
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adsorption of polyelectrolytes to like-charged substrates induced by multivalent counterions as exemplified by poly(styrene sulfonate) and silica†
Alberto
Tiraferri
ab,
Plinio
Maroni
b and
Michal
Borkovec
*b
aDepartment of Environment, Land, and Infrastructure Engineering, Polytechnic University of Turin, Torino, Italy
bDepartment of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, 30 Quai Ernest-Ansermet, 1205 Geneva, Switzerland. E-mail: michal.borkovec@unige.ch
First published on 23rd March 2015
Abstract
The present study demonstrates that multivalent counterions trigger adsorption of polyelectrolytes on a like-charged substrate. In particular, adsorption of polystyrene sulfonate on silica is studied experimentally in NaCl, MgCl2, and LaCl3 solutions by optical reflectivity. While adsorption is negligible in the presence of Na+, the polyelectrolyte adsorbs in the presence of Mg2+ and La3+. The adsorbed amount of the polyelectrolyte goes through a maximum as a function of the salt concentration. This maximum increases with increasing valence and shifts to lower salt concentrations. At low salt concentration, the adsorption is negligible. At intermediate salt level, ripening and multilayer formation leads to continuous growth of the adsorbed layer. At higher salt level, blocking and formation of a monolayer lead to saturation. These results are tentatively interpreted in terms of a charge reversal of the polyelectrolyte–metal complex. The molecular mass of the polyelectrolyte has an important effect on the adsorption behavior, whereby the tendency towards ripening becomes more pronounced at large molecular mass.
Polyelectrolytes adsorb readily to oppositely charged substrates, and this process is exploited in various applications, including water treatment, papermaking, formulation of foods, or multilayer coatings.1–7 The adsorption process is mainly driven electrostatically. Thereby, attraction between the polyelectrolyte and the oppositely charged substrate induces rapid formation of the adsorbed layer, while repulsion between the equally charged polyelectrolyte segments leads to surface saturation and blocking.
Given the importance of electrostatic interactions in the adsorption process, one might be tempted to think that polyelectrolytes should not adsorb on like-charged substrates. However, numerous applications rely on polyelectrolytes carrying the same charge as the substrates, for example, DNA imaging, water treatment, or formulation of particle slurries.7–9 Moreover, several studies suggest that polyelectrolytes actually do adsorb to like-charged substrates.9–16 In such situations, salts containing multivalent cations are often added (e.g., Ca2+, Ni2+, Fe3+, Al3+).7,12,13 Specific interaction of polyacrylates with calcite might involve similar processes, since their adsorption leads to calcite dissolution and release of Ca2+ ions.17
A potential like-charge adsorption mechanism stresses the importance of non-electrostatic forces, such as van der Waals or hydrophobic interactions.15,16,18–20 This mechanism seems particularly pronounced for comb-like or block-copolymers, whereby the hydrophobic part of the polymer is mainly responsible for the adsorption. An alternative mechanism came to focus recently, especially in the theoretical community. Computational studies suggest that polyelectrolytes adsorb to like-charged substrates through electrostatic forces only.18,21–23 A simplified interpretation of these findings involves a charge reversal through the binding of multivalent ions to the substrate or the polyelectrolyte. This situation may again lead to electrostatically induced adsorption, similar to the case of oppositely charged polyelectrolytes.
The effect of multivalent ions on polyelectrolyte adsorption to like-charged substrates was so far only studied for DNA and mica in detail.9–11 Imaging by atomic force microscopy (AFM) was used to demonstrate that anionic DNA does not adsorb to negatively charged mica in monovalent salt solutions, but that adsorption occurs from solutions containing divalent and trivalent cations. AFM was also used to show that Mg2+ ions induce adsorption of poly(styrene sulfonate) (PSS) on mica.16 Classical batch experiments also suggest that Ca2+ and Mg2+ ions enhance the adsorption of anionic polyacrylates to negatively charged alumina and titania particles.12,13 With the exception of DNA, however, the available experimental data base remains weak.
For these reasons, we report experimental findings demonstrating that multivalent counterions strongly promote adsorption of negatively charged PSS on like-charged silica in aqueous solutions. Our technique of choice is optical reflectivity,4,5 as it permits to study in real time adsorption to a planar substrate in situ in the impinging-jet geometry with excellent sensitivity down to few μg m−2. Experimental details are given in the ESI.†
The silica substrate was first flushed with pure electrolyte solution of pH 6.0, and subsequently a solution of the sodium salt of PSS of a molecular mass of 30 kg mol−1 of a concentration of 10 mg L−1 in the same electrolyte of the same pH was injected. After a period of time, the substrate was again rinsed by the pure electrolyte solution. The recorded traces of the adsorbed amount are shown in Fig. 1a. As one expects, there is very little adsorption in the presence of Na+. Adsorption already occurs in the presence of Mg2+, while for La3+ the adsorbed amount is substantial. The latter two ions were chosen due to negligible hydrolysis.24 Appreciable concentrations of hydroxide complexes MgOH+ form above pH 10, and LaOH2+ above pH 8. After rinsing the adsorbed layer with pure electrolyte solution, only minor desorption could be evidenced.
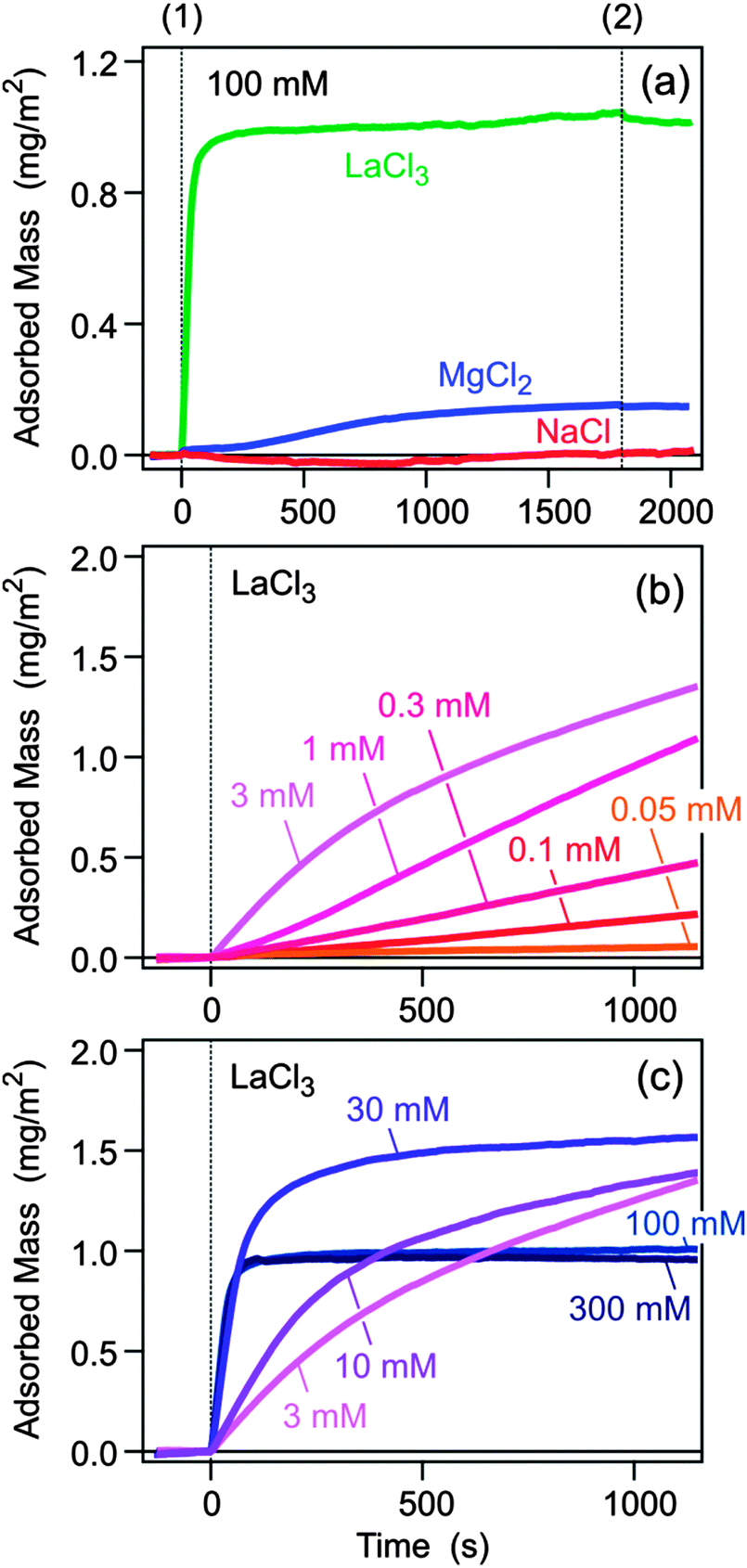 | ||
| Fig. 1 Adsorbed mass of negatively charged polyelectrolyte PSS on like-charged silica substrate in various electrolytes measured by optical reflectivity. The substrate is first rinsed with pure electrolyte solution and then the PSS solution is injected at time (1). The PSS concentration is 10 mg L−1 and solution pH 6.0. Molecular mass of PSS is 30 kg mol−1 unless indicated otherwise. (a) Adsorption in electrolyte solution with various counterions Na+, Mg2+, and La3+ at a concentration of 100 mM. At time (2) the substrate was flushed with pure electrolyte solution. Adsorption traces for (b) low and (c) high LaCl3 concentrations. The different concentrations are labeled. | ||
The adsorbed amount was studied for different salt concentrations, and the result is shown in Fig. 2a. In the presence of monovalent counterions Na+, adsorption remains negligible. For Mg2+ one finds a maximum adsorbed amount of 0.2 mg m−2 at a salt level around 300 mM, while for La3+ the maximum exceeds 1.5 mg m−2 and shifts to lower concentrations around 3 mM. The present data suggest that the adsorbed amount goes through a maximum, and that its position decreases with the valence of the counterion. A similar maximum was predicted by computer simulations for the adsorption of polyacrylates to silica in the presence of Ca2+ ions, albeit at lower salt concentrations.18
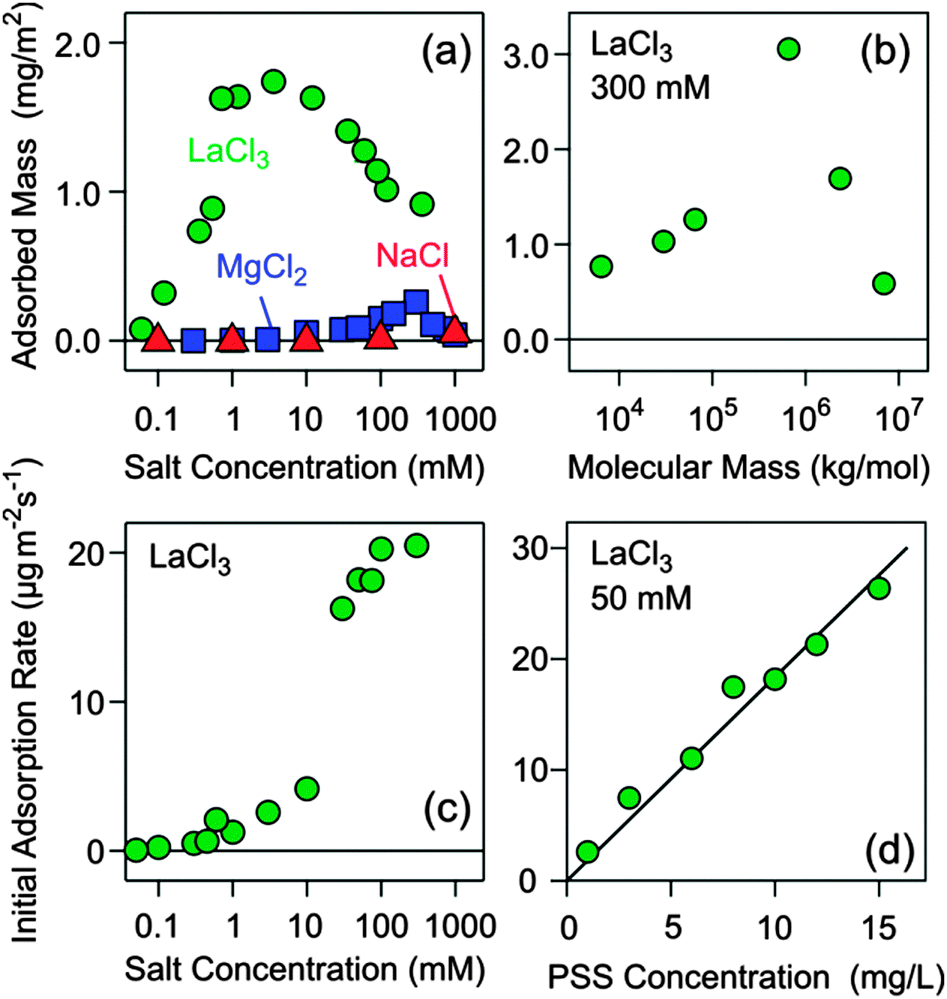 | ||
| Fig. 2 Adsorbed mass and initial adsorption rates for PSS on like-charged silica at pH 6.0. The PSS concentration is 10 mg L−1 and the molecular mass of PSS is 30 kg mol−1, unless indicated otherwise. (a) Amount adsorbed after 30 min and subsequent desorption during 10 min with the same electrolyte solution. (b) Adsorbed mass after 30 min versus the molecular mass. Initial adsorption rate versus (c) the concentration of the LaCl3 salt and (d) the polyelectrolyte concentration in 50 mM LaCl3 solution. | ||
To obtain better insight into the adsorption mechanism of PSS to silica in the presence of La3+, a detailed kinetic study was carried out. The initial adsorption rates were also studied.4 The respective traces are shown in Fig. 1b and c and the corresponding initial adsorption rates in Fig. 2c. At a concentration of La3+ <0.1 mM, the adsorption is negligible. However, the adsorbed amount increases quickly with increasing concentration. While the initial adsorption rate remains low, the adsorbed amount starts to increase after an initial induction period. More surprisingly, however, the adsorbed amount continues to increase, without showing any signs of saturation. In some experiments, adsorbed amounts even exceed about 10 mg m−2, but these conditions were not analyzed in detail due to eventual non-linearity of the detector. This situation indicates that the initial adsorption step of the polyelectrolyte to the surface is unfavorable, but once few polyelectrolyte chains are adsorbed, they serve as nucleating sites for further adsorption. In this regime, the surface undergoes ripening, whereby a polyelectrolyte multilayer is being formed.
A saturation of the surface sets in about at concentrations >3 mM, and this blocking process becomes fully evident at higher salt concentrations. The observed saturation amount of about 1.0 mg m−2 roughly corresponds to a dense polyelectrolyte monolayer. In this regime, the initial adsorption kinetics is fast and first order with respect to the polymer concentration (Fig. 2d). The resulting adsorption rate coefficient is 1.8 μm s−1. When one assumes perfect sink conditions at the surface, one can evaluate the transport controlled rate coefficient of the impinging-jet geometry.4,26 By approximating the diffusion coefficient of PSS to be comparable to the one in a monovalent salt solution of the same ionic strength,25 one obtains a sticking coefficient of about 0.2. These features resemble adsorption of polyelectrolytes to oppositely charged substrates.3,4
The molecular mass of the polyelectrolyte has an important effect on the adsorption of PSS on silica as induced by La3+ ions. This effect was studied in 300 mM LaCl3 solutions. As illustrated in Fig. 2b, the adsorbed amount goes through a maximum around 3.2 mg m−2 near 700 kg mol−1. At low molecular mass, the adsorption saturates, while at higher molecular mass, a gradual increase of the adsorbed amount can be evidenced without a clear onset of saturation. The actual adsorption traces shown in the supplement suggest that ripening becomes more important with increasing molecular mass.
We propose the following tentative interpretation of these results. PSS is a highly charged and hydrophobic polyelectrolyte, and therefore we suspect that it binds La3+ ions, forming a strong metal–polyelectrolyte complex. At low La3+ concentrations, the number of the bound La3+ ions is small and the polyelectrolyte–metal complex remains negatively charged. With increasing La3+ concentrations, however, the number of bound La3+ ions increases. At a particular La3+ concentration, the polyelectrolyte–metal complex will be neutral, probably around 1 mM. Increasing the La3+ concentration further, additional La3+ ions are bound by the polyelectrolyte, leading to a positively charged metal–polyelectrolyte complex. The fact that such a charge reversal might occur was also suggested by computer simulations.27,28 This charge reversal is also consistent with the observed precipitation of PSS by La3+ ions, which occurs at intermediate La3+ concentrations, while at low and high concentrations, the polyelectrolyte remains soluble.29,30 These precipitation experiments were carried out at much higher PSS concentrations than the ones used for the present experiments. We have no evidence of precipitation in the PSS solutions prepared here. On the other hand, we surmise that silica substrate remains negative. Divalent ions were shown to lead to a weak charge reversal of silica in weakly basic solutions at very high salt concentrations.31 Silica is less charged at the pH used suggesting that charge reversal of the substrate by La3+ is unlikely.
Based on this charge reversal scenario, the observed results can be interpreted as follows (see Fig. 3a–c). At low La3+ concentrations, the negatively charged metal–polyelectrolyte complex does not adsorb to the negatively charged substrate due to electrostatic repulsion. At intermediate La3+ concentrations around 1 mM, the hydrophobic forces acting between the neutral polyelectrolyte–metal complexes induce a continuous surface deposition, leading to ripening, and the formation of a thick multilayer. At higher La3+ concentrations, the positively charged polyelectrolyte–metal complex adsorbs rapidly due to electrostatic attraction to the negatively charged silica surface. The adsorption process continues until the electrostatic repulsion between the adsorbed complexes becomes sufficiently important such that the surface saturates. At this point, the adsorbed polyelectrolyte monolayer develops a positive charge, which prevents further adsorption of the positively charged polyelectrolyte–metal complexes. The latter situation resembles the adsorption of a polyelectrolyte to an oppositely charged substrate. While the present interpretation in terms of the charge reversal of the polyelectrolyte seems suggestive to us, other possible mechanisms may include aggregation of polyelectrolytes in solution, surface induced precipitation, or contributions of hydrophobic interactions.
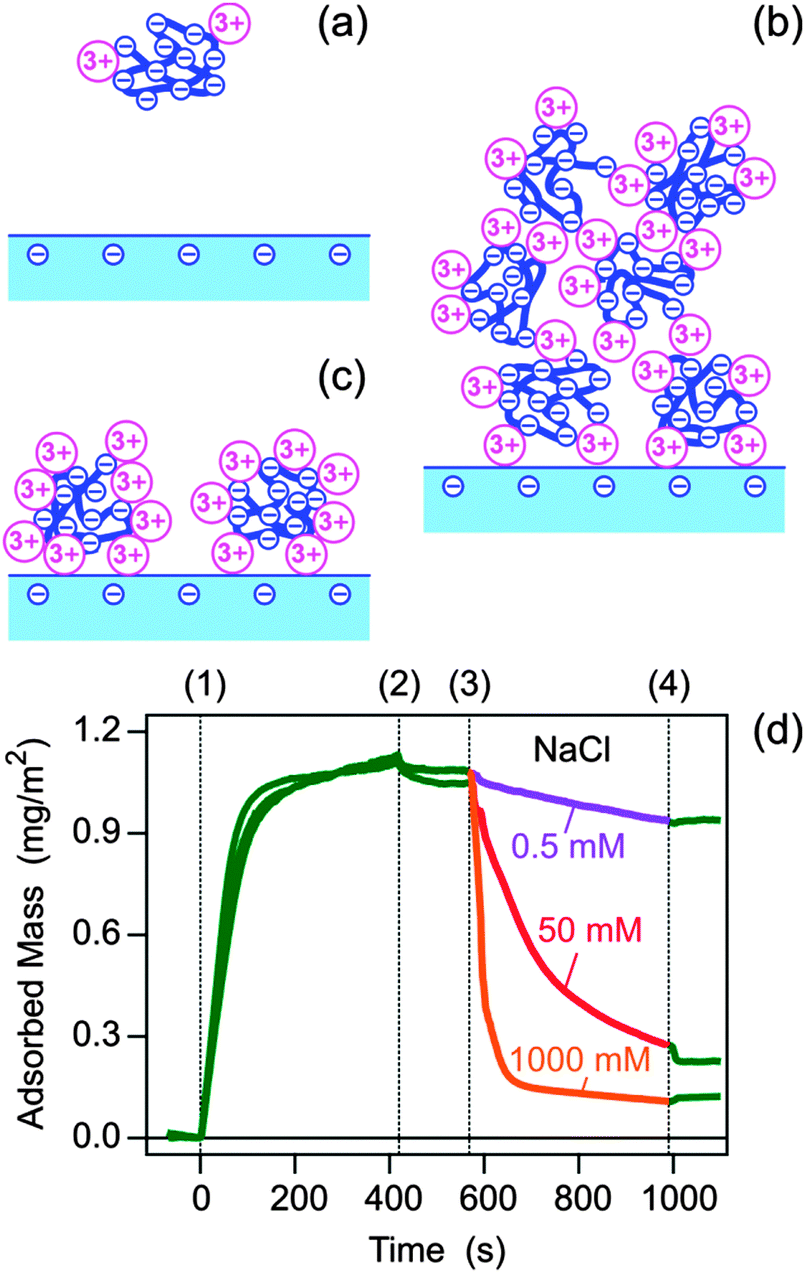 | ||
| Fig. 3 Proposed mechanism of the polyelectrolyte adsorption on like-charge substrates mediated by multivalent counterions. (a) No adsorption at low salt concentration, (b) ripening involving multilayer formation at intermediate concentrations, and (c) monolayer formation and blocking at high salt concentration. (d) Adsorption of PSS in LaCl3 solution and desorption with NaCl at pH 6.0. The substrate is flushed with 50 mM LaCl3 solution, then at time (1) PSS solution of 10 mg L−1 in the same LaCl3 solution is introduced, at time (2) the substrate is rinsed with pure LaCl3 solution, at time (3) a pure NaCl solution of the indicated concentration is introduced, and at time (4) the substrate is finally rinsed with pure LaCl3 solution. The different traces shown between (1) and (3) are repetitions of the same adsorption experiment, and illustrate the good reproducibility of these experiments. | ||
The fact that this mechanism based on the charge reversal is incomplete is further underlined by the non-monotonous molecular mass dependence discussed above (Fig. 2b). While the increase in the adsorbed mass is consistent with the initial increase in size of the polyelectrolyte–metal complex, the decrease at larger molecular mass and the tendency of the adsorbed layer towards ripening suggests the possible onset of different processes. They could involve the formation of pearl-necklace structures within the polyelectrolyte–metal complex or the formation of polyelectrolyte aggregates in solution mentioned above.32,33 The deposition of such aggregates may lead to the observed ripening of the adsorbed layer. The formation of these structures could be mediated by complexation of multivalent ions by the polyelectrolytes, and could resemble the effect on the aggregation of colloidal particles due to charge reversal induced by adsorbing polyelectrolytes or multivalent ions.3,34 The presently proposed adsorption mechanism remains speculative for the moment.
We remarked that rinsing the layer with the pure electrolyte solution containing multivalent ions does not lead to any appreciable polyelectrolyte desorption, which indicates irreversible adsorption within the experimental time window. However, desorption can be readily induced by rinsing with a solution containing monovalent counterions (Fig. 3d). Desorption becomes increasingly rapid with increasing concentration of NaCl, indicating that bound La3+ in the adsorbed layer is exchanged with Na+, and the resulting polyelectrolyte layer containing Na+ becomes unstable and desorbs. Desorption process can be stopped by reintroducing the LaCl3 solution, whereby Na+ ions are exchanged back with La3+ ions.
To conclude, we provide experimental evidence with optical reflectivity that highly charged polyelectrolytes adsorb to a like-charged substrate in the presence of multivalent counterions. The adsorbed amount of the polyelectrolyte versus the salt concentration goes through a maximum, which becomes higher with increasing valence and shifts to lower concentrations. At low concentration of multivalent ions, no adsorption is observed. At intermediate ionic levels, the adsorbed layer forms slowly, but continues to grow due to ripening and multilayer formation. At higher ionic levels, the adsorption saturates, probably leading to blocking and formation of a monolayer. The tendency towards ripening increases with increasing molecular mass of the polyelectrolyte. A tentative interpretation of these findings is based on the formation of a polyelectrolyte–metal complex between the anionic polymer chains and the multivalent cations, which undergoes a charge neutralization and subsequent charge reversal. Since computer simulations have shown that such adsorption of polyelectrolytes to like-charged substrates to be governed by electrostatic forces only, we suspect that the present findings could be generic and possibly applicable to a wide variety of systems. Specific interactions may of course modify the behavior of individual systems, for example, hydrophobic interactions may act together with the electrostatic interactions discussed here. While the extension of the present results to the practically more relevant multivalent cations Fe3+ and Al3+ would be important, such systems are probably complicated by the extensive hydrolysis reactions of these ions.24
Acknowledgements
This work was supported by a Marie Curie Intra-European Fellowship to A.T. within the Seventh European Community Framework Programme (PIEF-GA-2012-327977), by the Swiss National Science Foundation, and by the University of Geneva.References
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4, 729–740 CrossRef CAS PubMed.
- A. Martin-Molina, G. Luque-Caballero, J. Faraudo, M. Quesada-Perez and J. Maldonado-Valderrama, Adv. Colloid Interface Sci., 2014, 206, 172–185 CrossRef CAS PubMed.
- I. Szilagyi, G. Trefalt, A. Tiraferri and M. Borkovec, Soft Matter, 2014, 10, 2479–2502 RSC.
- I. Popa, B. P. Cahill, P. Maroni, G. Papastavrou and M. Borkovec, J. Colloid Interface Sci., 2007, 309, 28–35 CrossRef CAS PubMed.
- A. Tiraferri, P. Maroni, D. C. Rodriguez and M. Borkovec, Langmuir, 2014, 30, 4980–4988 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, Environ. Sci. Technol., 2008, 42, 7607–7614 CrossRef CAS.
- B. Bolto and J. Gregory, Water Res., 2007, 41, 2301–2324 CrossRef CAS PubMed.
- B. J. Lee, M. A. Schlautman, E. Toorman and M. Fettweis, Water Res., 2012, 46, 5696–5706 CrossRef CAS PubMed.
- O. Pietrement, D. Pastre, S. Fusil, J. Jeusset, M. O. David, F. Landousy, L. Hamon, A. Zozime and E. Le Cam, Langmuir, 2003, 19, 2536–2539 CrossRef CAS.
- D. Pastre, L. Hamon, F. Landousy, I. Sorel, M. O. David, A. Zozime, E. Le Cam and O. Pietrement, Langmuir, 2006, 22, 6651–6660 CrossRef CAS PubMed.
- C. Rivetti, M. Guthold and C. Bustamante, J. Mol. Biol., 1996, 264, 919–932 CrossRef CAS PubMed.
- L. Dupont, A. Foissy, R. Mercier and B. Mottet, J. Colloid Interface Sci., 1993, 161, 455–464 CrossRef CAS.
- J. Sun, L. Bergstrom and L. Gao, J. Am. Ceram. Soc., 2001, 84, 2710–2712 CrossRef CAS PubMed.
- N. G. Hoogeveen, M. A. Cohen Stuart and G. J. Fleer, J. Colloid Interface Sci., 1996, 182, 133–145 CrossRef CAS.
- P. Maroni, F. J. Montes Ruiz-Cabello and A. Tiraferri, Soft Matter, 2014, 10, 9220–9225 RSC.
- A. Gromer, M. Rawiso and M. Maaloum, Langmuir, 2008, 24, 8950–8953 CrossRef CAS PubMed.
- C. Geffroy, J. Persello, A. Foissy, B. Cabane and F. Tournilhac, Rev. Inst. Fr. Pet., 1997, 52, 183–190 CAS.
- M. Turesson, C. Labbez and A. Nonat, Langmuir, 2011, 27, 13572–13581 CrossRef CAS PubMed.
- T. Abraham, D. Christendat, Z. Xu, J. Masliyah, J. F. Gohy and R. Jerome, AIChE J., 2004, 50, 2613–2626 CrossRef CAS.
- C. P. Whitby, P. J. Scales, F. Grieser, T. W. Healy, G. Kirby, J. A. Lewis and C. F. Zukoski, J. Colloid Interface Sci., 2003, 262, 274–281 CrossRef CAS.
- R. Messina, C. Holm and K. Kremer, J. Chem. Phys., 2002, 117, 2947–2960 CrossRef CAS PubMed.
- L. Wang, H. J. Liang and J. Z. Wu, J. Chem. Phys., 2010, 133, 044906 CrossRef PubMed.
- G. Luque-Caballero, A. Martin-Molina and M. Quesada-Perez, J. Chem. Phys., 2014, 140, 174701 CrossRef PubMed.
- C. F. Baes and R. E. Mesmer, The Hydrolysis of Cations, Krieger Publishing, Malabar, 1976 Search PubMed.
- N. Borochov and H. Eisenberg, Macromolecules, 1994, 27, 1440–1445 CrossRef CAS.
- Z. Adamczyk, B. Siwek and P. Warszynski, J. Colloid Interface Sci., 2002, 248, 244–254 CrossRef CAS PubMed.
- M. Deserno, F. Jimenez-Angeles, C. Holm and M. Lozada-Cassou, J. Phys. Chem. B, 2001, 105, 10983–10991 CrossRef CAS.
- K. M. Wu, Y. F. Wei and P. Y. Hsiao, Electrophoresis, 2011, 32, 3348–3363 CrossRef CAS PubMed.
- M. O. de la Cruz, L. Belloni, M. Delsanti, J. P. Dalbiez, O. Spalla and M. Drifford, J. Chem. Phys., 1995, 103, 5781–5791 CrossRef PubMed.
- I. Sabbagh and M. Delsanti, Eur. Phys. J. E: Soft Matter Biol. Phys., 2000, 1, 75–86 CrossRef CAS.
- F. H. J. van der Heyden, D. Stein, K. Besteman, S. G. Lemay and C. Dekker, Phys. Rev. Lett., 2006, 96, 224502 CrossRef.
- J. Jeon and A. V. Dobrynin, Macromolecules, 2007, 40, 7695–7706 CrossRef CAS.
- M. O. Khan and B. Jonsson, Biopolymers, 1999, 49, 121–125 CrossRef CAS.
- P. Sinha, I. Szilagyi, F. J. Montes Ruiz-Cabello, P. Maroni and M. Borkovec, J. Phys. Chem. Lett., 2013, 4, 648–652 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional adsorption traces. See DOI: 10.1039/c5cp00910c |
| This journal is © the Owner Societies 2015 |