
−0.085 |
−0.068 |
−0.243 |
0.086 |
0.071 |
0.083 |
|
20
|
0.011 |
−0.315 |
−0.022 |
−0.073 |
−0.024 |
−0.081 |
−0.035 |
−0.082 |
|
21
|
0.014 |
0.048 |
0.102 |
−0.101 |
0.043 |
0.132 |
0.150 |
0.121 |
|
22
|
0.023 |
0.021 |
−0.122 |
0.122 |
0.097 |
−0.156 |
−0.182 |
−0.148 |
|
23
|
0.067 |
−0.072 |
0.051 |
−0.050 |
0.065 |
0.044 |
0.049 |
0.054 |
|
24
|
−0.068 |
0.074 |
−0.055 |
0.054 |
−0.065 |
−0.052 |
−0.052 |
−0.057 |
|
25
|
0.187 |
0.126 |
0.286 |
−0.281 |
0.348 |
0.311 |
0.342 |
0.267 |
|
26
|
−0.113 |
0.315 |
−0.503 |
0.505 |
0.332 |
−0.526 |
−0.625 |
−0.506 |
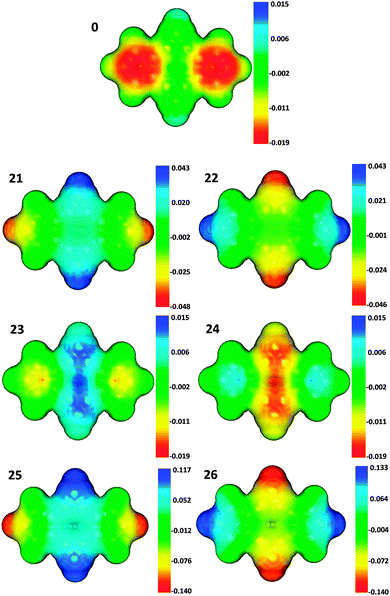 |
| | Fig. 3 Molecular electrostatic potential (MEP) of bicalicene (0) and the difference in the MEP of electrically perturbed bicalicene structures (21–26) with respect to 0. | |
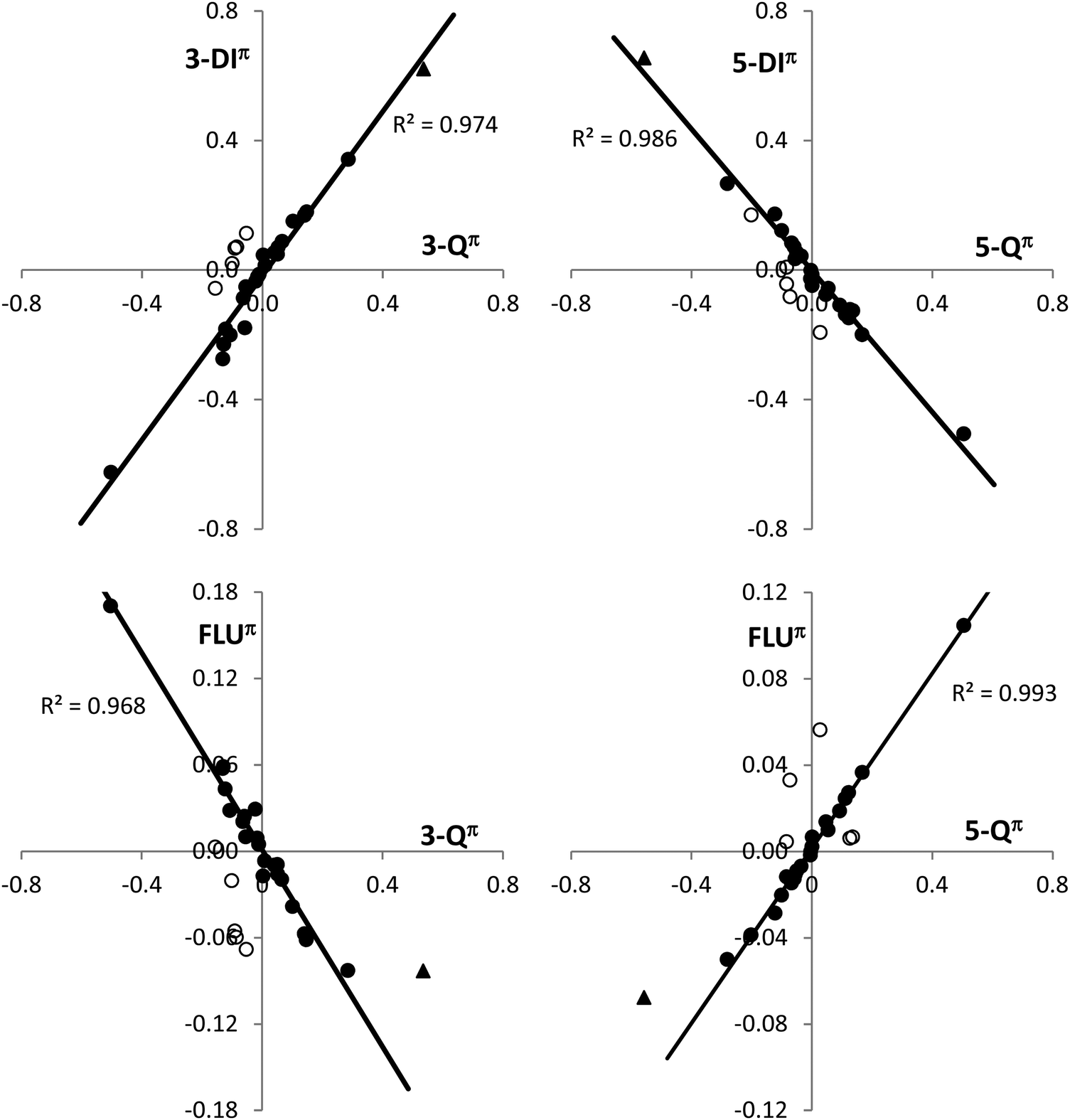
Calculation of 3-center and 5-center delocalization indices, 3-DI and 5-DI, allows determining the effect of the charge polarization on the electron delocalization within the cyclopropene and cyclopentadiene rings (Table 2). Unsubstituted bicalicene displays a π 3-center delocalization index, 3-DIπ, of 1.136 and a π 5-center delocalization index, 5-DIπ, of 0.836, which are significant values but far from those displayed by the cyclopropene cation (1.758) and cyclopentadiene anion (1.491). However, changes in 3-Qπ and 5-Qπ, by effect of the different groups or the external electric perturbations correlate quite well with the values of 3-DIπ and 5-DIπ. Thus, a good linear correlation between both magnitudes have been found for all the derivatives and electrically perturbed bicalicenes (Fig. 4). Only slight deviations from the fitting have been found for the rings containing the groups –OH, –OCH3, and –F. This may be due to the fact that these groups exert both inductive and resonance effects with opposite result, whereas –CN, –CHO, –CH3 and –Li only exert significant resonance effects in the former two and inductive in the latter two. In the case of –NO2, which also displays resonance and inductive effects, they do not counteract each other since both are negative and the resonance is expected to be clearly dominant. It must be remarked that isolated rings of cyclopropene cation and cyclopentadiene anion perfectly fit in the correlations shown in Fig. 4. This definitely reflects that polarization approaches the electron delocalization of the cyclopropene and cyclopentadiene rings in bicalicene to that of isolated cyclopropene cation and cyclopentadiene anion rings enhancing their aromaticity. On the other hand, using total delocalization indices, 3-DI and 5-DI, the correlation with the charges disappear for the cyclopropene ring even at qualitative level due to the large σ 3-center electron delocalization within this ring (see differences between 3-DI and 3-DIπ data in Table 2). As will be discussed later, the large sigma electron delocalization within the cyclopropene ring is also reflected in the magnetic response.
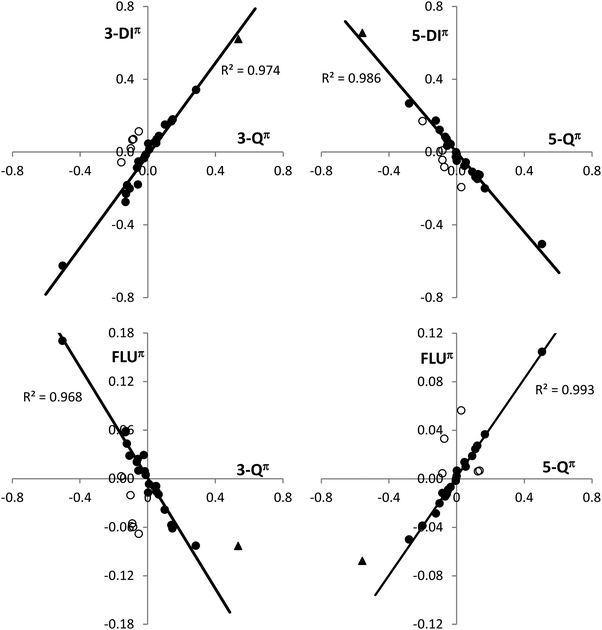 |
| | Fig. 4 Representation of the π 3-center delocalization index, 3-DIπ, and π 5-center delocalization index, 5-DIπ, (top plots) and the FLUπ index (bottom plots) for the cyclopropene and cyclopentadiene rings versus the corresponding π ring charges (3-Qπ and 5-Qπ) for the bicalicene derivatives and electrically perturbed bicalicene structures. In the representations are also included the cyclopropene cation and cyclopentadiene anion rings (represented by bold triangles). The values are given relative to the bicalicene molecule (0). The outliers, represented by empty dots, correspond to the rings substituted with –OH, –OCH3 and –F (see text for details). | |
A good correlation with the ring charges is also found for the FLUπ index (Fig. 4), with the exception of the points mentioned above. The FLUπ for these points, and for the isolated cyclopropene cation and cyclopentadiene anion, reflect a larger deviation with respect to the fitting line than the multicenter indices. In Table 3 the values of the FLUπ obtained for the cyclopropene and cyclopentadiene rings are collected together with the values of the HOMA index. The FLUπ index always reflects a larger aromaticity in the rings of the most stable conformation, i.e. with EDGs stabilizing the positively charged cyclopropene and EWGs the negatively charged cyclopentadiene rings with the only exception of the cyclopropene ring in the pair 9–10 with R = F (notice that smaller FLUπ reflects larger aromaticity). In the case of the HOMA index, the exceptions involve the pair 13–14 for the cyclopentadiene ring and the pairs 1–2, 5–6, 13–14 and 19–20 for the cyclopropene, although these exceptions reduces to only 5–6 and 13–14 when comparing the total HOMA for the molecule. This is not a surprising result, since the geometric criteria is expected to be more influenced by sigma bonding and steric factors associated to the nature of the substituent group and unconnected with the π aromaticity.
Table 3 HOMA and FLUπ indices for the cyclopropene (3-HOMA and 3-FLUπ) and cyclopentadiene rings (5-HOMA and 5-FLUπ) in the bicalicene structures. Total-HOMA and Total-FLUπ represent the summation of the corresponding values for the individual rings in each molecule
| |
3-FLUπ |
5-FLUπ |
Total-FLUπ |
3-HOMA |
5-HOMA |
Total-HOMA |
|
0
|
0.083 |
0.068 |
0.302 |
0.934 |
0.944 |
3.757 |
|
1
|
0.015 |
0.057 |
0.143 |
0.920 |
0.949 |
3.738 |
|
2
|
0.142 |
0.124 |
0.532 |
0.930 |
0.926 |
3.712 |
|
3
|
0.067 |
0.066 |
0.266 |
0.933 |
0.941 |
3.748 |
|
4
|
0.111 |
0.092 |
0.407 |
0.933 |
0.930 |
3.726 |
|
5
|
0.028 |
0.055 |
0.165 |
0.912 |
0.954 |
3.733 |
|
6
|
0.092 |
0.072 |
0.329 |
0.934 |
0.951 |
3.769 |
|
7
|
0.022 |
0.029 |
0.101 |
0.980 |
0.967 |
3.895 |
|
8
|
0.086 |
0.074 |
0.319 |
0.814 |
0.918 |
3.463 |
|
9
|
0.076 |
0.056 |
0.265 |
0.935 |
0.954 |
3.777 |
|
10
|
0.063 |
0.068 |
0.260 |
0.833 |
0.947 |
3.561 |
|
11
|
0.026 |
0.039 |
0.129 |
0.983 |
0.957 |
3.881 |
|
12
|
0.104 |
0.074 |
0.356 |
0.935 |
0.913 |
3.695 |
|
13
|
0.073 |
0.081 |
0.309 |
0.933 |
0.928 |
3.723 |
|
14
|
0.107 |
0.086 |
0.387 |
0.935 |
0.932 |
3.735 |
|
15
|
0.066 |
0.061 |
0.253 |
0.944 |
0.948 |
3.784 |
|
16
|
0.088 |
0.070 |
0.315 |
0.934 |
0.941 |
3.751 |
|
17
|
0.063 |
0.074 |
0.276 |
0.933 |
0.938 |
3.741 |
|
18
|
0.140 |
0.104 |
0.489 |
0.835 |
0.923 |
3.516 |
|
19
|
0.023 |
0.053 |
0.153 |
0.923 |
0.955 |
3.756 |
|
20
|
0.112 |
0.101 |
0.426 |
0.930 |
0.940 |
3.739 |
|
21
|
0.045 |
0.047 |
0.184 |
|
|
|
|
22
|
0.126 |
0.095 |
0.443 |
|
|
|
|
23
|
0.074 |
0.059 |
0.265 |
|
|
|
|
24
|
0.093 |
0.078 |
0.341 |
|
|
|
|
25
|
0.000 |
0.018 |
0.036 |
|
|
|
|
26
|
0.253 |
0.172 |
0.851 |
|
|
|
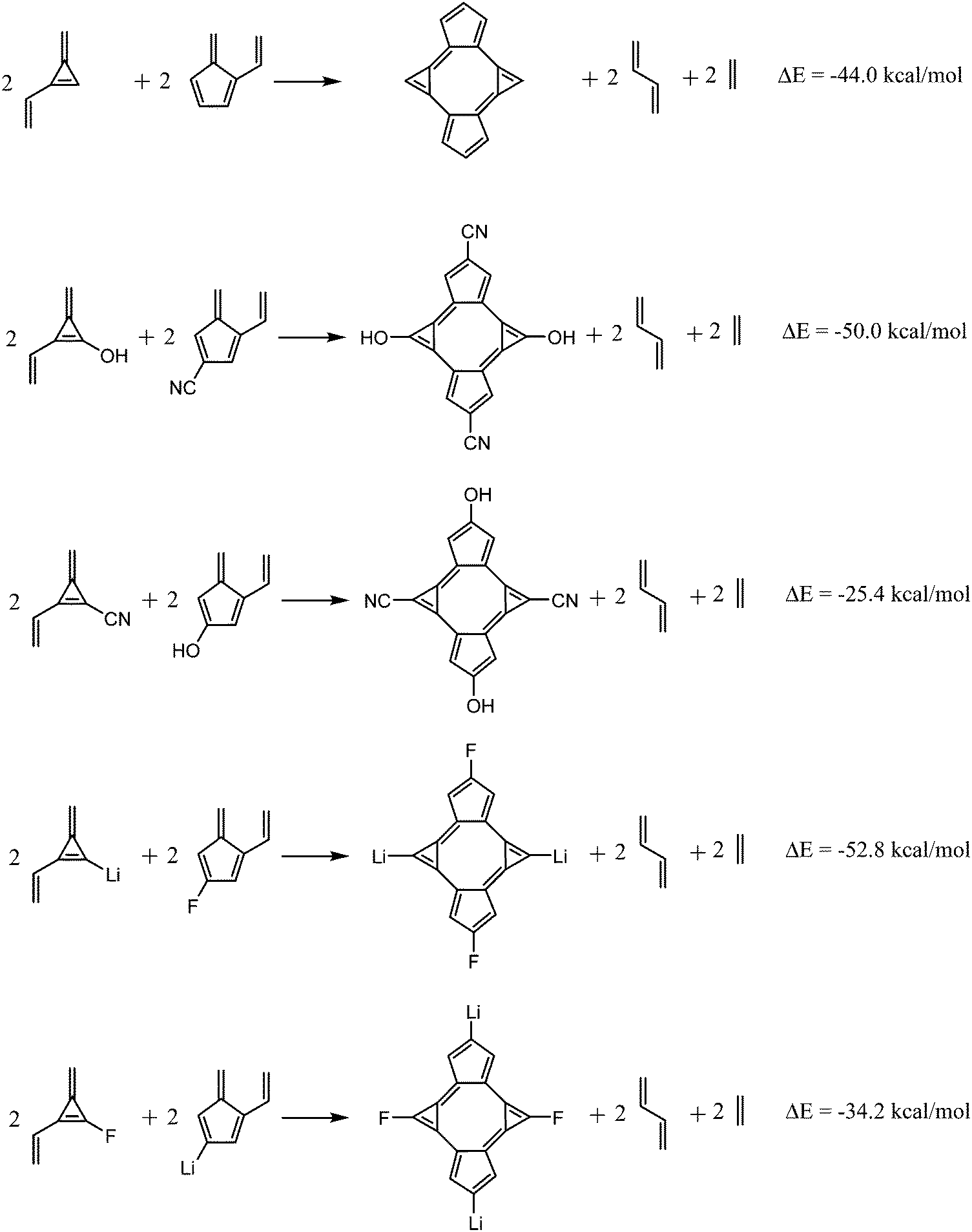
Another proof of the changes exerted by the EDGs and EWGs in the aromatic stability of bicalicene is given by the energies of the homodesmotic reactions shown in Fig. 5. As one can see, when the groups –CN and –F are linked to cyclopentadiene rings and –OH and –Li groups to cyclopropene the reactions are more exothermic than that of bicalicene. The contrary happens when the same groups are interchanged between the rings. This reflects the aromatic stabilization energy of bicalicene, accounted for by the first homodesmotic reaction of Fig. 5, increases or decreases depending on the ring substitution and its effect on the ring electron charges.
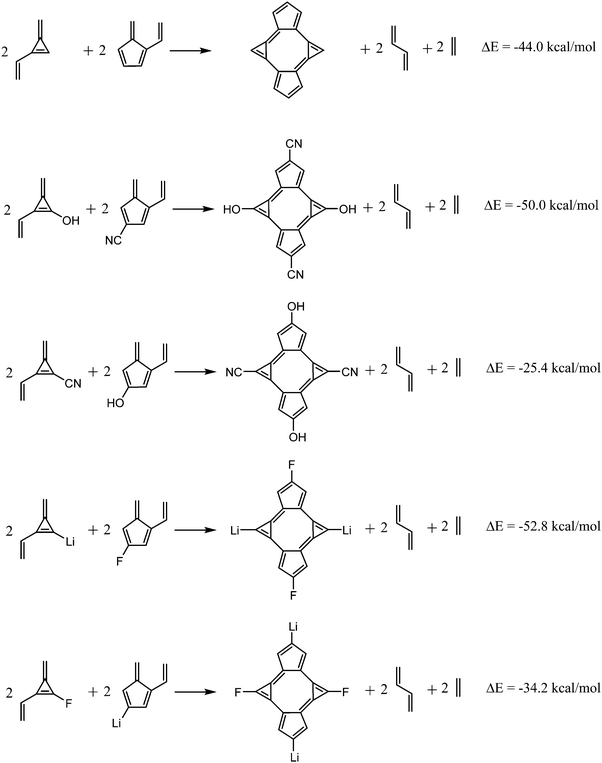 |
| | Fig. 5 Homodesmotic reactions involving bicalicene and selected bicalicene derivatives (1, 2, 7, 8). | |
Magnetic criteria of aromaticity also supports the aromaticity is larger in the bicalicene derivatives and electrically perturbed structures where the π electron charge is pushed from cyclopropene rings to cyclopentadiene. Thus, both total NICSzz(1) and NICSπzz(1) values collected in Table 4 are more negative in these structures with respect to their counterparts with the exception of the pair 5–6 (R = OH) and 19–20 (R = NO2) for NICSzz(1). Comparing ring by ring one finds more exceptions which also affects pairs 13–14 (R = CHO) for NICSzz(1) and 9–10 (R = F) for NICSπzz(1). Comparison with the unsubstituted bicalicene structure (0) reflects that the calculated NICS values are more negative for the structures 21 and 23 and less negative for 22 and 24.26 This is the expected trend, nevertheless, in the case of bicalicene derivatives the strong anisotropic effects associated to the resonance groups –CN and –OH do not only affect the strength of the ring currents and the NICS value can be significantly affected by other factors. Thus, comparison of the NICS values in these structures with those in structure 0 may not reflect properly the changes in the electron delocalization.
Table 4 NICSzz(1) and NICSπzz(1) values for the cyclopropene (3-NICS) and cyclopentadiene rings (5-NICS) in the bicalicene structures. Total-NICSzz (1) and Total-NICSπzz(1) represent the summation of the corresponding values for the individual rings in each molecule
| |
3-NICSzz(1) |
5-NICSzz(1) |
Total-NICSzz(1) |
3-NICSπzz(1) |
5-NICSπzz(1) |
Total-NICSπzz(1) |
|
0
|
−16.02 |
−21.77 |
−75.58 |
−1.41 |
−18.02 |
−38.86 |
|
1
|
−13.12 |
−20.02 |
−66.27 |
−2.31 |
−16.56 |
−37.74 |
|
2
|
−12.34 |
−18.94 |
−62.58 |
1.13 |
−16.22 |
−30.18 |
|
3
|
−17.37 |
−21.42 |
−77.58 |
−2.59 |
−17.98 |
−41.14 |
|
4
|
−14.28 |
−20.85 |
−70.26 |
−0.69 |
−17.07 |
−35.52 |
|
5
|
−11.66 |
−19.80 |
−62.91 |
−0.94 |
−16.05 |
−33.98 |
|
6
|
−13.97 |
−19.49 |
−66.93 |
0.52 |
−16.86 |
−32.68 |
|
7
|
−19.13 |
−21.69 |
−81.63 |
−1.32 |
−19.26 |
−41.16 |
|
8
|
−9.60 |
−19.60 |
−58.42 |
−0.52 |
−15.23 |
−31.50 |
|
9
|
−14.97 |
−20.35 |
−70.65 |
−0.31 |
−17.69 |
−36.00 |
|
10
|
−10.35 |
−19.77 |
−60.25 |
−1.13 |
−15.87 |
−34.00 |
|
11
|
−19.93 |
−23.30 |
−86.47 |
−2.20 |
−19.74 |
−43.88 |
|
12
|
−15.23 |
−21.78 |
−74.02 |
−0.79 |
−17.56 |
−36.70 |
|
13
|
−17.27 |
−20.46 |
−75.47 |
−2.63 |
−17.56 |
−40.38 |
|
14
|
−14.24 |
−21.21 |
−70.91 |
−0.85 |
−17.36 |
−36.42 |
|
15
|
−15.28 |
−21.58 |
−73.72 |
−1.17 |
−18.00 |
−38.34 |
|
16
|
−15.22 |
−20.40 |
−71.24 |
−0.80 |
−17.42 |
−36.44 |
|
17
|
−17.78 |
−20.02 |
−75.60 |
−3.02 |
−17.36 |
−40.76 |
|
18
|
−11.21 |
−19.88 |
−62.18 |
−0.68 |
−16.27 |
−33.90 |
|
19
|
−11.30 |
−19.67 |
−61.93 |
−0.79 |
−16.04 |
−33.66 |
|
20
|
−13.68 |
−19.29 |
−65.94 |
0.75 |
−16.72 |
−31.94 |
|
21
|
−17.41 |
−22.36 |
−79.54 |
|
|
|
|
22
|
−14.43 |
−21.36 |
−71.58 |
|
|
|
|
23
|
−16.65 |
−22.81 |
−78.91 |
|
|
|
|
24
|
−15.34 |
−20.70 |
−72.07 |
|
|
|
An important difference between multicenter delocalization indices and the rest is the degree of aromaticity predicted for the cyclopropene and cyclopentadiene rings. According to the 3-DIπ and 5-DIπ values, cyclopropene ring is more aromatic than cyclopentadiene. On the contray, FLUπ and HOMA indicate the cyclopentadiene ring is slightly more aromatic. NICSπzz(1) values indicate a scarce ring current in the cyclopropene ring, in qualitative agreement with previous calculations of π ring currents plots in bicalicene.8 On the contrary, NICSzz(1), which includes σ orbitals, is largely negative for cyclopropene but less than cyclopentadiene, reflecting an important contribution of the σ orbitals to the total NICS value, which can be related to a significant σ ring current. This is in agreement with the large σ electron delocalization reflected by the 3-DI values. It must be clarified that the σ part of the 3-DI is a negative value, but the sign of the multicenter delocalization index derives from topological factors27,28 and is not related with possible antiaromatic character. Antiaromatic structures display almost null multicenter electron delocalization12 and the index is then not able to distinguish a priori between antiaromatic and nonaromatic systems. Origin of the differences between multicenter electron delocalization measurements and magnetic criteria of aromaticity has been extensively analysed and explained in the literature.29–33 The fact that multicenter indices reflect a larger aromaticity for the cyclopropene rings may be related to the nature of the renormalization employed and introduced in ref. 16. This renormalization does not warrant the comparison between multicenter indices of different order and just allows putting the indices in a more or less similar scale.
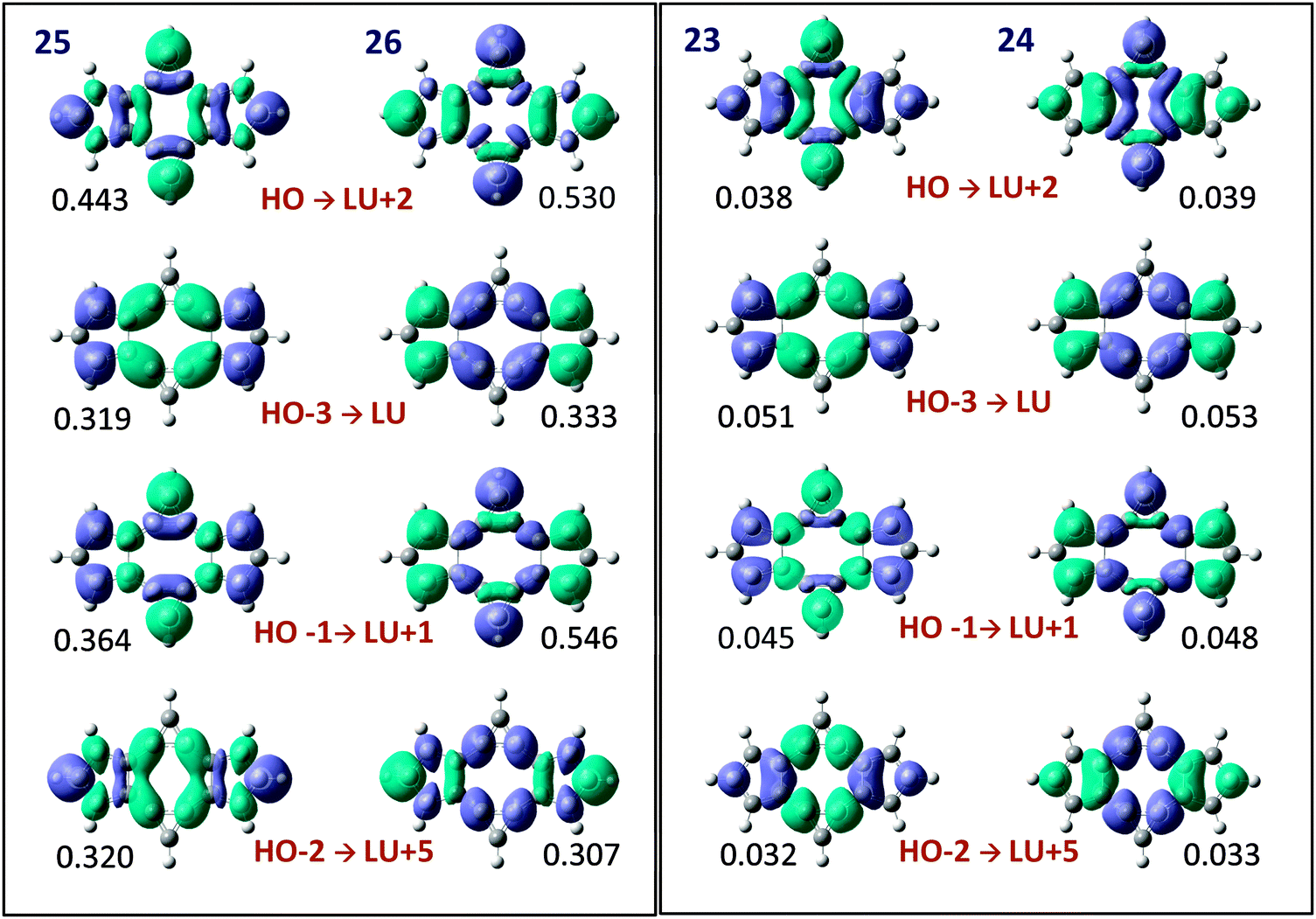
We have finally identified the main occupied and virtual π molecular orbitals involved in the π charge transfer between cyclopropene and cyclopentadiene rings. These molecular orbitals and the electron transfer associated to the combination of them were analysed with the help of the electron deformation orbitals (EDOs) induced by the electric perturbations. As can be seen in Fig. 6, the combinations HOMO → LUMO+2, HOMO−1 → LUMO+1, HOMO−2 → LUMO+5 and HOMO−3 → LUMO are the main responsible for the π electron transfer. The interesting picture reported by the deformation orbitals is that the electron density associated to the EDOs has almost identical shape in all the structures, despite depending on the orientation of the electric perturbation a net electron transfer occurs from the cyclopropene to the cyclopentadiene rings (structures 23 and 25) or in the opposite direction (structures 24 and 26). This reflects the orbital interactions upon the perturbation are identical even for opposite electric fields, so that the changes in the π electron delocalization are just originated in the electron transfer between rings and cannot be related to any fundamental difference in the molecular orbital nature or occupied–virtual orbital mixing.
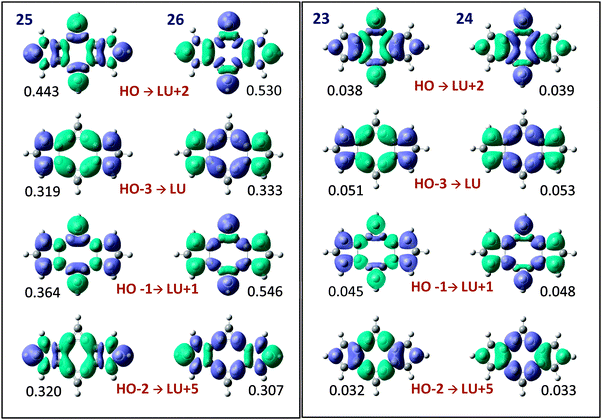 |
| | Fig. 6 Electron density redistributions associated to the main π EDOs for the electrically perturbed bicalicene structures, 23–26 (structures 21–22 provide similar pictures). The associated eigenvalues are shown below the images. In the figure are mentioned the MOs involved in the formation of the EDOs. Positive and negative densities are represented by lilac and light-blue colors, respectively. | |
Conclusions
In this work it has been shown how the aromatic stabilization of bicalicene can be tuned by addition of EDGs and EWGs to the cyclopropene and cyclopentadiene rings or applying external electric perturbations. A clear link between local aromaticity of these rings and the number of π electrons is established and the role played by tetraionic forms definitely revealed for these compounds. Energetic, geometric, electron delocalization and magnetic criteria investigated in this work support the conclusions recently obtained from rings current calculations. In addition, it is demonstrated that the local aromaticity of cyclopropene and cyclopentadiene rings in bicalicene is similar to that of cyclopropene cation and cyclopentadiene anion rings, reaching the same electron delocalization when the number of π electrons approaches 2 and 6, respectively. The results presented here may be interesting for synthetic purposes using polycalicene structures since it is shown how the aromatic stabilization in these systems is easily controlled electrically or by addition of different functional groups.
References
- Z. Yoshida, Pure Appl. Chem., 1982, 54, 1059–1074 CrossRef CAS.
- S. Yoneda, M. Shibata, S. Kida, Z. Yoshida, Y. Kai, K. Miki and N. Kasai, Angew. Chem., Int. Ed. Engl., 1984, 23, 63–64 CrossRef PubMed.
- T. Sugimoto, M. Shibata, S. Yoneda, Z. Yoshida, Y. Kai, K. Miki, N. Kasai and T. Kobayashi, J. Am. Chem. Soc., 1986, 108, 7032–7038 CrossRef CAS.
- T. Sugimoto, M. Sakaguchi, H. Ando, Y. Arai, T. Tanaka, Z. Yoshida, J. Yamauchi, A. A. Bothner-By and M. Mark Lisicki, J. Am. Chem. Soc., 1991, 113, 3193–3195 CrossRef CAS.
- J. Aihara, Bull. Chem. Soc. Jpn., 1991, 64, 3454–3456 CrossRef CAS.
- J. Aihara, J. Chem. Soc., Faraday Trans., 1995, 91, 237–239 RSC.
- J. Aihara, J. Chem. Soc., Perkin Trans. 2, 1996, 2185–2195 RSC.
- W. P. Oziminski, M. Palusiak, J. Dominikowska, T. Krygowski, R. W. A. Havenith, C. M. Gibson and P. W. Fowler, Phys. Chem. Chem. Phys., 2013, 15, 3286–3293 RSC.
- T. Sugimoto and S. Yoshida, Pure Appl. Chem., 1990, 62, 551–556 CrossRef CAS.
- J. Yamauchi, T. Sugimoto, Y. Arai, M. Sakaguchi and Z. Yoshida, J. Chem. Soc., Perkin Trans. 2, 1991, 1959–1962 RSC.
- T. Sugimoto, M. Shibata, A. Sakai, H. Ando, Y. Arai, M. Sakaguchi, Z. Yoshida, Y. Kai, N. Kanehisa and N. Kasai, Angew. Chem., Int. Ed. Engl., 1991, 30, 446–448 CrossRef PubMed.
- M. Mandado, M. González-Moa and R. A. Mosquera, J. Comput. Chem., 2007, 28, 127–136 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- M. Mandado, A. M. Graña and I. Pérez-Juste, J. Chem. Phys., 2008, 129, 164114 CrossRef PubMed.
- M. Mandado and R. Ponec, J. Phys. Org. Chem., 2009, 22, 1225–1232 CrossRef CAS PubMed.
- M. Giambiagi, M. S. de Giambiagi and K. C. Mundim, Struct. Chem., 1990, 1, 423–427 CrossRef CAS.
- C. G. Bollini, M. Giambiagi, M. S. de Giambiagi and A. P. de Figueiredo, J. Math. Chem., 2000, 28, 71–81 CrossRef CAS.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS PubMed.
- M. Mandado and N. Ramos-Berdullas, J. Comput. Chem., 2014, 35, 1261–1269 CrossRef CAS PubMed.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 36, 3839–3842 CrossRef.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed , and references therein.
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 014109 CrossRef PubMed.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001 Search PubMed.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- Calculations of the magnetic shielding tensor including electric quadrupole fields (structures 25 and 26) led to unmeaning results. Also, analysis of the molecular orbital components to the magnetic shielding using the NBO program in structures 21–24 (including point charges in the calculation) just reflected the orbital contributions in the unperturbed bicalicene (0), so that the NICSπzz(1) values for these structures were not included.
- A. B. Sannigrahi and T. Kar, J. Mol. Struct., 2000, 496, 1–17 CrossRef CAS.
- R. Ponec and I. Mayer, J. Phys. Chem. A, 1997, 101, 1738–1741 CrossRef CAS.
- S. Fias, P. W. Fowler, J. L. Delgado, U. Hahn and P. Bultinck, Chem. – Eur. J., 2008, 14, 3093–3099 CrossRef CAS PubMed.
- S. Fias, S. Van Damme and P. Bultinck, J. Comput. Chem., 2008, 29, 358–366 CrossRef CAS PubMed.
- S. Fias, S. Van Damme and P. Bultinck, J. Comput. Chem., 2010, 31, 2286–2293 CAS.
- M. Mandado, Theor. Chem. Acc., 2009, 126, 339–349 CrossRef.
- N. Otero, S. Fias, S. Radenković, P. Bultinck, A. M. Graña and M. Mandado, Chem. – Eur. J., 2011, 17, 3274–3286 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp00990a |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a