
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protein motions and dynamic effects in enzyme catalysis
Louis Y. P.
Luk
,
E. Joel
Loveridge
and
Rudolf K.
Allemann
*
School of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK. E-mail: allemannrk@cardiff.ac.uk
First published on 9th April 2015
Abstract
The role of protein motions in promoting the chemical step of enzyme catalysed reactions remains a subject of considerable debate. Here, a unified view of the role of protein dynamics in dihydrofolate reductase catalysis is described. Recently the role of such motions has been investigated by characterising the biophysical properties of isotopically substituted enzymes through a combination of experimental and computational analyses. Together with previous work, these results suggest that dynamic coupling to the chemical coordinate is detrimental to catalysis and may have been selected against during DHFR evolution. The full catalytic power of Nature's catalysts appears to depend on finely tuning protein motions in each step of the catalytic cycle.
Introduction
The biophysical basis of the enormous catalytic power of enzymes and the relationship between protein motions and enzyme catalysis, and how this may have evolved, are currently hotly debated and a matter of scientifically fruitful contention. Compared to man-made catalysts enzymes are relatively large and flexible structures with typical motions on milli- to femtosecond time scales. Conformational transitions are often essential for certain aspects of catalysis such as ligand association and dissociation and it has been proposed that such conformational changes might be coupled to the chemical coordinate.1 Indeed, the coupling of non-equilibrium fluctuations has been conceptualised as a beneficial evolutionary trait that promotes product formation in enzyme catalysis.2–6 Other investigations have been interpreted to indicate that enzymatic chemical transformation occur in relatively static environments.7–13 The apparent contradiction between some of these studies means that the relationship between enzyme motions and catalysis remains intensely investigated.Dihydrofolate reductase (DHFR) has been widely used in studies of the relationship between enzyme structure, motions and catalysis.8–30 DHFR transfers a hydride from NADPH and a proton from the solvent to C-6 and N-5 of 7,8-dihydrofolate (DHF) to generate 5,6,7,8-tetrahydrofolate (THF) (Fig. 1). DHFR from Escherichia coli (EcDHFR) contains a number of mobile segments including the M20 (residues 9–24), FG (residues 116–132) and GH (residues 142–149) loops and switches between a closed and an occluded conformation during the catalytic cycle (Fig. 2).16,31 Upon binding of substrate and cofactor, the M20 loop closes over the active site by forming stable hydrogen bonds with the FG loop16 to create an optimal electrostatic environment conducive to hydride transfer.13,16 Once the products have formed, the M20 loop releases the nicotinamide ring of the oxidized cofactor and occludes part of the active site by forming an alternative hydrogen bonding pattern with the GH loop,16 which triggers the exchange of NADP+ and NADPH. Finally, the product THF is released from the active site in the rate-limiting step at pH 7 and the enzyme returns to the closed conformation and another reduced cofactor enters the active site.16
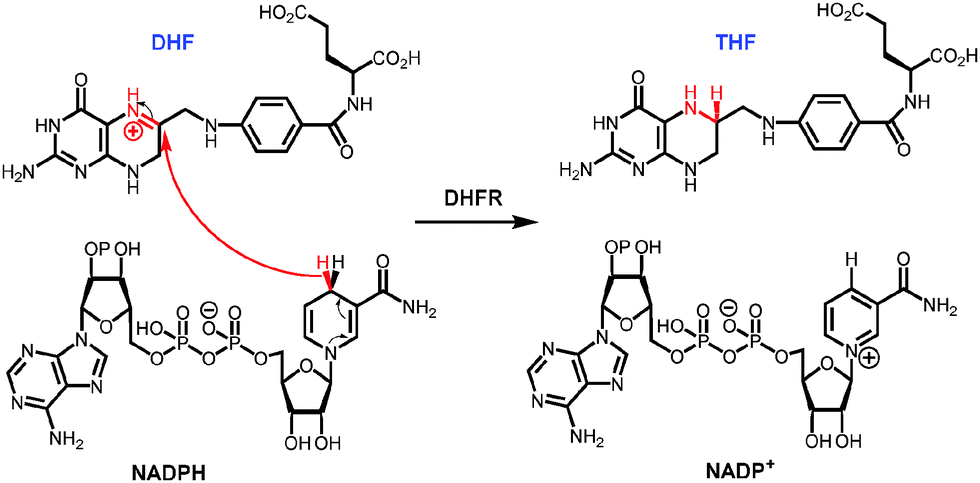 | ||
| Fig. 1 Chemical reaction catalysed by dihydrofolate reductase. | ||
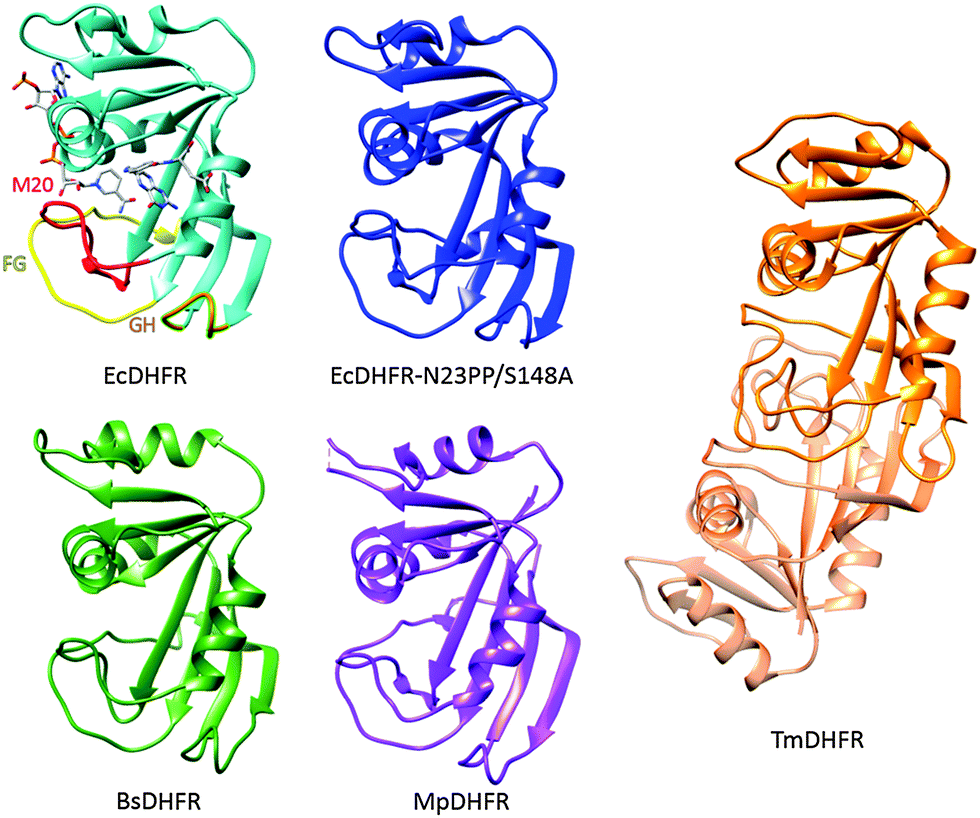 | ||
| Fig. 2 Cartoon representations of EcDHFR (1RX2),16 EcDHFR-N23PP/S148A (3QL0),24 MpDHFR (3IA5),54 TmDHFR (1D1G)39 and BsDHFR (1ZDR).35 NADP+ and folic acid serve as ligands in EcDHFR; the M20 (red), FG (yellow) and GH (orange) loops are highlighted. | ||
A number of DHFRs have been shown to follow similar catalytic cycles to EcDHFR, including DHFR from humans32 as well as those from bacteria including Lactobacillus casei (LcDHFR)33 and the psychrophile Moritella profunda (MpDHFR).34 In all these cases, and for DHFR from the thermophilic bacterium Geobacillus stearothermophilus (BsDHFR),35,36 the steady state turnover at pH 7 is limited by a physical step rather than the actual chemical step of hydride transfer. In contrast, catalytic turnover under steady-state conditions in DHFR from the hyperthermophilic bacterium Thermotoga maritima (TmDHFR) is at least partially limited by hydride transfer;37,38 TmDHFR has dramatically lower hydride transfer rate constants than other characterised chromosomal DHFRs.38 Unlike these DHFRs, TmDHFR relies on its dimeric structure for activity and with a melting temperature of 81 °C is the most thermostable DHFR characterised.39–41
The kinetic isotope effects (KIE) on the DHFR-catalysed reaction have been measured chiefly by two experimental techniques. For many DHFRs, the turnover number kcat at pH 7 reports mainly on product release14,32–34 and transient kinetic techniques such as stopped flow must be used to extract information about the chemical step of the catalytic cycle. These have the advantage that they can be performed under conditions where hydride transfer makes little or no contribution to kcat.42 We have made widespread use of single turnover stopped flow kinetics at pH 7 to study the DHFR-catalysed reaction. However, the observed single-turnover (or burst phase) stopped flow rate constants do not report exclusively on hydride transfer, but are complicated by typically small contributions from physical events.42 On the other hand, hydride transfer becomes increasingly rate limiting when the pH is elevated, so kcat at pH > 8.5 reports predominantly on hydride transfer with small contributions from physical steps.14 Competitive multiple-KIE measurements at elevated pH allow extraction of intrinsic KIEs, relatively free from kinetic complexity and reporting solely on the hydride transfer step, but such measurements are not ideal because the protonation state and conformational preference of the enzyme are altered, and the catalytic behaviour of the enzyme may not mirror that under physiological conditions42,43 and most computational investigations of the EcDHFR catalysed reaction have employed X-ray structures determined under conditions of neutral pH.18,21,44–46 The intrinsic KIE of EcDHFR has been estimated at various pH values showing that the degree of kinetic complexity depends on a number of factors, including reaction temperature and enzyme conformational behaviour as well as the protonation state of the enzyme.42 It has also been shown that the intrinsic KIE of EcDHFR at pH 7 is different to that at pH 9.47 Accordingly, single turnover KIEs under physiological and intrinsic KIEs at elevated pH must both be interpreted with caution.
The role of DHFR motions
The occluded conformation
The switch between the closed and occluded conformations is essential for the EcDHFR catalytic cycle, but this conformational transition has not been found in other DHFRs.28,48 For example, MpDHFR follows a similar catalytic cycle to EcDHFR but it does not form an occluded conformation since the crucial residue Ser148 in EcDHFR (Fig. 3) is replaced by a proline in MpDHFR.48 Ser148 forms two hydrogen bonds to the M20 loop in the occluded conformation of EcDHFR,16 neither of which can be formed by proline. M20 loop motions generally do not appear to play a significant role in MpDHFR, which likely remains in a closed conformation for all complexes in the catalytic cycle.49,50 In TmDHFR, the FG loop is buried in the dimer interface, apparently locking the enzyme in an open conformation (Fig. 2).39 Progression through the catalytic cycle has also been studied through the use of infrared probes.51–53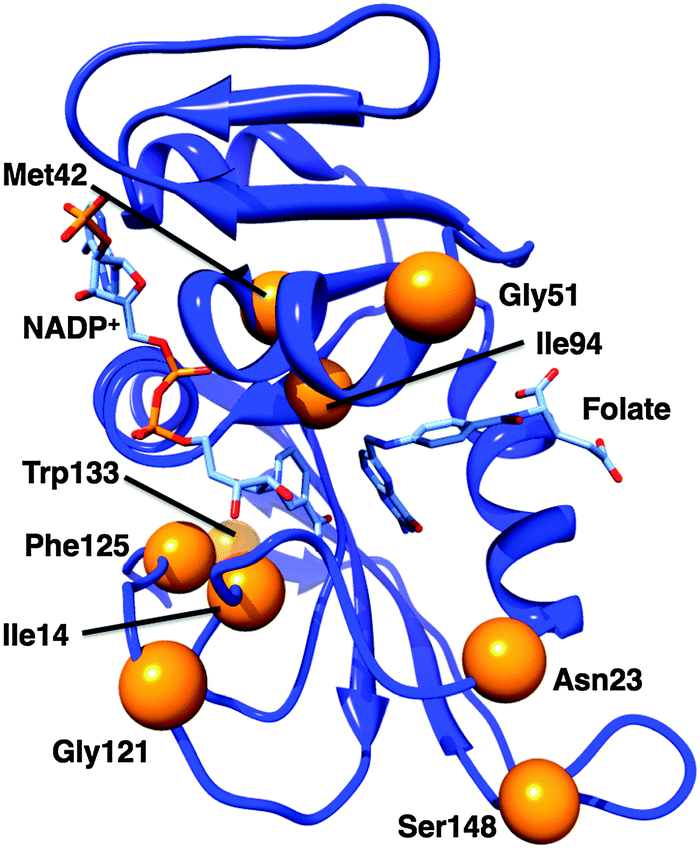 | ||
| Fig. 3 Cartoon representation of EcDHFR (1RX2).24 Bound ligands are shown as sticks; the α-carbons of the residues discussed in the text are marked with orange spheres. | ||
Hydride transfer is simply not possible in the occluded conformation as the reactants are not sufficiently close to one another. More generally, the ability to form the occluded conformation does not affect the chemical step of the catalytic cycle directly, as shown both by the similarity of the single turnover rate constants of EcDHFR and MpDHFR at pH 712,48 and by the existence of EcDHFR variants such as EcDHFR-S148A that are incapable of forming an occluded conformation yet maintain wild-type-like single turnover rate constants.43,44 It is likely that only DHFRs with a Ser148 equivalent are capable of adopting an occluded conformation.43,44 However, even when large conformational changes are not present, protein motions can still be involved in progression through the catalytic cycle, as demonstrated by the changes in millisecond motions in different EcDHFR product complexes,31 all of which adopt the occluded conformation.16 In agreement with the observation that only certain DHFRs are likely to form an occluded conformation, and that this does not affect the overall turnover rates, it has also been shown that native-like protein equilibrium motions are not required for the function of beta-lactamases.55
As discussed elsewhere,56 such equilibrium motions are the consequence of the free energy surface and modulation of this surface by ligand exchange is well known. Enzyme motions clearly affect the electrostatic environment by changing the relative positions of the interacting atoms. However, electrostatic effects determine the form of the free energy surface, and motions of the enzyme simply reflect the change in interactions between the atoms as the enzyme moves across it; they do not actively alter the free energy surface. Therefore a change in the motions of the enzyme is always a consequence of a change to the electrostatics of the enzyme, not the other way round. This does not diminish the importance of studies of conformational and other equilibrium motions, as they greatly aid the understanding of the free energy landscape. The importance of the free energy landscape in tuning conformational changes across multiple steps of an enzyme's catalytic cycle is well known.57 Enzyme motions, or rather enzyme conformational flexibility, have also been suggested to be important for the evolution of new enzymatic functions.58
Binding of antifolates to EcDHFR dampens millisecond conformational motions,59 consistent with the EcDHFR catalytic cycle involving millisecond motions that gate ligand binding and release.31 Enzyme-inhibitor interactions will be more favourable than those obtained in alternative conformational states (placing the enzyme in what has been referred to as a ‘dynamic straightjacket’59), imposing changes to the free energy surface that cause these motions to be dampened. However, measuring the effect on the enzyme motions will potentially provide a quantitative measure of how strong an interaction needs to be to overcome conformational switching.
While bacterial DHFRs typically have a mobile M20 loop, mammalian ones are more rigid due to the presence of a proline-rich sequence at the end of that loop.24,28,29 Incorporation of a diproline repeat into the M20 loop of EcDHFR to form EcDHFR-N23PP (Fig. 3) prevented formation of the occluded conformation and caused a general loss of the millisecond motions found in the wild type Michaelis complex; it has therefore been proposed that such motions could be linked to the chemical step.24 A subsequent computational study suggested that the impaired catalytic activity results from changes to the reorganisation free energy of the reaction brought about by changes to the electrostatic preorganisation within the active site rather than directly from changes to the motional behaviour of the enzyme.8 An experimental investigation supported the dominant effect of changes to the activation entropy on the single turnover rate constant at pH 7.13 Although the temperature dependence of the intrinsic KIE differed considerably between the wild type and variant enzymes, which was interpreted as being due to changes in the values and distribution of the donor–acceptor distance,47 such changes are an equilibrium property of the enzyme, affected by changes to the active site electrostatics.
It was later shown that the extension of the M20 loop rather than the presence of proline per se was responsible for the loss of conformational flexibility,28 presumably by disrupting the ability of the M20 loop to form stabilising hydrogen bonds to the GH loop. Additionally, it was shown that an insertion in the folate-binding region, acquired earlier in evolutionary history than the proline-rich region, is necessary for high catalytic activity in mammalian DHFRs.29 EcDHFR-N23PP/G51PEKN (Fig. 3) has a comparable rate constant for hydride transfer to wild type human DHFR,29 and the temperature dependence of the intrinsic KIE at pH 9 is similar to that of EcDHFR.47 Hence, despite the similarities in protein architectures, and even where the kinetics of the chemical step are highly similar, the conformational landscapes of different DHFRs may be quite different.
Networks of coupled motions
EcDHFR contains a network of residues whose motions couple to one another;17,18 this network is different in the closed and occluded conformations.17 Consequently, a network of motions that promote hydride transfer was postulated.18,46 Mutations of Gly121 or Met42 (Fig. 3) have large effects on the kinetics and stability of EcDHFR,15,60–62 and it was proposed that such mutations disrupt this network of promoting motions.46 Gly121 is found in the FG loop and Met42 in the adenosine-binding domain; both are rather far from the active site and both are highly conserved. The intrinsic KIE on hydride transfer at pH 9 is elevated for EcDHFR-G121V compared to the wild type enzyme with a slightly greater temperature dependence,63 while that of EcDHFR-M42W is less elevated but more temperature dependent,19 and EcDHFR-G121V/M42W has a greatly increased temperature dependence of the KIE.64 The single turnover rate constants and observed KIEs at pH 7 are also affected by the G121V mutation.65 On the basis of synergistic effects of mutations on the temperature dependence of the intrinsic KIE at pH 9, Phe125 (Fig. 3) was also identified as a member of the network, while Trp133 (Fig. 3) was excluded from it.66 Fitting of the intrinsic KIEs to a phenomenological modified Marcus model led to the proposal that these distal mutations alter the conformational sampling (based on equilibrium thermal motions) that leads to an optimal active site configuration in the wild type enzyme,67 altering the hydride transfer donor–acceptor distance and the ability of the enzyme to optimally control this distance.66 Interestingly, however, computational studies indicated that the donor–acceptor distance is similar in the wild type and variant enzymes at the transition state itself.68The G121V mutation causes a change to the ground-state structure of EcDHFR including alterations to the active site itself.65,69–71 Evidence from NMR measurements indicated that EcDHFR-G121V adopts an occluded conformation in the ground state of the Michaelis complex72 and that conformational fluctuations sample a state other than the closed conformation.71 This is unsurprising as the isopropyl side chain introduced by the conversion of Gly to Val is likely to point toward the interior of the enzyme16 leading to unfavourable steric interactions with the M20 loop and destabilisation of the hydrogen bonds between the FG and M20 loops that are necessary for stabilisation of the catalytically competent closed conformation,65,73 as well as consequent disruption of correlated motions between these loops. Networks of coupled motions have been identified in TmDHFR74 and in DHFR from Bacillus subtilis,75 although differences between these and the network in EcDHFR were seen, likely reflecting differences in their conformational landscapes. Indeed, the G123V mutation in MpDHFR has little effect on the single turnover rate constant or its observed KIE at pH 7, consistent with the proposal that interactions between the FG and M20 loops do not play a significant role in MpDHFR catalysis.49
Binding of NADPH and the inhibitor methotrexate to EcDHFR-G121V forms a putative mimic of the transition state and causes the enzyme to adopt a closed conformation,26 confirming that EcDHFR-G121V is capable of forming this conformation for hydride transfer. However, the mutation leads to aberrant millisecond conformational switching of the M20 and FG loops,26 as would be expected when the closed conformation is strongly destabilised, suggesting that these motions are anti-catalytic and destabilise the optimum active site configuration.26 The EcDHFR-M42W complex with NADPH and methotrexate also forms a closed conformation and shows slower millisecond motion than the wild-type enzyme.76 Interestingly, the effects of both the G121V and M42W mutations on the ps–ns motions are small,26,76 although side chain methyl fluctuations are sensitive to the M42W mutation with many showing increased rigidity and a smaller number showing greater flexibility.76 Computational studies indicate that EcDHFR-G121V/M42W has reduced M20 loop conformational motions and reduced flexibility at the transition state, related to an increased entropic barrier. This is in contrast to EcDHFR-N23PP/S148A, where increased flexibility at the transition state77 and in the Michaelis complex73 was observed despite the loss of millisecond conformational motions.24
These results indicate a role for networks of coupled motions in ‘promoting’ the chemical step in DHFR. However, they do so through thermal motions that are directly affected by changes to the active site electrostatics brought about by the mutations, however minor. This again is an equilibrium property and a manifestation of changes to the free energy surface of the enzyme. As such these motions are not the same as ‘promoting motions’ or ‘promoting vibrations’ that have been proposed to modulate the reaction coordinate by actively reducing the barrier height and/or width.3,78–87 Indeed, computational studies of EcDHFR indicate that motions are mostly thermally dissipated before the chemical transformation takes place,8,27,45,68,77,88 and the influence of mutations at Gly121 in EcDHFR has been explained in terms of the effect on the free energy surface.21,89,90 Additionally, while statistical motions may be coupled over large distances, true dynamic correlations (i.e. involving non-statistical motions) only exist over very small distances and non-local, long-range dynamics therefore appear not to have a significant role in EcDHFR catalysis.10 Formation of an optimal ‘reaction-ready’ active site configuration could be interpreted either as an enzyme rearrangement following substrate binding or as part of the ascent on the free energy surface towards the transition state. In either case coupled motions and their proposed effect on hydride transfer are a manifestation of movement on the enzyme's energy surface rather than an effect in their own right; these motions do not ‘couple’ directly to hydride transfer.
Active site volume and conformational sampling
Computational studies have suggested that side chain rotations of Ile14 and Ile94 of EcDHFR (Fig. 3) bring the reactants closer towards the ‘reaction-ready’ active site configuration.91 Subsequently, based on measurements of the intrinsic KIE at pH 9 and data fitting to a phenomenological modified Marcus model,67 it was proposed that changes to the active site volume brought about by altering Ile14 to less bulky residues affects the hydride transfer donor–acceptor distance in a similar manner to the distal mutations discussed above.25Similar effects of active site volume have been proposed for enzymes such as morphinone reductase,92 soybean lipoxygenase93,94 and alcohol dehydrogenase95 and the effect of pressure on hydride transfer kinetics has been investigated and related to ‘promoting vibrations’ in the active site.96–98 Changes in active site volume and in pressure do however not cause true ‘dynamic’ effects, but alter the equilibrium conformational ensemble of the enzyme. It has been shown that pressure does not directly compress the free energy surface,99 and more generally that barrier compression would lead to reduced tunnelling and reduced KIEs,21,99 in contrast to recent claims that shorter donor–acceptor distances lead to reduced KIEs due to increased deuterium tunnelling.47
The use of organic co-solvents to probe DHFR catalysis
A major theme of research in our laboratory has been an investigation of the effect of the addition of organic co-solvents on DHFR catalysis. Changing the composition of the solvent affects parameters such as the dielectric constant and the viscosity of the medium, both of which affect protein motions. Increasing solvent viscosity will dampen protein motions directly by opposing any motions that require rearrangement of the solvent,100 while reducing the dielectric constant of the solvent inhibits protein motions by decreasing the shielding effect of the medium on dipole–dipole interactions, thereby strengthening the H-bonding network and making the protein more stable but less flexible.101 While changes in the dielectric constant affect mainly the surface of a protein, leaving the interior motions essentially unaltered,102 the effects of viscosity changes can reach deep into the protein interior.100 In addition, co-solvents may affect the hydration layer on the surface of the protein and so alter protein motions “slaved” to this layer.103 In either case, changes to the motions can be considered a consequence of changes to the free energy surface for the protein.104,105For TmDHFR, EcDHFR, MpDHFR and BsDHFR, viscosity had no effect on the single turnover rate constants at pH 7 and solvent composition did not affect the KIE.11,12,22,36 In contrast, the dielectric constant of the medium had a pronounced effect on both the single turnover and steady state rate constants at pH 7. Although no viscosity effect on the steady state rate constant was observed for TmDHFR,22 MpDHFR12 or BsDHFR,36 solvents with similar dielectric constants reveal a small, but consistent, effect from viscosity in the steady state for EcDHFR.11 This is consistent with the rate-determining step in the catalytic cycle of EcDHFR, but not those of the other three DHFRs, involving a substantial conformational change (vide supra).
We initially interpreted our results for TmDHFR in terms of an environmentally coupled tunnelling model of catalysis22 but it became clear that this model could not adequately explain the effect of co-solvents on catalysis by other DHFRs.11,12 The dominant effect of the dielectric constant provides strong support for an overriding role for electrostatics in controlling catalysis, as proposed for enzymes in general104,105 and DHFR specifically.21,53,90 Changes to the solvent composition will affect the free energy surface for the protein and so affect the reorganisation energy of the catalysed reaction. In contrast, the lack of a viscosity effect provides strong evidence against long-range motions on any timescale being involved in the chemical step.
‘Heavy’ DHFRs
Most recently, efforts in our laboratory have turned to the use of isotope labelling to probe the effect of protein motions on DHFR catalysis. While isotope labelling has long been employed in protein NMR experiments, it has also recently found use in kinetic studies.27,30,77,82,106–111 ‘Heavy’ enzymes are produced in minimal media containing appropriate labelled ingredients such as 15NH4Cl, U-13C,2H-glucose, and 2H2O.27 Purification in 1H2O-based buffers leads to exchange of labile deuterons for protons. Increases of ∼10% in an enzyme's molecular weight corresponding to >98% heavy isotope incorporation at non-exchangeable positions are typically achieved in this way. As bond vibrational frequencies and other motions of enzymes can be slowed by isotope substitution, the entire profile of motions of an enzyme can be altered by isotope labelling. Furthermore, given that within the Born–Oppenheimer approximation the electrostatic potential of an enzyme is independent of atomic mass, the biophysical differences between the ‘heavy’ enzyme and the ‘light’ enzyme (with isotopes of natural abundance) originate predominantly from changes in the profile of motions.106,107 Accordingly, characterising these biophysical differences provides valuable insight into the role of the conformational landscape in enzyme catalysis.The effect of protein isotope labelling is assessed by comparing the reaction rate constants for the light enzyme, kLE, and heavy enzyme, kHE, to give an ‘enzyme KIE’, kLE/kHE, where the reactants are not isotopically labelled. In contrast to primary hydrogen KIEs, where extensive literature exists for the interpretation of their temperature dependence,13,21,44,93,112–114 the effect of temperature on the enzyme KIE has not yet been fully analysed. To investigate dynamic (i.e. non-statistical) contributions to the hydride transfer rate constant ensemble-averaged variational transition-state theory (EA-VTST) calculations were performed that incorporate a correction to the quasi-classical rate constant, the transmission coefficient, which takes into account dynamic and tunnelling contributions to the reaction:115–117
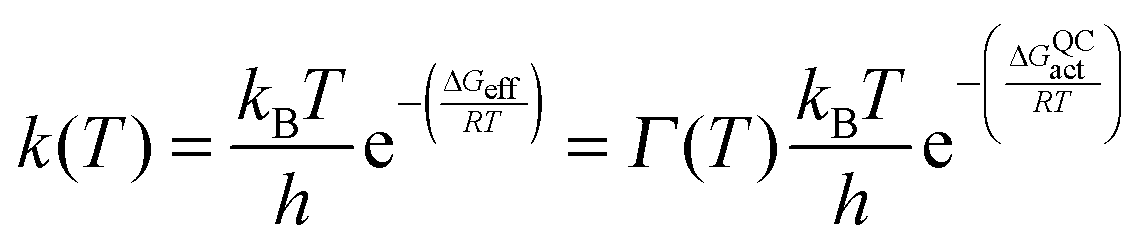
| Γ(T) = γ(T)·κ(T) |
To date, we have performed ‘heavy’ enzyme studies on four DHFRs, namely EcDHFR, EcDHFR-N23PP/S148A, the thermophilic BsDHFR and the hyperthermophilic TmDHFR. Under pH 7 steady state conditions, EcDHFR gave a small, temperature-dependent enzyme KIEcat (kLEcat/kHEcat) (Fig. 4),27 while for EcDHFR-N23PP/S148A kcat was not affected by isotope labelling.77 As the rate-limiting product release step in EcDHFR catalysis involves movement of the M20 and FG loops,31 whereas in EcDHFR-N23PP/S148A NADP+ release is rate limiting and is not coupled to a significant conformational change,24 a temperature-dependent enzyme KIEcat is likely an indication of the involvement of a conformational change in the rate-limiting step of the catalytic cycle.27,77 ‘Heavy’ BsDHFR gave a relatively large, temperature independent enzyme KIEcat of ∼2.6 (Fig. 4). As the temperature independence of KIEcat suggests no involvement of a large-scale conformational change in kcat, the measured kinetic difference was attributed to the inherent flexibility of the enzyme.111
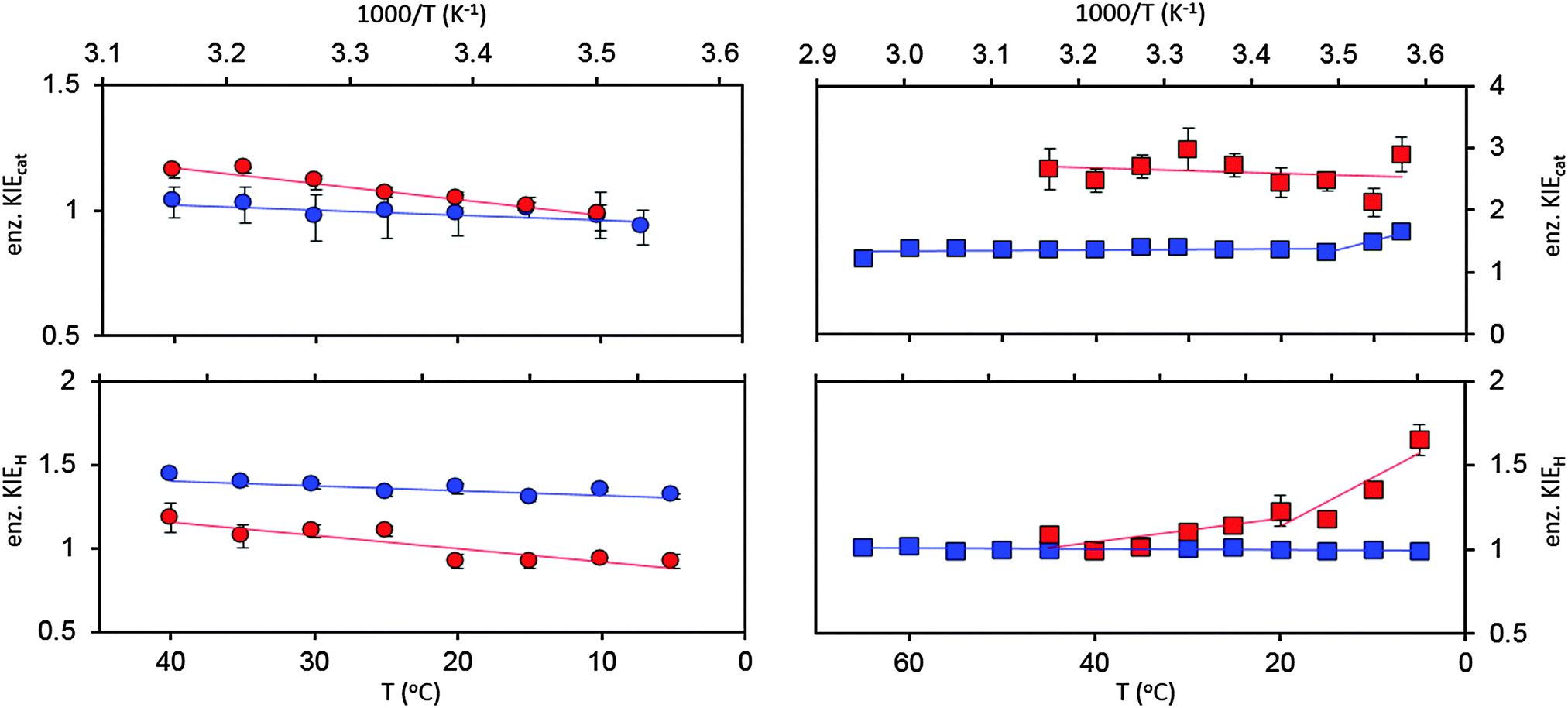 | ||
Fig. 4 Enzyme KIEs at pH 7 for EcDHFR ( ),27 EcDHFR-N23PP/S148A ( ),27 EcDHFR-N23PP/S148A ( ),77 TmDHFR ( ),77 TmDHFR ( )110 and BsDHFR ( )110 and BsDHFR ( )111 under steady state (top) and pre-steady state (bottom) conditions. )111 under steady state (top) and pre-steady state (bottom) conditions. | ||
At pH 7 the enzyme KIEH (kLEH/kHEH, where kH is the single turnover rate constant that reports predominantly on hydride transfer) for EcDHFR was slightly inverse at 5 °C but increased with temperature, passing through zero and increasing to give a small, normal value at 40 °C (Fig. 4).27 It was demonstrated that the data for EcDHFR could be fit to a single-conformer kinetic model, without the need to invoke multiple catalytically competent conformations.27,119,120 A weakly temperature dependent enzyme KIE was observed for hydride transfer catalysed by EcDHFR-N23PP/S148A, but with greater magnitude than for the wild type enzyme.77
EA-VTST calculations indicated that the tunnelling coefficient remains unchanged on isotope substitution and is not significantly different between the wild type and variant enzymes,27,77 in agreement with a previous computational study that found no role for ‘promoting motions’, including those in the network of coupled motions (vide supra), in driving barrier passage in EcDHFR.121 Instead, the observed difference in the single turnover rate constant is due to a small but significant difference between the recrossing coefficients in the ‘light’ and ‘heavy’ enzymes. It appears that the reaction in the ‘heavy’ enzymes is more prone to dynamic recrossing because the response from the environmental motions along the reaction coordinate is slowed.27,77 The non-productive recrossing trajectories were enhanced in the N23PP/S148A variant,77 suggesting that additional protein vibrations are incorporated into the transition state. Hence, although EcDHFR-N23PP/S148A is a ‘dynamic knock-out’ on the millisecond timescale, it is actually a ‘dynamic knock-in’ on the timescale of the chemical step, which contributes to the reduction in the efficiency of hydride transfer.77 While similar concepts have been discussed in previous computational studies,7,21 this report provided the first experimental evidence for the existence of ‘demoting vibrations’.77
The enzyme KIEH for the BsDHFR catalysed reaction showed biphasic behaviour, being relatively low (∼1.1) and temperature independent above 20 °C, but increasing sharply with decreasing temperature (Fig. 4).111 As observed previously,27,77 only the recrossing coefficients, not the tunnelling coefficients, were affected by isotope labeling,111 again in disagreement with the proposals of barrier modulation and the involvement of ‘promoting’ motions. The singly deuterium-labelled and doubly 15N and 13C-labelled BsDHFRs were also prepared to investigate the biphasic temperature dependence. Since the molecular weights of these enzymes increased by ∼5%, instead of ∼10% as observed for the triply 15N, 13C, 2H-labelled, fully ‘heavy’ BsDHFR, the enzyme KIEs measured with these partially labelled enzymes would reveal clues about the intensity of the dynamic coupling.111 Above 25 °C, the enzyme KIEH measured with these labelled enzymes were statistically the same as those with the fully ‘heavy’ enzyme, suggesting that dynamic coupling is limited.111 This also confirmed that the observed enzyme KIE is caused by alteration of the protein's dynamics rather than other mass-induced effects such as a change in van der Waals radii or hydrophobicity due to deuterium labelling.111 In contrast, at 5 °C the enzyme KIEH measured with the fully ‘heavy’ enzyme was noticeably higher than those of the singly and doubly labelled enzymes. The thermal melting temperatures of the ‘light’ and ‘heavy’ BsHDFRs also showed no significant difference.
In contrast to EcDHFR, EcDHFR-N23PP/S148A and BsDHFR, protein isotope labelling exerted no effect on the single-turnover rate constants for TmDHFR at pH 7 resulting in an enzyme KIEH of unity for all temperatures investigated (Fig. 4). Accordingly, mass-dependent protein dynamics do not appear to exert any detectable effect on the chemical step of TmDHFR catalysis. It may therefore be possible that TmDHFR has gained a small catalytic benefit by eliminating dynamic recrossing trajectories from the transition state dividing surface.110 It had previously been postulated that active site motions may be detrimental to catalysis and that this observation becomes more pronounced in enzymes from thermophilic species.122 Our studies however indicate that TmDHFR is able to overcome this, by eliminating dynamic coupling at high temperatures. Under steady-state conditions at pH 7 the enzyme KIEcat for TmDHFR remained at ∼1.35 from 15 °C to 65 °C, but increased sharply below 15 °C (Fig. 4). The temperature-independent region likely relates to the absence of significant conformational changes in the TmDHFR catalytic cycle, whereas the temperature-dependent region suggests a change in the conformational equilibrium favourable for reaction. It should be noted that wild type TmDHFR revealed subtle biphasic behaviour under pH 7 steady state conditions, but TmDHFR variants with disrupted dimeric interface did not show this behaviour.112,123 Hence, this hypothetical switch in conformational equilibrium could result from a change in inter-subunit interactions.110
Of further interest is the fact that KIEH increases with increasing temperature for EcDHFR27 (and to a lesser extent EcDHFR-N23PP/S148A77), is temperature independent (and negligible) for TmDHFR,110 but decreases with increasing temperature for BsDHFR.111 For BsDHFR, nine pairs of recrossing coefficients were calculated to investigate the temperature dependence of KIEH. The recrossing coefficient was shown to absorb all non-statistical effects; in particular, at low temperature it is noticeably lower in the ‘heavy’ enzyme than in the ‘light’ enzyme.111In silico studies also indicated that BsDHFR is more flexible on the nanosecond timescale,111 supporting earlier experimental studies.124–126 The reverse temperature dependence of the enzyme KIEH in BsDHFR relative to EcDHFR may be related to this intrinsic flexibility of BsDHFR.111 Heavy isotope labelling raises the enthalpic barrier for electrostatic preorganisation and the reorganisation energy for reorienting the substrates; consequently, the transition state experiences additional friction in the enzyme active site. For BsDHFR, this is more pronounced at low temperatures, because the enzyme lacks the energy required to adopt an ideal configuration, leading to an increased recrossing contribution in the ‘heavy’ enzyme and a strong enzyme KIEH. However, most of this enthalpic barrier can be overcome at physiological temperatures, allowing hydride transfer to proceed in an ideal electrostatic configuration with minimal dynamic effects. This was also evident from the activation parameters; the higher activation enthalpy (ΔH‡) in ‘heavy’ BsDHFR is accompanied by a smaller magnitude of the activation entropy (ΔS‡). The activation entropy that is contributed by the temperature-dependent recrossing coefficient (ΔS‡γ) can be expressed as:111
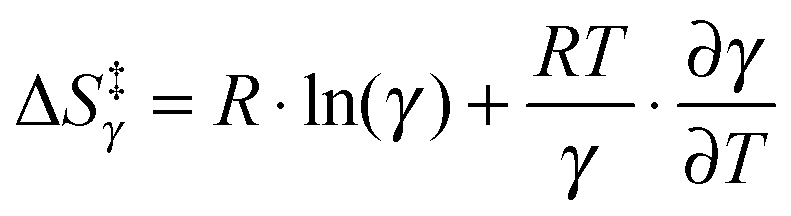
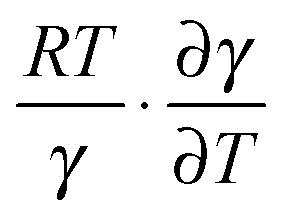 can counteract this due to the enhanced protein flexibility at high temperature. In EcDHFR, the weakly temperature-dependent increase of the enzyme KIEH can be accounted for by the dominance of R·ln(γ), while for EcDHFR-N23PP/S148A, ΔS‡ is approximately the same in the ‘light’ and ‘heavy’ enzymes, most likely because there is a compensating effect between R·ln(γ) and
can counteract this due to the enhanced protein flexibility at high temperature. In EcDHFR, the weakly temperature-dependent increase of the enzyme KIEH can be accounted for by the dominance of R·ln(γ), while for EcDHFR-N23PP/S148A, ΔS‡ is approximately the same in the ‘light’ and ‘heavy’ enzymes, most likely because there is a compensating effect between R·ln(γ) and 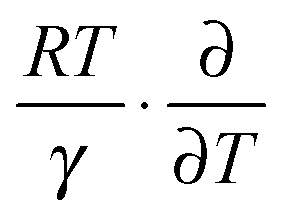 . Lastly, the activation entropy contributed by dynamic effects is likely insignificant in TmDFHR due to its extreme rigidity
. Lastly, the activation entropy contributed by dynamic effects is likely insignificant in TmDFHR due to its extreme rigidity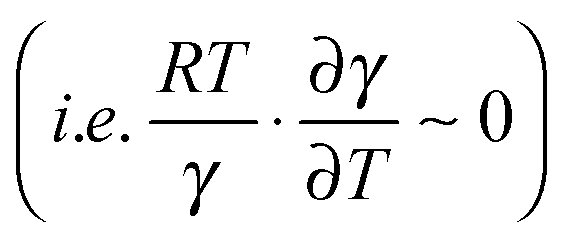 .
.
Recently, a complementary experimental analysis of ‘heavy’ EcDHFR was reported.30 Differences in thermal melting temperatures and binding kinetics of certain ligands were observed, leading to the conclusion that the conformational ensemble is altered on protein isotope labelling.30 Furthermore, the intrinsic primary KIE at pH 9 was found to be unchanged by enzyme isotopic substitution at temperatures above 20 °C, but to diverge at low temperatures, giving a ∼2-fold difference between the ‘light’ and ‘heavy’ enzymes at 5 °C.30 Based on a phenomenological modified Marcus model,67 it was suggested that the average distance between hydride donor and acceptor increases at low temperatures in the ‘heavy’ enzyme.30 However, all of these reports conclude that dynamic coupling is not a dominant factor in the DHFR-catalysed reaction.27,30,77,110,111 HIV-1 protease, purine nucleoside phosphorylase, alanine racemase and pentaerythritol tetranitrate reductase have also been used for protein isotope substitution studies.82,106–108 The kinetics were affected differently among these enzymes, implying that there is no universal relationship between the conformational and chemical coordinates. Nevertheless, these experiments demonstrate that protein isotope labelling in combination with computational studies, provide a pragmatic approach to the study of the role of protein dynamics in enzyme catalysis.
Conclusions
Protein motions have a range of roles in DHFR catalysis. Equilibrium thermal motions are involved in progressing the physical steps of the catalytic cycle28,31 even in the absence of major conformational changes. In the Michaelis complex such motions allow water access to protonate N5 of the substrate127,128 and subtle rearrangements generate the reaction-ready active site configuration.To define the direct relation between enzyme motions and the chemical step of catalysis is more challenging. Both BsDHFR111,124,125 and MpDHFR12 are more flexible than EcDHFR, yet at pH 7 the three enzymes have similar single turnover rate constants.12,36 EcDHFR-N23PP/S148A has reduced thermal motion on the millisecond timescale, which impedes the conformational changes required for optimal progression through the catalytic cycle.24 The increased dynamic coupling to the reaction on the femtosecond–picosecond timescale is detrimental to the chemical step as it increases the proportion of unproductive trajectories on the transition state dividing surface.77 Phenomenological models of enzyme catalysis that interpret the KIE in terms of a distribution of donor–acceptor distances cannot account for alternative explanations for these data.67 Dynamic coupling to the reaction coordinate is effectively minimised in TmDHFR, which should provide some benefit, but its catalytic performance is poor due to the dominance of other factors.110 Thermal motions prior to the chemical step may ‘promote’ it by providing an optimal reaction-ready active site configuration, but correlations between motions, and between motions and kinetics, must be interpreted with caution. Although this is most obvious when the timescales of the motions and the chemical events are different, it has recently been demonstrated that many motions in DHFR do not affect the chemical step despite occurring on similar timescales to it.129 Furthermore, even where motions are truly coupled to the reaction coordinate, it does not necessarily follow that these motions drive catalysis since they are controlled by the same free energy landscape.
In the majority of the cases discussed here, the motions are equilibrium fluctuations. ‘Well tuned’ equilibrium motions are a consequence of well-tuned electrostatics. However, this does not subtract from the importance of studies of protein motions in enzyme catalysis. Although statistical motions are a consequence of the free energy landscape of the enzyme rather than a cause of it, and non-statistical dynamics can be affected by statistical motions, their effects can provide valuable information about the physical effects of the free energy surface and changes to it.
It appears that efficient enzymes minimise dynamic coupling in their transition states. The chemical step is a fast step of catalysis and so selective pressure cannot easily act on it. Although most DHFRs give a measurable steady-state KIE at pH 7, the typically >10-fold difference between kcat and kH indicates that the small benefit obtained from eliminating dynamic coupling is unlikely sufficient for selective pressure to act on. Evolution-based arguments may therefore appear inappropriate on first examination. However, evolution does appear to have managed both to minimise dynamic coupling to the reaction coordinate and to maintain this favourable state. One possibility for how this occurred is that enzyme dynamics were optimised early in history, when the chemistry was rate limiting, and neutral drift has not overcome this even when physical steps became rate limiting. This seems an unsatisfactory explanation as it relies on the absence of neutral drift. A more likely alternative may be that coupling of fast dynamics to the reaction coordinate is tied to other factors that selective pressure can act on, such as conformational sampling or other equilibrium motions on millisecond timescales. As all motions are affected by the same free energy surface this seems reasonable, although the question remains how the local non-statistical dynamics, which appear to be uncoupled from motions in the surrounding environment,10,121 are affected by the free energy surface.
These findings lead to the question of why dynamic coupling exists, if it is detrimental to catalysis. Indeed, it is a relatively common phenomenon with a number of enzymes showing a measurable enzyme KIE at or near physiological conditions.27,30,82,106,130 At least in the case of DHFR catalysis, maximal catalytic power appears to be achieved by carefully tuning the involvement of protein motions. Conformational transitions couple to the physical steps, but an ideal reaction-ready active site configuration should provide a static environment, where hydride transfer can proceed efficiently. The dynamic effects observed in DHFR are likely ‘residual’ motions from the reorganisation needed to facilitate the charge transfer in forming the transition state species. As the system climbs the energy barrier towards the transition state, degrees of freedom are progressively lost. This constraint of the atomic positions is likely to translate to a loss of non-statistical motions. Dynamic coupling in DHFR is therefore an undesired by-product that has been imperfectly eliminated. It may be that since the chemical transformation catalysed by DHFR is relatively simple with few charge transfers involved, dynamic effects are automatically minimised so long as the enzyme samples an ideal reaction ready configuration. For more complex, multi-step chemical transformations, dynamic coupling may be less well suppressed. These findings may find uses in designing enzymes with new functionality. To test the validity of the current proposal, investigations of further enzyme systems will be required.
Acknowledgements
Work on DHFR catalysis in our laboratory has been funded by the UK's Biotechnology and Biological Sciences Research Council (BBSRC) through grants 6/B15285, BB/E008380/1, BB/J005266/1 and BB/L020394/1, by the UK's Engineering and Physical Sciences Research Council through various PhD studentships, by the University of Birmingham and by Cardiff University.Notes and references
- M. Karplus and J. A. McCammon, Annu. Rev. Biochem., 1984, 52, 263 CrossRef PubMed.
- J. Basran, M. J. Sutcliffe and N. S. Scrutton, Biochemistry, 1999, 39, 3218 CrossRef PubMed.
- D. Antoniou, S. Caratzoulas, C. Kalyanaraman, J. S. Mincer and S. D. Schwartz, Eur. J. Biochem., 2002, 269, 3103 CrossRef CAS.
- M. J. Sutcliffe and N. S. Scrutton, Eur. J. Biochem., 2002, 269, 3096 CrossRef CAS.
- M. J. Knapp and J. P. Klinman, Eur. J. Biochem., 2002, 269, 3113 CrossRef CAS.
- J. P. Klinman, J. Phys. Org. Chem., 2010, 23, 606 CrossRef CAS.
- Q. Cui and M. Karplus, J. Phys. Chem. B, 2002, 106, 7927 CrossRef CAS.
- A. J. Adamczyk, J. Cao, S. C. L. Kamerlin and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14115 CrossRef CAS PubMed.
- A. V. Pisliakov, J. Cao, S. C. L. Kamerlin and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17359 CrossRef CAS PubMed.
- N. Boekelheide, R. Salomón-Ferrer and T. F. Miller, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16159 CrossRef CAS PubMed.
- E. J. Loveridge, L.-H. Tey and R. K. Allemann, J. Am. Chem. Soc., 2010, 132, 1137 CrossRef CAS PubMed.
- E. J. Loveridge, L.-H. Tey, E. M. Behiry, W. M. Dawson, R. M. Evans, S. B.-M. Whittaker, U. L. Gunther, C. Williams, M. P. Crump and R. K. Allemann, J. Am. Chem. Soc., 2011, 133, 20561 CrossRef CAS PubMed.
- E. J. Loveridge, E. M. Behiry, J. Guo and R. K. Allemann, Nat. Chem., 2012, 4, 292 CrossRef CAS PubMed.
- C. A. Fierke, K. A. Johnson and S. J. Benkovic, Biochemistry, 1987, 26, 4085 CrossRef CAS.
- K. Gekko, K. Yamagami, Y. Kunori, S. Ichihara, M. Kodama and M. Iwakura, J. Biochem., 1993, 113, 74 CAS.
- M. R. Sawaya and J. Kraut, Biochemistry, 1997, 36, 586 CrossRef CAS PubMed.
- J. L. Radkiewicz and C. L. Brooks III, J. Am. Chem. Soc., 2000, 122, 225 CrossRef CAS.
- P. K. Agarwal, S. R. Billeter, P. T. R. Rajagopalan, S. J. Benkovic and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2794 CrossRef CAS PubMed.
- L. Wang, N. M. Goodey, S. J. Benkovic and A. Kohen, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1307 CrossRef CAS PubMed.
- R. K. Allemann, R. M. Evans, L.-H. Tey, G. Maglia, J. Pang, R. Rodriguez, P. J. Shrimpton and R. S. Swanwick, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1317 CrossRef CAS PubMed.
- H. Liu and A. Warshel, J. Phys. Chem. B, 2007, 111, 7852 CrossRef CAS PubMed.
- E. J. Loveridge, R. M. Evans and R. K. Allemann, Chem. – Eur. J., 2008, 14, 10782 CrossRef CAS PubMed.
- R. K. Allemann, R. M. Evans and E. J. Loveridge, Biochem. Soc. Trans., 2009, 37, 349 CrossRef CAS PubMed.
- G. Bhabha, J. Lee, D. C. Ekiert, J. Gam, I. A. Wilson, H. J. Dyson, S. J. Benkovic and P. E. Wright, Science, 2011, 332, 234 CrossRef CAS PubMed.
- V. Stojković, L. L. Perissinotti, D. Willmer, S. J. Benkovic and A. Kohen, J. Am. Chem. Soc., 2011, 134, 1738 CrossRef PubMed.
- R. V. Mauldin, P. J. Sapienza, C. M. Petit and A. L. Lee, PLoS One, 2012, 7, e33252 CAS.
- L. Y. P. Luk, J. J. Ruiz-Pernia, W. M. Dawson, M. Roca, E. J. Loveridge, D. R. Glowacki, J. N. Harvey, A. J. Mulholland, I. Tuñón, V. Moliner and R. K. Allemann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16344 CrossRef CAS PubMed.
- G. Bhabha, D. C. Ekiert, M. Jennewein, C. M. Zmasek, L. M. Tuttle, G. Kroon, H. J. Dyson, A. Godzik, I. A. Wilson and P. E. Wright, Nat. Struct. Mol. Biol., 2013, 20, 1243 CAS.
- C. T. Liu, P. Hanoian, J. B. French, T. H. Pringle, S. Hammes-Schiffer and S. J. Benkovic, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10159 CrossRef CAS PubMed.
- Z. Wang, P. N. Singh, C. M. Czekster, A. Kohen and V. L. Schramm, J. Am. Chem. Soc., 2014, 136, 8333 CrossRef CAS PubMed.
- D. D. Boehr, D. McElheny, H. J. Dyson and P. E. Wright, Science, 2006, 313, 1638 CrossRef CAS PubMed.
- J. R. Appleman, W. A. Beard, T. J. Delcamp, N. J. Prendergast, J. H. Freisheim and R. L. Blakley, J. Biol. Chem., 1990, 265, 2740 CAS.
- J. Andrews, C. A. Fierke, B. Birdsall, G. Ostler, J. Feeney, G. C. K. Roberts and S. J. Benkovic, Biochemistry, 1989, 28, 5743 CrossRef CAS.
- R. M. Evans, E. M. Behiry, L.-H. Tey, J. Guo, E. J. Loveridge and R. K. Allemann, ChemBioChem, 2010, 11, 2010 CrossRef CAS PubMed.
- H. S. Kim, S. M. Damo, S. Y. Lee, D. Wemmer and J. P. Klinman, Biochemistry, 2005, 44, 11428 CrossRef CAS PubMed.
- J. Guo, L. Y. P. Luk, E. J. Loveridge and R. K. Allemann, Biochemistry, 2014, 53, 2855 CrossRef CAS PubMed.
- G. Maglia and R. K. Allemann, J. Am. Chem. Soc., 2003, 125, 13372 CrossRef CAS PubMed.
- G. Maglia, M. H. Javed and R. K. Allemann, Biochem. J., 2003, 374, 529 CrossRef CAS PubMed.
- T. Dams, G. Auerbach, G. Bader, U. Jacob, T. Ploom, R. Huber and R. Jaenicke, J. Mol. Biol., 2000, 297, 659 CrossRef CAS PubMed.
- T. Dams, G. Bohm, G. Auerbach, G. Bader, H. Schuring and R. Jaenicke, Biol. Chem., 1998, 379, 367 CAS.
- V. Wilquet, J. A. Gaspar, M. van de Lande, M. van de Casteele, C. Legrain, E. M. Meiering and N. Glansdorff, Eur. J. Biochem., 1998, 255, 628 CAS.
- E. J. Loveridge and R. K. Allemann, ChemBioChem, 2011, 12, 1258 CrossRef CAS PubMed.
- Y.-Q. Chen, J. Kraut and R. Callender, Biophys. J., 1997, 72, 936 CrossRef CAS.
- J. Pu, S. Ma, J. Gao and D. G. Truhlar, J. Phys. Chem. B, 2005, 109, 8551 CrossRef CAS PubMed.
- M. Garcia-Viloca, D. G. Truhlar and J. Gao, Biochemistry, 2003, 42, 13558 CrossRef CAS PubMed.
- L. B. Watney, P. K. Agarwal and S. Hammes-Schiffer, J. Am. Chem. Soc., 2003, 125, 3745 CrossRef PubMed.
- K. Francis, V. Stojković and A. Kohen, J. Biol. Chem., 2013, 288, 35961 CrossRef CAS PubMed.
- E. M. Behiry, L. Y. P. Luk, S. M. Matthews, E. J. Loveridge and R. K. Allemann, Biochemistry, 2014, 53, 4761 CrossRef CAS PubMed.
- E. M. Behiry, R. M. Evans, J. Guo, E. J. Loveridge and R. K. Allemann, Biochemistry, 2014, 53, 4769 CrossRef CAS PubMed.
- E. M. Behiry, L. Y. P. Luk, S. M. Matthews, E. J. Loveridge and R. K. Allemann, Biochemistry, 2014, 53, 4761 CrossRef CAS PubMed.
- M. C. Thielges, D. A. Case and F. E. Romesberg, J. Am. Chem. Soc., 2008, 130, 6597 CrossRef CAS PubMed.
- D. Groff, M. C. Thielges, S. Cellitti, P. G. Schultz and F. E. Romesberg, Angew. Chem., Int. Ed., 2009, 48, 3478 CrossRef CAS PubMed.
- C. T. Liu, J. P. Layfield, R. J. Stewart, J. B. French, P. Hanoian, J. B. Asbury, S. Hammes-Schiffer and S. J. Benkovic, J. Am. Chem. Soc., 2014, 136, 10349 CrossRef CAS PubMed.
- S. Hay, R. M. Evans, C. Levy, E. J. Loveridge, X. Wang, D. Leys, R. K. Allemann and N. S. Scrutton, ChemBioChem, 2009, 10, 2348 CrossRef CAS PubMed.
- S. M. C. Gobeil, C. M. Clouthier, J. Park, D. Gagné, A. M. Berghuis, N. Doucet and J. N. Pelletier, Chem. Biol., 2014, 21, 1330 CrossRef CAS PubMed.
- S. C. L. Kamerlin and A. Warshel, Proteins: Struct., Funct., Bioinf., 2010, 78, 1339 CAS.
- J. Villali and D. Kern, Curr. Opin. Chem. Biol., 2010, 14, 636 CrossRef CAS PubMed.
- N. Tokuriki and D. S. Tawfik, Science, 2009, 324, 203 CrossRef CAS PubMed.
- R. V. Mauldin, M. J. Carroll and A. L. Lee, Structure, 2009, 17, 386 CrossRef CAS PubMed.
- K. Gekko, Y. Kunori, H. Takeuchi, S. Ichihara and M. Kodama, J. Biochem., 1994, 116, 34 CAS.
- C. E. Cameron and S. J. Benkovic, Biochemistry, 1997, 36, 15792 CrossRef CAS PubMed.
- E. Ohmae, Y. Fukumizu, M. Iwakura and K. Gekko, J. Biochem., 2005, 137, 643 CrossRef CAS PubMed.
- L. Wang, S. Tharp, T. Selzer, S. J. Benkovic and A. Kohen, Biochemistry, 2006, 45, 1383 CrossRef CAS PubMed.
- L. Wang, N. M. Goodey, S. J. Benkovic and A. Kohen, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15753 CrossRef CAS PubMed.
- R. S. Swanwick, P. J. Shrimpton and R. K. Allemann, Biochemistry, 2004, 43, 4119 CrossRef CAS PubMed.
- P. Singh, A. Sen, K. Francis and A. Kohen, J. Am. Chem. Soc., 2014, 136, 2575 CrossRef CAS PubMed.
- D. Roston, C. M. Cheatum and A. Kohen, Biochemistry, 2012, 51, 6860 CrossRef CAS PubMed.
- D. Roston, A. Kohen, D. Doron and D. T. Major, J. Comput. Chem., 2014, 35, 1411 CrossRef CAS PubMed.
- K. Gekko, Y. Tamura, E. Ohmae, H. Hayashi, H. Kagamiyama and H. Ueno, Protein Sci., 1996, 5, 542 CrossRef CAS PubMed.
- I. F. Thorpe and C. L. Brooks III, Proteins: Struct., Funct., Bioinf., 2004, 57, 444 CrossRef CAS PubMed.
- D. D. Boehr, J. R. Schnell, D. McElheny, S.-H. Bae, B. M. Duggan, S. J. Benkovic, H. J. Dyson and P. E. Wright, Biochemistry, 2013, 52, 4605 CrossRef CAS PubMed.
- R. P. Venkitakrishnan, E. Zaborowski, D. McElheny, S. J. Benkovic, H. J. Dyson and P. E. Wright, Biochemistry, 2004, 43, 16046 CrossRef CAS PubMed.
- H. van den Bedem, G. Bhabha, K. Yang, P. E. Wright and J. S. Fraser, Nat. Methods, 2013, 10, 896 CrossRef CAS PubMed.
- J. Y. Pang, J. Z. Pu, J. L. Gao, D. G. Truhlar and R. K. Allemann, J. Am. Chem. Soc., 2006, 128, 8015 CrossRef CAS PubMed.
- J. B. Watney and S. Hammes-Schiffer, J. Phys. Chem. B, 2006, 110, 10130 CrossRef CAS PubMed.
- R. V. Mauldin and A. L. Lee, Biochemistry, 2010, 49, 1606 CrossRef CAS PubMed.
- J. J. Ruiz-Pernia, L. Y. P. Luk, R. García-Meseguer, S. Martí, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, J. Am. Chem. Soc., 2013, 135, 18689 CrossRef CAS PubMed.
- S. D. Schwartz and V. L. Schramm, Nat. Chem. Biol., 2009, 5, 552 CrossRef PubMed.
- D. Antoniou and S. D. Schwartz, Philos. Trans. R. Soc. London, Ser. B, 2006, 361, 1433 CrossRef PubMed.
- J. S. Mincer and S. D. Schwartz, J. Chem. Phys., 2004, 120, 7755 CrossRef CAS PubMed.
- S. Caratzoulas and S. D. Schwartz, J. Chem. Phys., 2001, 114, 2910 CrossRef CAS PubMed.
- C. R. Pudney, A. Guerriero, N. J. Baxter, L. O. Johannissen, J. P. Waltho, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2013, 135, 2512 CrossRef CAS PubMed.
- S. Hay and N. S. Scrutton, Nat. Chem., 2012, 4, 161 CrossRef CAS PubMed.
- S. Hay, C. R. Pudney, M. J. Sutcliffe and N. S. Scrutton, Angew. Chem., Int. Ed., 2008, 47, 537 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2006, 128, 14053 CrossRef CAS PubMed.
- J. Basran, R. J. Harris, M. J. Sutcliffe and N. S. Scrutton, J. Biol. Chem., 2003, 278, 43973 CrossRef CAS PubMed.
- M. J. Sutcliffe and N. S. Scrutton, Eur. J. Biochem., 2002, 269, 3096 CrossRef CAS.
- Y. Fan, A. Cembran, S. Ma and J. Gao, Biochemistry, 2013, 52, 2036 CrossRef CAS PubMed.
- T. H. Rod, J. L. Radkiewicz and C. L. Brooks III, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6980 CrossRef CAS PubMed.
- H. Liu and A. Warshel, Biochemistry, 2007, 46, 6011 CrossRef CAS PubMed.
- K. Arora and C. L. Brooks III, J. Am. Chem. Soc., 2009, 131, 5642 CrossRef CAS PubMed.
- C. R. Pudney, L. O. Johannissen, M. J. Sutcliffe, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2010, 132, 11329 CrossRef CAS PubMed.
- M. J. Knapp, K. Rickert and J. P. Klinman, J. Am. Chem. Soc., 2002, 124, 3865 CrossRef CAS PubMed.
- S. Hu, S. C. Sharma, A. D. Scouras, A. V. Soudackov, C. A. M. Carr, S. Hammes-Schiffer, T. Alber and J. P. Klinman, J. Am. Chem. Soc., 2014, 136, 8157 CrossRef CAS PubMed.
- B. J. Bahnson, T. D. Colby, J. K. Chin, B. M. Goldstein and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12797 CrossRef CAS.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 507 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, C. Levy, J. Pang, M. J. Sutcliffe, D. Leys and N. S. Scrutton, J. Am. Chem. Soc., 2009, 131, 17072 CrossRef CAS PubMed.
- S. Hay, L. O. Johannissen, P. Hothi, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2012, 134, 9749 CrossRef CAS PubMed.
- S. C. L. Kamerlin, J. Mavri and A. Warshel, FEBS Lett., 2010, 584, 2759 CrossRef CAS PubMed.
- R. Walser and W. F. van Gunsteren, Proteins: Struct., Funct., Genet., 2001, 42, 414 CrossRef CAS.
- D. S. Hartsough and K. M. Merz, J. Am. Chem. Soc., 1993, 115, 6529 CrossRef CAS.
- R. Affleck, C. A. Haynes and D. S. Clark, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5167 CrossRef CAS.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047 CrossRef CAS PubMed.
- A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 444 CrossRef CAS.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210 CrossRef CAS PubMed.
- R. G. Silva, A. S. Murkin and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 18661 CrossRef CAS PubMed.
- D. R. Kipp, R. G. Silva and V. L. Schramm, J. Am. Chem. Soc., 2011, 133, 19358 CrossRef CAS PubMed.
- M. D. Toney, J. N. Castro and T. A. Addington, J. Am. Chem. Soc., 2013, 135, 2509 CrossRef CAS PubMed.
- K. Świderek, J. Javier Ruiz-Pernía, V. Moliner and I. Tuñón, Curr. Opin. Chem. Biol., 2014, 21, 11 CrossRef PubMed.
- L. Y. P. Luk, E. J. Loveridge and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 6862 CrossRef CAS PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernía, W. M. Dawson, E. J. Loveridge, I. Tuñón, V. Moliner and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 17317 CrossRef CAS PubMed.
- E. J. Loveridge and R. K. Allemann, Biochemistry, 2010, 49, 5390 CrossRef CAS PubMed.
- R. S. Sikorski, L. Wang, K. A. Markham, P. T. R. Rajagopalan, S. J. Benkovic and A. Kohen, J. Am. Chem. Soc., 2004, 126, 4778 CrossRef CAS PubMed.
- D. Roston, Z. Islam and A. Kohen, Molecules, 2013, 18, 5543 CrossRef CAS PubMed.
- S. Glasstone,K. J. Laidler and H. Eyring, The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena, McGraw-Hill, New York, 1941 Search PubMed.
- J. C. Keck, In Advances in Chemical Physics, John Wiley & Sons, Inc., 2007, p. 85 Search PubMed.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771 CrossRef CAS.
- J. Pu, J. Gao and D. G. Truhlar, Chem. Rev., 2006, 106, 3140 CrossRef CAS PubMed.
- D. R. Glowacki, J. N. Harvey and A. J. Mulholland, Biochem. Soc. Trans., 2012, 40, 515 CrossRef CAS PubMed.
- D. R. Glowacki, J. N. Harvey and A. J. Mulholland, Nat. Chem., 2012, 4, 169 CrossRef CAS PubMed.
- M. Dametto, D. Antoniou and S. D. Schwartz, Mol. Phys., 2012, 110, 531 CrossRef CAS PubMed.
- M. Elias, G. Wieczorek, S. Rosenne and D. S. Tawfik, Trends Biochem. Sci., 2014, 39, 1 CrossRef CAS PubMed.
- E. J. Loveridge, R. J. Rodriguez, R. S. Swanwick and R. K. Allemann, Biochemistry, 2009, 48, 5922 CrossRef CAS PubMed.
- L. Meinhold, D. Clement, M. Tehei, R. Daniel, J. L. Finney and J. C. Smith, Biochem. J., 2008, 94, 4812 CAS.
- O. A. Oyeyemi, K. M. Sours, T. Lee, A. Kohen, K. A. Resing, N. G. Ahn and J. P. Klinman, Biochemistry, 2011, 50, 8251 CrossRef CAS PubMed.
- O. A. Oyeyemi, K. M. Sours, T. Lee, K. A. Resing, N. G. Ahn and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10074 CrossRef CAS PubMed.
- P. Shrimpton and R. K. Allemann, Protein Sci., 2002, 11, 1442 CrossRef CAS PubMed.
- I. V. Khavrutskii, D. J. Price, J. Lee and C. L. Brooks III, Protein Sci., 2007, 16, 1087 CrossRef CAS PubMed.
- C. T. Liu, L. Wang, N. M. Goodey, P. Hanoian and S. J. Benkovic, Biochemistry, 2013, 52, 5332 CrossRef CAS PubMed.
- D. R. Kipp, J. S. Hirschi, A. Wakata, H. Goldstein and V. L. Schramm, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6543 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |