
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge transfer, bonding conditioning and solvation effect in the activation of the oxygen reduction reaction on unclustered graphitic-nitrogen-doped graphene†
Adolfo
Ferre-Vilaplana
ab and
Enrique
Herrero
*c
aInstituto Tecnológico de Informática, Ciudad Politécnica de la Innovación, Camino de Vera s/n, E-46022 Valencia, Spain
bDepartamento de Sistemas Informáticos y Computación, Escuela Politécnica Superior de Alcoy, Universidad Politécnica de Valencia, Plaza Ferrándiz y Carbonell s/n, E-03801 Alcoy, Spain
cInstituto de Electroquímica, Universidad de Alicante, Apdo. 99, E-03080 Alicante, Spain. E-mail: herrero@ua.es
First published on 26th May 2015
Abstract
The monodentate associative chemisorption of molecular oxygen on unclustered graphitic-nitrogen-doped graphene requires two nitrogen dopants per activated molecule. Significant charge transfers from regions corresponding to distant nitrogen-dopants, the presence of a nitrogen-dopant adjacent to the carbon atom acting as an active site, which favours its transition from a sp2 hybridization state to sp3, and the solvation effect turn the investigated mechanism to a favourable process.
Experimental results show that nitrogen-doped graphitic materials, such as carbon nanotubes1–3 and graphene,4–6 develop activity towards the oxygen reduction reaction (ORR), for which reason they may eventually replace the much more costly and scarce platinum based electrodes as cathodes in fuel cells. However, the nature of the sites responsible for the observed activity, as well as the mechanisms driving the molecular oxygen activation and further evolution are still under discussion. In the considered materials, the ORR activity has been mainly attributed both to graphitic5,6 and pyridinic5,7,8 nitrogen, but also to the presence of metal impurities,9 at the same time that it has been discarded for both graphitic10 and pyridinic11 nitrogen. Associative12 and dissociative13 molecular oxygen chemisorptions have been investigated as activation mechanisms. And, two-3,5,14 and four-electron3,5,14 evolution processes have been described. A comprehensive review on the subject is available.15
To further investigate the considered reaction, a detailed picture of the substrates would be desirable. Lastly, techniques enabling the unambiguous characterization of the as-synthesized nitrogen-doped graphitic materials are being applied.16–20 For moderate levels of nitrogen doping, unclustered substitutional graphitic-nitrogen-dopants perfectly integrated in the graphene lattice have been visualized.18 Pyridinic nitrogen-dopants have also been unambiguously identified in edges, though they could also take part in high energy defects involving vacancies in cluster form. However, the precise control of the nitrogen-dopant clustering during the synthesis process is still a difficult task.19 The relatively isolated substitutional graphitic-nitrogen defect would have the lowest formation energy in graphene.19 Therefore, in the absence of a process specifically targeted to synthesize a particular kind of defect, the unclustered graphitic nitrogen-dopant would be the most abundant nitrogen-dopant for moderate levels of doping. For this reason, it seems that effects involving graphitic nitrogen-dopants would have to play a role in explaining the activity observed on the investigated materials. In this work, we focus on the effect that unclustered graphitic-nitrogen-dopants could have on the activation mechanism of molecular oxygen on the basal plane of graphene. Given that fundamental effects existing on this prototypical material may also exist on more complex nitrogen-doped graphitic materials, their identification may guide future developments.
For the ORR on nitrogen-doped graphitic materials, both associative12 and dissociative13 chemisorptions of molecular oxygen have been explored as activation mechanisms. Because the monodentate associative chemisorption seems to be a much more simple process than the dissociative one, we decided initially to explore the plausibility of the simplest process as the activation mechanism on the basal plane of unclustered moderately graphitic-nitrogen-doped graphene. The monodentate associative chemisorption of molecular oxygen on nitrogen-doped graphitic materials has been explained involving complex configurations of Stone–Wales defects.1 Also, it has been described as an unfavourable process.10 And, it has been linked to high doping levels.12 Very recently, after discarding other mechanisms, such as metal impurities and processes involving edges, the long range charge transfer from the electrode surface to the outer Helmholtz plane has been proposed as the activation mechanism.6 The new mechanism is postulated after establishing that the activity of the investigated samples towards the ORR should be explained by graphitic-nitrogen dopants in the basal plane, and assuming that a conventional activation mechanism based on chemisorption on that material would not be favourable enough. In the following, we provide computational evidence that the monodentate associative chemisorption of molecular oxygen on specific sites of the basal plane of unclustered moderately graphitic-nitrogen-doped graphene becomes a favourable enough process under certain conditions.
With the goal of determining the eventual role played by a distant graphitic-nitrogen-dopant in a site locally influenced by the other one, using a periodic model of nitrogen-doped graphene of size 8 × 4 which includes a pair of distant graphitic-nitrogen-dopants, a hypothetical chemisorbed state of an oxygen molecule located on top of a carbon atom adjacent to a nitrogen-dopant, in the monodentate associative configuration, was searched for. Moreover, knowing that the solvation effects could become determinant,12 an explicit water molecule in addition to the COSMO21 continuum model was initially adopted for solvation effect treatment (Fig. 1A). A stationary state, corresponding to a minimum, was reached by the optimization process (Fig. 1A). Therefore, the speculated chemisorbed state exists, at least at the level of the chemistry used in the model (details of the methods are given in the ESI†). As can be seen in Fig. S1 (ESI†), the carbon acting as an active site in such a chemisorbed state is displaced out of the plane, presumably evolving from a sp2 hybridization state to sp3. A bond between the active carbon and the nearest atom of the oxygen molecule (O1) is formed (ca. 1.46 Å). The O1–O2 bond of the molecule is significantly elongated, from ca. 1.21 Å to ca. 1.44 Å. And, an atypically short (ca. 1.64 Å) hydrogen bond between O2 and the nearest hydrogen atom of the explicit water molecule is observed. To qualitatively understand the eventual contribution of the distant nitrogen-dopant to the stability of the found chemisorbed state, the electrostatic potential mapped on an electron isodensity surface was visualized for the density value ρ = 0.01 e Å−3 (Fig. 1B). Surprisingly, a very segregated charge distribution can be inferred from Fig. 1B. Though the analysis of partial charges strongly depends on the formalism adopted, for guidance, the corresponding Mulliken population analysis assigns ca. −0.25 and ca. −0.54 to O1 and O2, respectively, whereas ca. 0.14 is assigned to each of the carbon atoms adjacent to nitrogen, with the exception of the carbon atom acting as an active site, and only ca. −0.29 is assigned to each of the nitrogen-dopants (a complete Mulliken population analysis is provided in Fig. S2, ESI†). Jointly, Fig. 1B and Fig. S2 (ESI†) clearly describe that a significant long range charge transfer from a distant region corresponding to a distant nitrogen-dopant mainly to the O2 atom of the oxygen molecule would contribute to the viability of the investigated chemisorbed state. To verify that the suggested long range charge transfer takes place, Fig. 1B was compared with the same kind of representation (the same density value and the same electrostatic potential mapping) corresponding to the adsorbent without the adsorbate complex (Fig. S3, ESI†). Also, the Mulliken population analysis corresponding to the last model is provided in Fig. S4 (ESI†). From Fig. 1B and Fig. S2–S4 (ESI†), the conjectured long range charge transfer, from the region corresponding to the distant dopant to the region corresponding to the active site, can be confirmed. The excess of charge located on O2 would be stabilized by spin pairing with the unpaired electron originating in O2, as a result of the double bond breaking, and by the solvation effect. To further prove the identified charge transfer effect, different distances between unclustered graphitic nitrogen-dopants were also considered, obtaining similar results. When additional unclustered graphitic nitrogen-dopants were included in the model, a collective contribution of charge was observed (Fig. S5, ESI†).
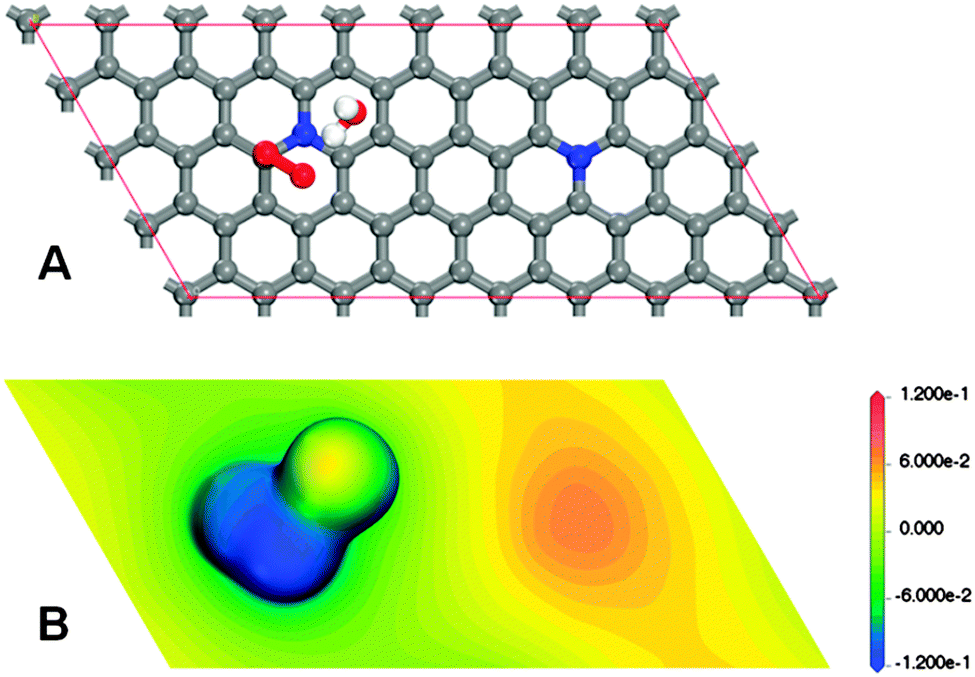 | ||
| Fig. 1 Monodentate associative chemisorbed state of an oxygen molecule on top of a carbon atom adjacent to a graphitic nitrogen-dopant, estimated using a graphitic periodic cell of size 8 × 4 including an additional distant graphitic-nitrogen dopant and an explicit water molecule in addition to the COSMO continuum model as solvation effect treatment. (A) Adsorbent–adsorbate–solvent configuration (additional geometric details can be obtained in Fig. S1, ESI†). (B) Electrostatic potential [Ha per e] mapped on an electron isodensity surface for a density value ρ = 0.01 e Å−3. | ||
In order to establish the relevance of the found chemisorbed state, the reaction path corresponding to the investigated mechanism was estimated performing several constrained optimizations. For each of them, the basic configuration and treatment were retained, but the distance between the carbon acting as an active site and the O1 atom of the oxygen molecule was varied and constrained during the optimization process. The considered C–O distances were limited to those in which each curve reaches their respective cusp. Thus, the final convergence to the alignment level of the respective references, at very long distances, is not shown. The reason for doing so is the following. When solvation effects are sufficiently captured, the relatively available charge, contributed by the regions corresponding to graphitic nitrogen-dopants, is assigned to the solvated oxygen molecule during the optimization process, giving rise to the superoxide anion, also when the oxygen molecule has not yet even come close to the surface. Because such a kind of large range charge transfer does not correspond to any physical feasible process, we decided to focus on relatively short C–O distances.
The curve in Fig. 2 that corresponds to the initial solvation effect treatment applied (one explicit water molecule in addition to the COSMO continuum model) suggests that the found chemisorbed state has certain relevance. It describes a relatively stable chemisorbed state, which is accessible by ascending a relatively moderate barrier. Searching for establishing the impact of the solvation effect and that of the treatment, in relation to the found chemisorbed state, different solvation effect treatments were explored. Using the above described approach, the reaction paths corresponding to different solvation effect treatments were estimated. From Fig. 2, it can be concluded that, depending on the solvation effect treatment, the assessment of the considered hypothetical chemisorbed state changes, from irrelevant, because under vacuum conditions the required stationary state does not exist, to highly relevant, because it is favorable enough and stable enough. Therefore, for the investigated process, treatments under vacuum conditions would not capture essential effects.1,10 Considering only a continuum model of solvation, without enabling potential to capture hydrogen bonding by means of the inclusion of explicit water molecules in the model, would be a completely insufficient treatment.6 At very least, the inclusion of an explicit molecule, in addition to the continuum model, as solvation effect treatment, would be required. And, for a more complete modeling, the explicit inclusion of the first hydration sphere should be considered.12
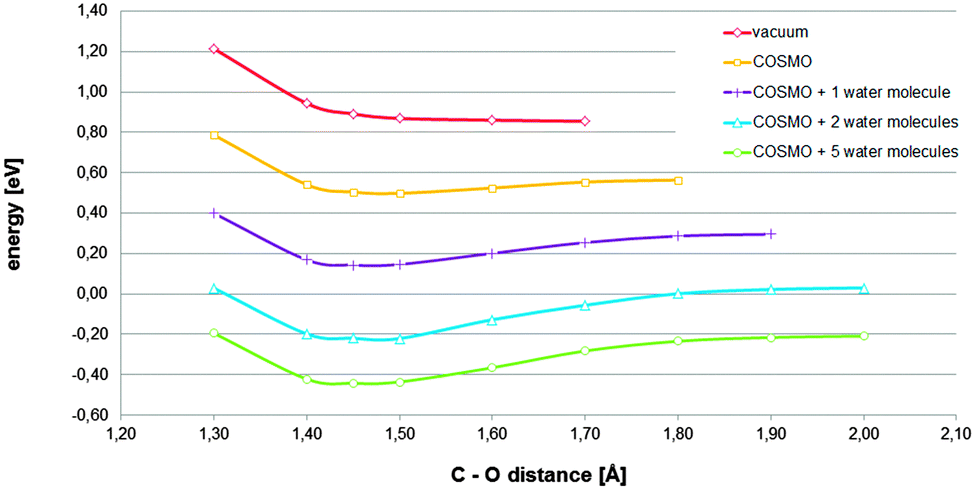 | ||
| Fig. 2 Total energies referring to their respective references (adsorbent energy plus that corresponding to the adsorbate complex in the bulk) for the adsorbent–adsorbate complex described in Fig. 1A at different constrained distances between the active site and the nearest oxygen atom of the oxygen molecule estimated under different solvation effect treatments (details of explicit water configurations are provided in Fig. S1, S6 and S7, ESI†). | ||
Assuming that the found chemisorbed state is relevant for the ORR, and taking into account the conclusions reached on the solvation effect treatment (five water molecules in addition to the COSMO continuum model as solvation effect treatment), the dependence of the viability of the investigated state of the availability of the charge provided by a region corresponding to a second nitrogen-dopant was explored. The reaction path corresponding to the model in which the most distant dopant from the active site was substituted by a carbon atom is displayed in Fig. 3. The paths corresponding to pristine graphene and the original model, in which two graphitic nitrogen-dopants are present, are also included, for comparison. In Fig. 3, it can be observed that the availability of the charge provided by the region corresponding to a second nitrogen-dopant is the determinant for the relevance of the investigated chemisorbed state.
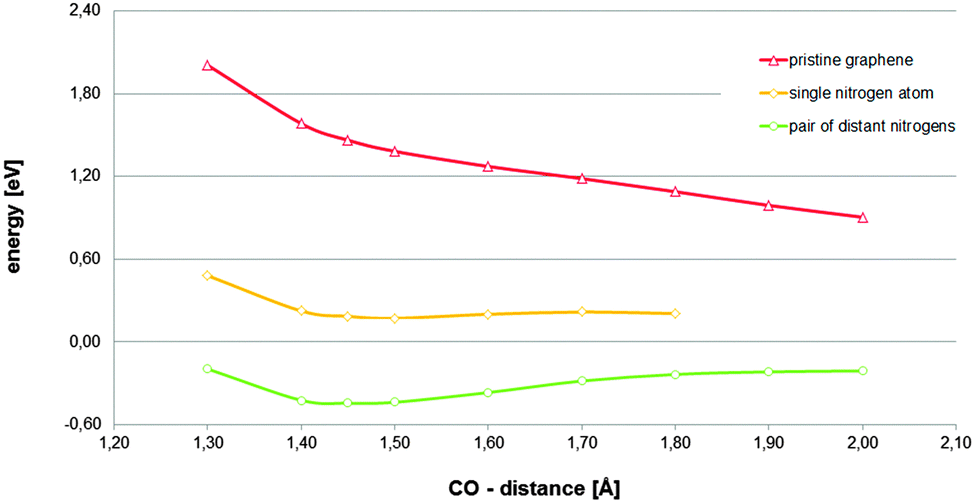 | ||
| Fig. 3 Total energies referring to their respective references (adsorbent energy plus that corresponding to the adsorbate complex in the bulk) for an oxygen molecule located on top of carbon atom at different constrained distances between the active site and the nearest oxygen atom of the oxygen molecule estimated on a 8 × 4 periodic cell of graphene with different graphitic-nitrogen doping levels. For nitrogen-doped graphene, the carbon active site is a carbon atom adjacent to a nitrogen atom. Five explicit water molecules in addition to the COSMO continuum model as solvation effect treatment have been always used (additional information can be obtained in Fig. S8 and S9, ESI†). | ||
In order to determine the role played by the nitrogen dopant adjacent to the carbon atom acting as an active site, a carbon atom located at a distance of two bonds from a graphitic nitrogen-dopant was tried as an active site (Fig. S10, ESI†). The reaction path corresponding to the last configuration (estimated including five water molecules in addition to the COSMO continuum model) and the comparison with pristine graphene and the original model are displayed in Fig. 4. It can be observed that, in spite of the fact that the unlocalized charge provided by two regions corresponding to two graphitic-nitrogen-dopants is still available, and that the solvation effect treatment is reasonably complete, the relevant stationary state, corresponding to the investigated chemisorbed state, has even disappeared, for the new carbon atom tested as an active site. Therefore, for the investigated material, the presence of a nitrogen-dopant adjacent to a carbon atom is crucial in its reactivity towards the ORR. Given that it has been established that long range charge transfers, from regions corresponding to nitrogen-dopants to locations where the transferred charge would lower the energy, are possible, the local effect of the graphitic-nitrogen-dopant on a hypothetical active site has to be interpreted in terms of bonding conditioning. The presence of a graphitic-nitrogen-dopant adjacent to a hypothetical active carbon atom would destabilize it in the sense that it would favour the transition of this from a sp2 hybridization state to an alternative sp3 hybridization state.
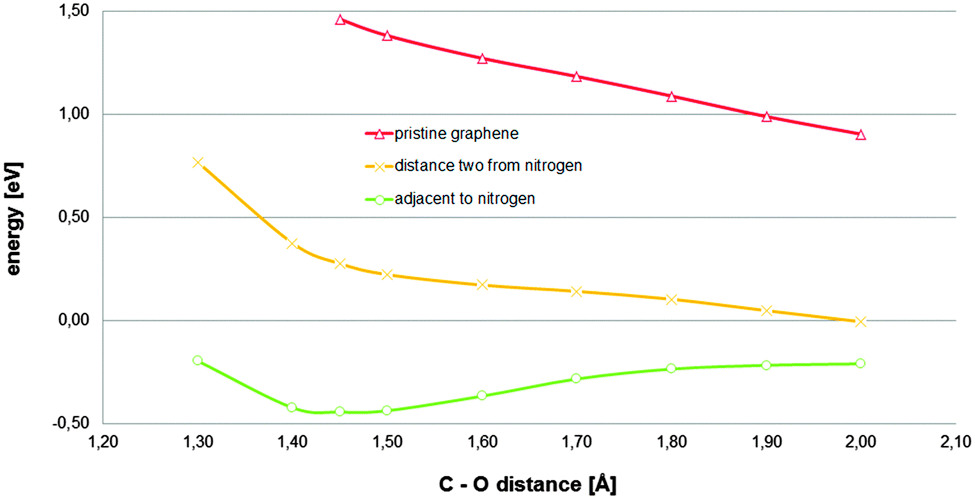 | ||
| Fig. 4 Total energies referring to their respective references (adsorbent energy plus that corresponding to the adsorbate complex in the bulk) for an oxygen molecule located on top of a carbon atom of the adsorbent model described in Fig. 1A at different constrained distances between the tested carbon atom and the nearest oxygen atom of the oxygen molecule estimated for different carbon atoms tested as active sites: -○- a carbon atom adjacent to a nitrogen-dopant and -×- a carbon atom located at a distance of two bonds from the dopant. The results corresponding to pristine graphene are also plotted for comparison. Five explicit water molecules in addition to the COSMO continuum model as solvation effect treatment was used (additional geometric details are provided in Fig. S10, ESI†). | ||
To better establish the relevance of the graphitic form of the nitrogen-dopant, in relation to the considered molecular oxygen activation mechanism, in the model including two graphitic nitrogen-dopants, the nitrogen contributing the charge was substituted by a pyridinic nitrogen dopant clustered with a vacancy defect (Fig. S11, ESI†). It was found that a pyridinic nitrogen plus the vacancy defect is not able to contribute transferable charge and therefore, the monodentate associative chemisorbed state is not favoured.
Finally, having established the roles played by the graphitic nitrogen-dopants, contributing charge and destabilizing carbon atoms, and having emphasized the relevance of the solvation effect, the viability of a hypothetical dissociative chemisorption route for the oxygen reduction reaction on these materials is considered. For such a purpose, a reaction model, in which a solvated oxygen molecule chemisorbed, in monodentate form, on a carbon atom adjacent to a graphitic nitrogen-dopant evolves into a bidentate adsorption mode eventually cleaving the O–O bond, is assumed. It can be reasoned that, in order to complete the above described reaction, the solvation shell formed during the monodentate chemisorption process should be removed, which imposes a large barrier to the whole of the reaction. For this reason, we consider that the dissociative route is less probable than the monodentate associative route.
Conclusions
Experimental results suggest that nitrogen-doped graphitic materials may eventually replace platinum based electrodes as cathodes in fuel cells. But, before this can ever happen, the considered materials would have to be optimized for such a purpose. Focusing on graphitic-nitrogen-dopants in graphene, and on the hypothetical monodentate associative chemisorption, as the molecular oxygen activation mechanism, in this communication, fundamental effects that can be useful for the further development of the considered materials have been identified. Under solvation conditions, the activation of molecular oxygen by means of the monodentate associative chemisorption on the adsorbent poses two conditions. On the one hand, a bistable site, capable of commuting from the bonding state in which it is integrated in the adsorbent to an alternative bonding state enabling it to bond to the nearest atom of the oxygen molecule, is required. On the other hand, an adsorbent capable of transferring charge to the oxygen atom presenting the unpaired electron in the molecule is also required, so that the energy of the system decreases sufficiently by means of the spin pairing and with the aid of the solvation effect. Because carbon atoms are integrated in the surface adopting a sp2 hybridization state, but they can be forced to change to the tetrahedral sp3 hybridization state (for instance in the hydrogenation by means of chemisorption), pristine graphitic materials meet the first of the abovementioned conditions. However, due to the collective nature of the sp2 hybridization state that carbon atoms present in these materials, these are, in certain sense, over-stabilized, and thus, they present a significant barrier to the transition to the sp3 hybridization state. Additionally, in pristine graphitic materials, there is no clear source of unlocalized charge available to transfer to the oxygen atom presenting the unpaired electron. Here, we have provided computational evidence that graphitic nitrogen-dopants would be capable of contributing relatively available charge, which could be easily transferable even to large distance, and that a single relatively isolated graphitic nitrogen-dopant would be capable of sufficiently destabilizing its adjacent carbon atoms, as for enabling the transition of any of them from the sp2 hybridization state to sp3, for the investigated activation mechanism, if enough charge is available on the surface. It has been shown that, the combination of both identified fundamental effects, aided by the solvation effect, whose relevance has also been emphasized, would have potential to turn the monodentate associative chemisorption of molecular oxygen on the basal plane of moderately graphitic-nitrogen-doped graphene to a favourable enough and stable enough process as to be considered as a relevant process. It seems to be plausible that manifestations of the here identified fundamental effects can become operative in most of the nitrogen containing graphitic materials, influencing the oxygen reduction reaction in any form and, in general, other electrophilic processes on the surface.Acknowledgements
This work has been financially supported by the MICINN (Spain) (project 2013-44083-P) and Generalitat Valenciana (project PROMETEOII/2014/013).Notes and references
- K. Gong, F. Du, Z. Xia and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- Z. Wang, R. Jia, J. Zheng, J. Zhao, L. Li, J. Song and Z. Zhu, ACS Nano, 2011, 5, 1677 CrossRef CAS PubMed.
- T. Sharifi, G. Hu, X. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS PubMed.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 CAS.
- C. H. Choi, H.-K. Lim, M. W. Chung, J. C. Park, H. Shin, H. Kim and S. I. Woo, J. Am. Chem. Soc., 2014, 136, 9070 CrossRef CAS PubMed.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622 CrossRef CAS.
- T. Xing, Y. Zheng, L. H. Li, B. C. C. Cowie, D. Gunzelmann, S. Z. Qiao, S. Huang and Y. Chen, ACS Nano, 2014, 8, 6856 CrossRef CAS PubMed.
- L. Wang, A. Ambrosi and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 13818 CrossRef CAS PubMed.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X.-F. Zhou, H. T. Wang, J.-J. Xu and E. G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155454 CrossRef.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038 RSC.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183 CrossRef CAS PubMed.
- D. W. Boukhvalov and Y.-W. Son, Nanoscale, 2012, 4, 417 RSC.
- S. Yasuda, L. Yu, J. Kim and K. Murakoshi, Chem. Commun., 2013, 49, 9627 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS.
- J. Zhou, J. Wang, H. Liu, M. N. Banis, X. Sun and T. K. Sham, J. Phys. Chem. Lett., 2010, 1, 1709 CrossRef CAS.
- J. C. Meyer, S. Kurasch, H. J. Park, V. Skakalova, D. Künzel, A. Groß, A. Chuvilin, G. Algara-Siller, S. Roth, T. Iwasaki, U. Starke, J. H. Smet and U. Kaiser, Nat. Mater., 2011, 10, 209 CrossRef CAS PubMed.
- L. Zhao, R. He, K. T. Rim, T. Schiros, K. S. Kim, H. Zhou, C. Gutiérrez, S. P. Chockalingam, C. J. Arguello, L. Pálová, D. Nordlund, M. S. Hybertsen, D. R. Reichman, T. F. Heinz, P. Kim, A. Pinczuk, G. W. Flynn and A. N. Pasupathy, Science, 2011, 333, 999 CrossRef CAS PubMed.
- R. Lv, Q. Li, A. R. Botello-Méndez, T. Hayashi, B. Wang, A. Berkdemir, Q. Hao, A. L. Elías, R. Cruz-Silva, H. R. Gutiérrez, Y. A. Kim, H. Muramatsu, J. Zhu, M. Endo, H. Terrones, J.-C. Charlier, M. Pan and M. Terrones, Sci. Rep., 2012, 2, 586 Search PubMed.
- R. Arenal, K. March, C. P. Ewels, X. Rocquefelte, M. Kociak, A. Loiseau and O. Stéphan, Nano Lett., 2014, 14, 5509 CrossRef CAS PubMed.
- B. Delley, Mol. Simul., 2006, 32, 117 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods and figures. See DOI: 10.1039/c5cp00918a |
| This journal is © the Owner Societies 2015 |