
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nuclear quantum tunnelling in enzymatic reactions – an enzymologist's perspective
Linus O.
Johannissen
,
Sam
Hay
* and
Nigel S.
Scrutton
*
SYNBIOCHEM, Manchester Institute of Biotechnology, Faculty of Life Sciences, The University of Manchester, Manchester M1 7DN, UK. E-mail: sam.hay@manchester.ac.uk; nigel.scrutton@manchester.ac.uk
First published on 27th March 2015
Abstract
Enzyme-catalysed H-transfer reactions are ubiquitous, yet fundamental details of these reactions remain unresolved. In this perspective, we discuss the roles of nuclear quantum tunnelling and (compressive) dynamics during these reactions. Evidence for the coupling of specific substrate and/or protein vibrations to the chemical coordinate is considered and a case is made for the combination of multiple experimental and computational/theoretical approaches when studying these reactions.
1. Introduction
The ability to design and control enzymatic reactions is essential as synthetic biology and industrial biotechnology become increasingly important for the production of sustainable fuels, pharmaceuticals and other chemicals.1–3 A thorough understanding of enzyme catalysis is therefore of paramount importance, yet after over a century of research, since the first enzyme lock-and-key model,4 the importance of certain fundamental principles remains unresolved. While it is well established that electrostatic stabilisation of the transition state is of key importance for rapid biochemical reactions,5–7 the roles of dynamics and nuclear quantum tunnelling (NQT) are more contentious. Since the first experimental evidence of NQT in an enzyme-catalysed reaction8 there has been a plethora of publications arguing both for and against the relative importance of this effect.9–25 Much of the discussion has arisen from the experimental characterisation of enzymatic H-transfer reactions (where H = hydride, hydrogen atom or proton), which exhibit a wide range of (kinetic) isotope effects (KIEs) on observed and/or intrinsic rate constants and steady state velocities, as well as on key thermodynamic parameters such as Arrhenius prefactors (i.e. AH/AD) and activation energies (ΔEa = EaD–EaH). New theory has been developed and adapted in concert with experiment to rationalise these data, and while classical reactions are governed by barrier height, the importance of barrier shape on NQT has become apparent. Consequently, it has been suggested that factors other than transition state stabilisation are important to these reactions, in particular the concept of compressive dynamics – defined below – which has been successful in rationalising a range of temperature- and pressure-dependent KIEs.19,26–30 Here, we will discuss our perspective on the potential role of (compressive) dynamics and NQT on selected enzymatic reactions.2. The tunnelling contribution and rate enhancement
While there is currently no direct experimental measure for the degree of NQT in a chemical reaction, the magnitude of the primary kinetic isotope effect (KIE) is likely to be a reasonable first approximation (e.g.Fig. 1B and C).31 Using computational techniques however, it is fairly straight-forward to calculate a reaction barrier and related parameters such as recrossing coefficients, zero point energies and tunnelling coefficients, which can then be benchmarked against experimental rate constants and KIEs.32–34 For NQT reactions, methods of note include ensemble-averaged variational transition state theory with multidimensional tunnelling corrections (EA-VTST/MT),11,35 the quantum classical path (QCP) method36,37 and the molecular dynamics with quantum transitions (MDQT) method.12 | ||
| Fig. 1 Effect of barrier shape on H-transfer in AADH with tryptamine; barriers were calculated for structures taken from QM/MM MD simulations. (A) Typical adiabatic potential energy barrier (solid line) and transfer probability, Q(V)Ptun(V) from eqn (3) for H (dashed line) and D (dotted line). The RTE is the representative tunnelling energy where the transfer probability is maximised and rtun is the tunnelling distance at this energy; (B, D) effect of tunnelling distance (rtun) and (C, E) effect of barrier height (V0). κ (for H; blue circles) and the KIE (red circles) are shown in panels B and C. kTST (black circles), ktun (open circles) and kobs (crosses) are shown in panels D and E (Adapted from ref. 31). | ||
Within the framework of semiclassical transition state theory (TST; eqn (1)), the rate of reaction depends exponentially on the energy difference between reactant and transition state,  , the barrier height, and depends linearly on the pre-exponential frequency term, A, which encompasses terms such as recrossing and the transmission coefficient.38
, the barrier height, and depends linearly on the pre-exponential frequency term, A, which encompasses terms such as recrossing and the transmission coefficient.38
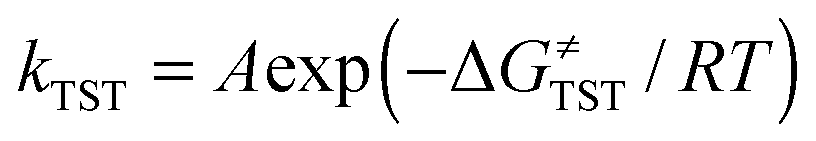 | (1) |
 : there is always a certain probability of NQT occurring through the classical barrier, particularly for transfer of a light particle such as the H nucleus whose de Broglie wavelength (λ = h/mν) is on the same order as a bond length. According to the Wentzel–Kramers–Brillouin (WKB) approximation, the probability of NQT at any point q along the reaction coordinate is of the form:31,39
: there is always a certain probability of NQT occurring through the classical barrier, particularly for transfer of a light particle such as the H nucleus whose de Broglie wavelength (λ = h/mν) is on the same order as a bond length. According to the Wentzel–Kramers–Brillouin (WKB) approximation, the probability of NQT at any point q along the reaction coordinate is of the form:31,39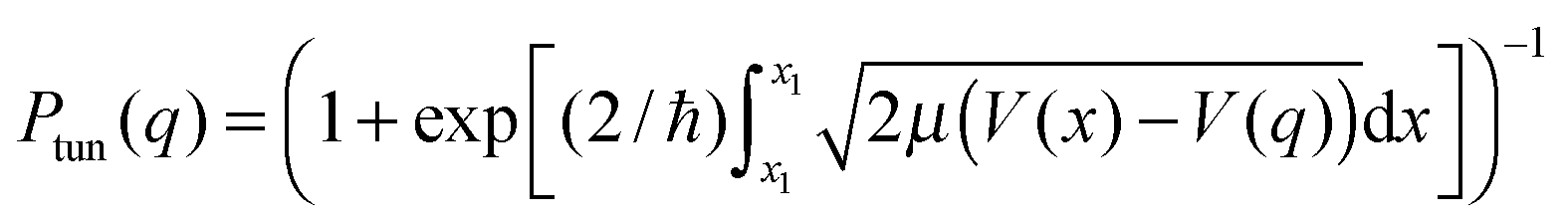 | (2) |
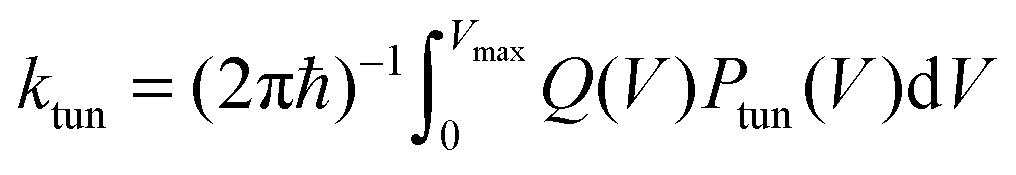 | (3) |
| κ = 1 + ktun/kTST | (4) |
In any case, evolutionary pressure does not operate on the degree of NQT but on absolute rates. It is often pointed out that NQT contributes at most 3 orders of magnitude to the rate enhancement,15 while 10–12 orders magnitude are achieved by lowering the free energy of activation.15 However, a rate enhancement of even one order of magnitude can be vitally important for cellular metabolism; e.g. if the chemical step is rate limiting, and the enzyme contributes to the flux through a metabolic pathway, then any rate enhancement will directly influence the rate of metabolism and thus cell viability. Given the ubiquitous nature of H-transfers in biology, any metabolic pathway will involve multiple H-transfer steps and thus NQT could in principle have a major effect on metabolic flux as these rate enhancements will become multiplicative. The importance of NQT in biology is therefore not limited to its contribution to the “catalytic effect” (see next Section) but its contribution to metabolic rates essential for life.41 Furthermore, the tunnelling contribution κ does not reflect the absolute amount of NQT, i.e. the rate at which the transferring nucleus tunnels from the reactant to product well, but the rate relative to that of classical over-barrier transfer. This is an important distinction, as only looking at κ can lead to the interpretation that NQT is in fact anticatalytic25 – after all, the rate enhancement due to NQT is typically smaller for a lower barrier.24,25,31,42 However, as is evident from eqn (2), the absolute tunnelling probability and hence ktun increases with a decrease in barrier height/width. This decreases the relative NQT contribution, since kTST increases more drastically, as illustrated in Fig. 1, which shows how ktun and kTST change as a function of barrier height as the distance between donor and acceptor atoms is altered for a model reaction.
3. The catalytic effect and evolution
One of the key arguments against the importance of enzymatic NQT is that the KIE and tunnelling contribution for enzymatic reactions are similar to the equivalent “reference” reaction in solution. There are a limited number of examples where experimental data is available for both reactions,43–45 as comparable uncatalysed reactions typically do not occur on tractable timescales. However, this is also the conclusion from most computational comparisons, where the reference reaction in water is also analysed.45–47 In the case of nitroalkane oxidase, where experimental evidence does exist,23 the NQT contribution increases 3-fold in the enzyme-catalysed reaction, although the NQT contribution in the enzyme is still relatively small (κ = 3.5). Therefore, while tunnelling can enhance the rate of enzymatic reactions relative to a classical over-barrier reaction, these data suggest this may not be a significant part of the catalytic effect, i.e. the rate enhancement over the uncatalysed reaction.These data raise another interesting question: why are the enzymatic KIEs and NQT contribution not significantly smaller? Given that enzymes considerably lower the barrier height, one would expect the enzyme-catalysed reactions to have significantly smaller NQT contributions. Therefore, it is entirely possible that while enzymes may not have evolved to increase NQT contributions relative to the uncatalysed reaction, evolution has favoured H-transfer reactions that, at least, maintain the same relative NQT contribution (κ). Furthermore, comparisons to the uncatalysed reaction are not necessarily useful for understanding the evolution of enzymes, as organisms did not start out utilising the uncatalysed reactions – these often have half-times approaching the age of the earth48 – so much of the chemistry that enzymes carry out today could not have been part of the metabolism of early life. From an enzymologist's perspective, it is therefore more interesting to ask how NQT contributions have changed during the course of evolution. This question could be addressed within the growing field of paleoenzymology,49–52 by comparing NQT in modern enzymes to those from less-evolved or extinct organisms, using e.g. ancestral reconstruction. When discussing evolutionary pressure, it is also important to consider the conditions under which organisms evolved. For example mammalian enzymes evolved under conditions of tight thermal regulation while many other organisms did not, and thus experience relatively larger temperature fluctuations. Further, one might expect thermophilic enzymes to have larger NQT contributions than meso- or psychrophilic enzymes, as thermophilic enzymes typically have larger barriers,53,54 which are likely to have relatively larger associated κ values. In the case of hydride transfer in DHFR, however, the mesophilic (EcDHFR),55 thermophilic (BsDHFR)56 and hyperthermophilic (TmDHFR)14 enzymes have near identical KIEs despite the wide range in activation energies (Ea(H) = 13, 23 & 50 kJ mol−1, respectively).†
4. Promoting vibrations and barrier compression
Atomistic understanding of the temperature-dependence of KIEs remains an important challenge in enzymology, and it is difficult to account for both temperature independent and strongly temperature dependent KIEs using semiclassical TST. The wide range in Arrhenius prefactor ratios observed in many cases18,57,58 cannot also be accounted for within the Bell correction model,59 which incorporates a relatively small NQT correction within a TST formalism. However, these data are readily accommodated within non-adiabatic, vibronic formalisms,13,14,42,57,58,60–67 which were initially introduced to deal with the strongly quantised (ℏω ≫ kBT) C–H bonds broken in many enzymatic H-transfers.68 Within such models, the NQT probability depends on the nuclear wavefunction overlap, 〈ϕν|ϕμ〉, between the reactant and product states, e.g.:58,68,69 | (5) |
Vibronic models rely on the non-adiabatic assumption of weak coupling between reactant and product states. However, it should be pointed out that this breaks down for very short transfer distances (near the top of the barrier from a TST correction perspective), so we tend to use these models phenomenologically to rationalise differences between very similar enzymatic reactions, e.g. in a range of enzyme variants64 or as a function of hydrostatic pressure.77,78 Nevertheless, it has been shown that a vibronic treatment gives the same general trend as the quantum classical path (QCP)36,37 method for H-transfer in dihydrofolate reductase (DHFR),42 and we have shown that our vibronic model performs well for proton transfer in aromatic amine dehydrogenase (AADH) when compared to the WKB model.31 This is not surprising, given the similarities between eqn (3) and (5): in both cases a classical activation term represents the Boltzmann probability of achieving a conformation at which tunnelling can occur, at which point the tunnelling probability depends on the width of the barrier at that conformation.
The concept of promoting vibrations remains highly contentious and, for example, it has been argued that catalytically important vibrational modes are the same in solution as in an enzyme.20 While this may be the case, a fundamental differences between enzymatic and solution chemistry is that enzyme catalysis typically involves a high degree of preorganisation, which minimises the reorganisation energy for individual chemical steps as well as the atomic fluctuations necessary for the transition between consecutive steps.7,79 The latter was elegantly illustrated in a recent computational study of serine esterases and idealised forms of their transition states,80 which found that the active site geometry is optimised to minimise the degree of rearrangement required between consecutive steps, and not necessarily that which is most optimal for a given chemical step. Enzymes therefore impose significant restrictions along most degrees of freedom; at the same time, however, effective barrier crossing requires facile motion along the H-coordinate. Molecules in solution can explore conformational space until relevant vibrational motion can carry the system from reactant to transition state (or TRC), while the enzyme-substrate complex is conformationally restricted. Efficient use of specific compressive modes/promoting vibrations might represent the best strategy for ensuring that dynamical excursions from the ground state are as productive as possible given the high degree of restriction. The nature of the dynamic coupling to the H-coordinate may involve utilising vibrational modes of active site residues,72,73 restraining the substrate in a conformation which favours specific modes,81 or simply binding the substrates in an orientation where inherent normal modes coincide with the H-coordinate (Fig. 2).77 For example, the conformation of the substrate in aromatic amine dehydrogenase was found to be crucial for the effectiveness of a promoting vibration: in a variant form of the enzyme, the alternative substrate conformation was such that the promoting vibration was occluded (Fig. 3), leading to an increase in barrier height and a 3-orders-of-magnitude decrease in the observed rate constant.81 If both catalysed and uncatalysed reactions utilise similar vibrational modes, the difference being that these are more likely to be productive in the enzyme, then the rate enhancement solely arises from preorganisation.6 Nevertheless, this does not diminish the importance of promoting vibrations to the enzymatic reaction or their effect on observable activation parameters.
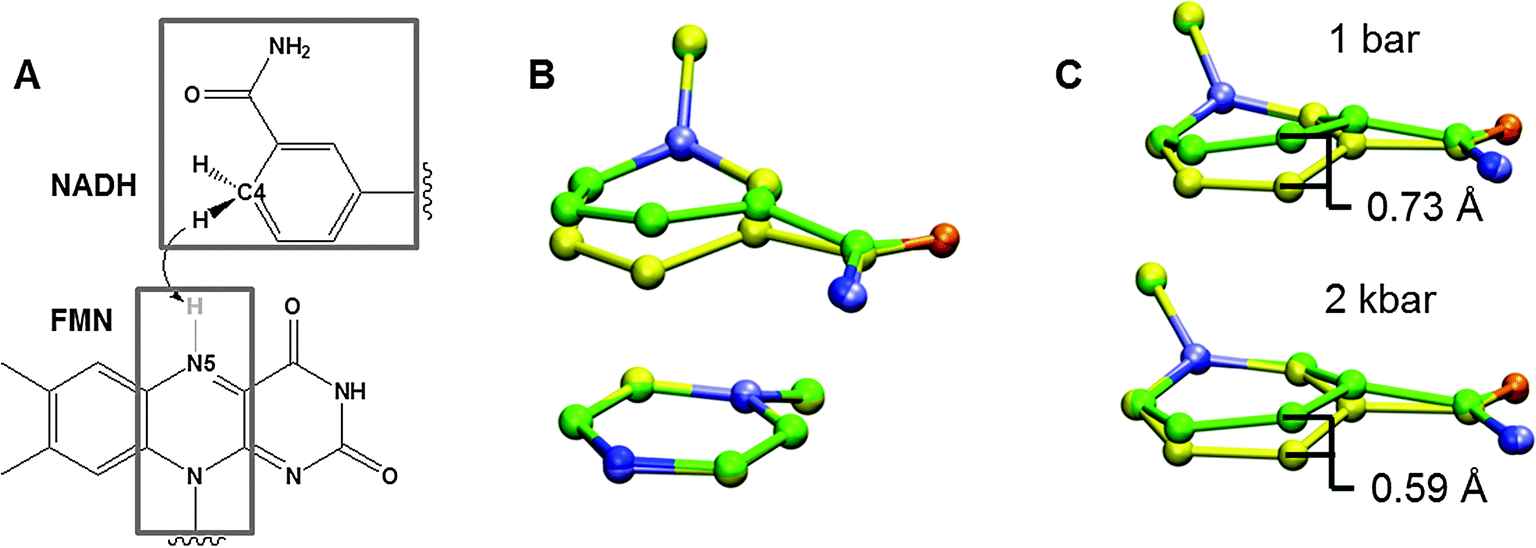 | ||
| Fig. 2 (A) Hydride transfer from NADH to FMN in Morphinone Reductase. (B) Promoting vibration illustrated for the heavy atoms in the grey boxes in (A), identified from constant pressure MD simulations and digital filtering by frequency deconvolution.106,107 The overlaid structures correspond to the average structures at the minimum and maximum displacements. (C) Effect of hydrostatic pressure on the degree of done-acceptor compression (Adapted from ref. 77). | ||
 | ||
| Fig. 3 Substrate conformations in AADH with bound tryptamine. (A) The promoting vibration illustrated by the curved arrow moves the system towards the representative structure for tunnelling (RTE structure, yellow carbon atoms) in the native enzyme, while (B) the same O–H compression in the isolated small subunit requires a significant degree of C–H stretching. (C, D) Representative adiabatic potential energy scans, spanning the full range of observed activation energies, with the reaction coordinate z = d(C–H)–d(O–H) for (C) the native enzyme and (D) the isolated small subunit. The stiffer C–H stretching in the small subunit leads to a narrower reactant well, which allows less fluctuation along the reaction coordinate (Adapted from ref. 81). | ||
In a recent attempt to explain the temperature-dependence of the KIE without invoking promoting vibrations, an alternative framework invoking conformational complexity was proposed where the KIE depends on the temperature-dependent equilibrium between multiple reactive conformations, each leading to a different KIE.82 Of course, conformational complexity does occur and can affect the observed KIE.83,84 However, while this model has successfully been used to fit the temperature-dependence of the KIE on several enzymatic H-transfers, it predicts that the magnitude of the KIE can either increase or decrease with temperature, depending on the relative free energy of each conformer. In enzymatic H-transfers, the magnitude of significantly temperature-dependent KIEs is always observed to decrease with increasing temperature (i.e. we are not aware of any enzymatic KIEs that significantly decrease with increasing temperature), a trend which does not arise within this model. On the other hand, it is well known that rapid D–A fluctuations modulate the barrier height such that the KIE will always decrease as distance sampling increases with increasing temperature.42,77,85
5. Experimental evidence for NQT and promoting vibrations?
There is currently no direct experimental evidence that the temperature-dependence of the KIE arises from vibrational modes coupled to NQT and this remains a major challenge in the field. However, there is compelling evidence from combined experimental and computational studies that this may be the case. Typically these focus on the temperature-dependence of the KIE (see above), but we have also looked at the effect of hydrostatic pressure.27,77,78,84,86,87 In the absence of NQT, KIEs arise from differences in zero-point energy and thus the stretching frequency of the transferred 1H, 2D or 3T, which are typically taken to be invariant to changes in pressure over the experimental range.88,89 Consequently, pressure-dependent KIEs have been used as evidence for the involvement of NQT.90,91 Within the vibronic framework (eqn (5)), pressure can perturb the KIE on the reaction by changing the equilibrium H-transfer distance and/or the frequency of (promoting) vibrational modes as other changes are likely to cancel in the KIE calculation.27 These perturbations can be modelled using MD simulations, and may be monitored experimentally in rare cases. In the case of hydride transfer in Morphinone Reductase (MR), MD simulations predicted a decrease in D–A distance with increasing pressure,77,92 which was confirmed experimentally through the observation of an increased strength in the charge-transfer (CT) complex between the donor and acceptor groups (nicotinamide of NADH and isoalloxazine of FMN, respectively).92 Experiments revealed that pressure causes an increase in the magnitude of the KIE with a concomitant increase in the observed rate of H-transfer, which can be rationalised if the decrease in D–A distance is accompanied by an increase in the frequency of the promoting vibration:29 although the average D–A distance decreases, the shorter, more productive pathways with higher tunnelling probabilities become more difficult to attain (Fig. 2C). The increase in promoting vibration force constant was confirmed through MD simulations both by the narrower distribution of D–A distances at high pressure92 and by spectral density72,93 analysis which revealed an increase in the putative promoting vibration frequency with pressure.77Pressure acts to anisotropically perturb enzyme structure94 and no simple trends have emerged between the pressure- and temperature-dependence of KIEs.95 Pressure experiments can nevertheless be highly informative on a case-by-case basis, and while phenomenological models have been derived for these reactions,27,90,91 structural data and/or molecular dynamics simulations are essential to understand the atomistic origin of observed effects. For example, proton transfer in AADH with phenylethylamine as substrate exhibits complex pressure-dependent H-transfer kinetics, which we were able to explain within our vibronic formulism after analysis of MD simulations. In contrast to MR, these revealed no change in equilibrium D–A distance, which is readily rationalised given that one key effect of hydrostatic pressure on proteins is that additional water molecules are forced into cavities96 and the cofactor in MR is much more solvent exposed than the substrate in AADH. On the other hand, interpreting pressure data for light-driven NQT in protochlorophyllide oxidoreductase (POR)95 or proton transfer during ascorbate oxidation by ferricyanide97 has proven difficult in the absence of structural data and MD simulations.
In recent years “heavy-protein” experiments have become popular for studying the link between dynamics and NQT: if dynamics are important, then isotopic substitution with 13C, 15N and 2H should perturb these dynamics and cause observable perturbations to H-transfer. We have performed experiments on the MR homologue pentaerythritol tetranitrate reductase (PETNR) where the temperature dependence of the KIE is much larger when H-transfer occurs from NADPH than from NADH.60 In triple-labelled “heavy” PETNR, the temperature dependence of the KIE on both reactions increased,98 consistent with an underlying promoting vibration involved in the NADH reaction that does not give rise to a strongly temperature-dependent KIE.62 Similar experiments have been carried out on other enzymes,99–103 and the rate of H-transfer tends to decrease in the heavy enzyme (though not always101). This can be attributed to a number of effects,104 including a small change in electrostatics due to slight difference in bond lengths.105 In the case of DHFR, EA-VTST calculations have suggested that the decrease in rate of the heavy protein is due to a decrease in the recrossing coefficient arising from an increase in coupling between protein motion and the reaction coordinate.100
6. Conclusions and future outlook
Since the majority of enzyme reactions involve at least one H-transfer step, which will involve some degree of NQT, it is essential to understand the factors that govern enzymatic NQT. While significant progress has been made in this field since NQT was first reported in an enzyme over 25 years ago,8 questions remain unanswered and controversies persist. Experimental probes for the involvement of NQT include deviations from classical behaviour for the temperature- and pressure-dependence of the primary KIE. It is more challenging to directly measure the role of dynamics in NQT reactions, although much insight has been gained from combined pressure and temperature-dependence measurements coupled with MD simulations and modelling. Perhaps the most promising new development in this field is the advent of “heavy-protein” experiments, which allows a direct and titratable perturbation of protein dynamics. From our perspective, it is clear that rapid compressive dynamics can play an important role in facilitating enzymatic NQT reactions, although the precise nature and ubiquity of such modes is not established: to what extent is the protein involved and how has evolution directed/selected these modes?Acknowledgements
This is a contribution from the BBSRC/EPSRC Centre for Synthetic Biology of Fine and Speciality Chemicals (SYNBIOCHEM). SH is a BBSRC David Phillips Research Fellow. NSS is an EPSRC Established Career Fellow.References
- M. T. Smith, K. M. Wilding, J. M. Hunt, A. M. Bennett and B. C. Bundy, FEBS Lett., 2014, 588, 2755–2761 CrossRef CAS PubMed.
- A. V. Bryksin, A. C. Brown, M. M. Baksh, M. G. Finn and T. H. Barker, Acta Biomater., 2014, 10, 1761–1769 CrossRef CAS PubMed.
- V. Singh, Gene, 2014, 535, 1–11 CrossRef CAS PubMed.
- E. Fischer, Ber. Dtsch. Chem. Ges., 1894, 27, 2985–2993 CrossRef CAS PubMed.
- Transition States of Biochemical Processes, ed. R. D. Gandour and R. L. Schowen, Plenum Press, New York, 1978 Search PubMed.
- A. Warshel, J. Biol. Chem., 1998, 273, 27035–27038 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- Y. Cha, C. J. Murray and J. P. Klinman, Science, 1989, 243, 1325–1330 CAS.
- B. J. Bahnson, T. D. Colby, J. K. Chin, B. M. Goldstein and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12797–12802 CrossRef CAS.
- J. Basran, M. J. Sutcliffe and N. S. Scrutton, Biochemistry, 1999, 38, 3218–3222 CrossRef CAS PubMed.
- C. Alhambra, J. Corchado, M. L. Snchez, M. Garcia-Viloca, J. Gao and D. G. Truhlar, J. Phys. Chem. B, 2001, 105, 11326–11340 CrossRef CAS.
- S. R. Billeter, S. P. Webb, P. K. Agarwal, T. Iordanov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2001, 123, 11262–11272 CrossRef CAS PubMed.
- M. J. Knapp and J. P. Klinman, Eur. J. Biochem., 2002, 269, 3113–3121 CrossRef CAS.
- G. Maglia and R. K. Allemann, J. Am. Chem. Soc., 2003, 125, 13372–13373 CrossRef CAS PubMed.
- M. Garcia-Viloca, J. Gao, M. Karplus and D. G. Truhlar, Science, 2004, 303, 186–195 CrossRef CAS PubMed.
- Z. X. Liang and J. P. Klinman, Curr. Opin. Struct. Biol., 2004, 14, 648–655 CrossRef CAS PubMed.
- Z. X. Liang, T. Lee, K. A. Resing, N. G. Ahn and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9556–9561 CrossRef CAS PubMed.
- L. Masgrau, A. Roujeinikova, L. O. Johannissen, P. Hothi, J. Basran, K. E. Ranaghan, A. J. Mulholland, M. J. Sutcliffe, N. S. Scrutton and D. Leys, Science, 2006, 312, 237–241 CrossRef CAS PubMed.
- Z. D. Nagel and J. P. Klinman, Chem. Rev., 2006, 106, 3095–3118 CrossRef CAS PubMed.
- M. H. Olsson, W. W. Parson and A. Warshel, Chem. Rev., 2006, 106, 1737–1756 CrossRef CAS PubMed.
- J. Z. Pu, J. L. Gao and D. G. Truhlar, Chem. Rev., 2006, 106, 3140–3169 CrossRef CAS PubMed.
- S. Hay, C. R. Pudney and N. S. Scrutton, FEBS J., 2009, 276, 3930–3941 CrossRef CAS PubMed.
- D. T. Major, A. Heroux, A. M. Orville, M. P. Valley, P. F. Fitzpatrick and J. L. Gao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20734–20739 CrossRef CAS PubMed.
- S. C. L. Kamerlin, J. Mavri and A. Warshel, FEBS Lett., 2010, 584, 2759–2766 CrossRef CAS PubMed.
- S. C. L. Kamerlin and A. Warshel, J. Phys. Org. Chem., 2010, 23, 677–684 CrossRef CAS PubMed.
- S. Hay and N. S. Scrutton, Nat. Chem., 2012, 4, 161–168 CrossRef CAS PubMed.
- S. Hay and N. S. Scrutton, Biochemistry, 2008, 47, 9880–9887 CrossRef CAS PubMed.
- S. Hay, C. Pudney, P. Hothi, L. O. Johannissen, L. Masgrau, J. Pang, D. Leys, M. J. Sutcliffe and N. S. Scrutton, Biochem. Soc. Trans., 2008, 36, 16–21 CrossRef CAS PubMed.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 507–512 CrossRef CAS PubMed.
- A. Kohen, Prog. React. Kinet. Mech., 2003, 28, 119–156 CrossRef CAS.
- S. Hay, L. O. Johannissen, M. J. Sutcliffe and N. S. Scrutton, Biophys. J., 2010, 98, 121–128 CrossRef CAS PubMed.
- J. Gao and D. G. Truhlar, Annu. Rev. Phys. Chem., 2002, 53, 467–505 CrossRef CAS PubMed.
- Chemical Modelling: Applications and Theory, ed. M. Springborg, 2006, vol. 4, pp. 1–528 Search PubMed.
- M. W. van der Kamp, K. E. Shaw, C. J. Woods and A. J. Mulholland, J. R. Soc., Interface, 2008, 5, S173–S190 CrossRef CAS PubMed.
- D. G. Truhlar, J. L. Gao, M. Garcia-Viloca, C. Alhambra, J. Corchado, M. L. Sanchez and T. D. Poulsen, Int. J. Quantum Chem., 2004, 100, 1136–1152 CrossRef CAS PubMed.
- J. K. Hwang and A. Warshel, J. Phys. Chem., 1993, 97, 10053–10058 CrossRef CAS.
- J. Mavri, H. Liu, M. H. Olsson and A. Warshel, J. Phys. Chem. B, 2008, 112, 5950–5954 CrossRef CAS PubMed.
- M. Garcia-Viloca, D. G. Truhlar and J. Gao, Biochemistry, 2003, 42, 13558–13575 CrossRef CAS PubMed.
- W. H. Miller, Science, 1986, 233, 171–177 CAS.
- P. Pechukas, Annu. Rev. Phys. Chem., 1981, 32, 159–177 CrossRef CAS.
- S. Hay and N. S. Scrutton, 22nd Solvay Conference on Chemistry: Quantum Effects in Chemistry and Biology, 2011, vol. 3 Search PubMed.
- H. Liu and A. Warshel, J. Phys. Chem. B, 2007, 111, 7852–7861 CrossRef CAS PubMed.
- K. M. Doll, B. R. Bender and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10877–10884 CrossRef CAS PubMed.
- K. M. Doll and R. G. Finke, Inorg. Chem., 2003, 42, 4849–4856 CrossRef CAS PubMed.
- I. Feierberg, V. Luzhkov and J. Aqvist, J. Biol. Chem., 2000, 275, 22657–22662 CrossRef CAS PubMed.
- I. H. Williams, J. Phys. Org. Chem., 2010, 23, 685–689 CrossRef CAS PubMed.
- M. H. M. Olsson, J. Mavri and A. Warshel, Philos. Trans. R. Soc., B, 2006, 361, 1417–1432 CrossRef CAS PubMed.
- R. Wolfenden and M. J. Snider, Acc. Chem. Res., 2001, 34, 938–945 CrossRef CAS PubMed.
- S. A. Benner, S. O. Sassi and E. A. Gaucher, Adv. Enzymol. Relat. Areas Mol. Biol., 2007, 75, 1–132 CAS , xi.
- B. G. Hall, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5431–5436 CrossRef CAS PubMed.
- R. Perez-Jimenez, A. Ingles-Prieto, Z. M. Zhao, I. Sanchez-Romero, J. Alegre-Cebollada, P. Kosuri, S. Garcia-Manyes, T. J. Kappock, M. Tanokura, A. Holmgren, J. M. Sanchez-Ruiz, E. A. Gaucher and J. M. Fernandez, Nat. Struct. Mol. Biol., 2011, 18, 592–596 CAS.
- J. W. Thornton, Nat. Rev. Genet., 2004, 5, 366–375 CrossRef CAS PubMed.
- G. Feller and C. Gerday, Nat. Rev. Microbiol., 2003, 1, 200–208 CrossRef CAS PubMed.
- K. S. Siddiqui and R. Cavicchioli, Annu. Rev. Biochem., 2006, 75, 403–433 CrossRef CAS PubMed.
- R. S. Sikorski, L. Wang, K. A. Markham, P. T. R. Rajagopalan, S. J. Benkovic and A. Kohen, J. Am. Chem. Soc., 2004, 126, 4778–4779 CrossRef CAS PubMed.
- H. S. Kim, S. M. Damo, S. Y. Lee, D. Wemmer and J. P. Klinman, Biochemistry, 2005, 44, 11428–11439 CrossRef CAS PubMed.
- A. Kohen, R. Cannio, S. Bartolucci and J. P. Klinman, Nature, 1999, 399, 496–499 CrossRef CAS PubMed.
- M. J. Knapp, K. Rickert and J. P. Klinman, J. Am. Chem. Soc., 2002, 124, 3865–3874 CrossRef CAS PubMed.
- R. P. Bell, The Tunnel Effect in Chemistry, Chapman and Hall, London, 1980, pp. 51–140 Search PubMed.
- J. Basran, R. J. Harris, M. J. Sutcliffe and N. S. Scrutton, J. Biol. Chem., 2003, 278, 43973–43982 CrossRef CAS PubMed.
- E. Hatcher, A. V. Soudackov and S. Hammes-Schiffer, J. Am. Chem. Soc., 2007, 129, 187–196 CrossRef CAS PubMed.
- L. O. Johannissen, S. Hay, N. S. Scrutton and M. J. Sutcliffe, J. Phys. Chem. B, 2007, 111, 2631–2638 CrossRef CAS PubMed.
- V. Stojkovic, L. L. Perissinotti, D. Willmer, S. J. Benkovic and A. Kohen, J. Am. Chem. Soc., 2012, 134, 1738–1745 CrossRef CAS PubMed.
- C. R. Pudney, L. O. Johannissen, M. J. Sutcliffe, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2010, 132, 11329–11335 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, C. Levy, J. Pang, M. J. Sutcliffe, D. Leys and N. S. Scrutton, J. Am. Chem. Soc., 2009, 131, 17072–17073 CrossRef CAS PubMed.
- M. P. Meyer, D. R. Tomchick and J. P. Klinman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1146–1151 CrossRef CAS PubMed.
- D. J. Heyes, M. Sakuma, S. P. de Visser and N. S. Scrutton, J. Biol. Chem., 2009, 284, 3762–3767 CrossRef CAS PubMed.
- A. M. Kuznetsov and J. Ulstrup, Can. J. Chem., 1999, 77, 1085–1096 CAS.
- L. O. Johannissen, T. Irebo, M. Sjöin, O. Johansson and L. Hammarström, J. Phys. Chem. B, 2009, 113, 16214–16225 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2006, 128, 14053–14058 CrossRef CAS PubMed.
- D. Antoniou and S. D. Schwartz, J. Phys. Chem. B, 2001, 105, 5553–5558 CrossRef CAS.
- S. Caratzoulas, J. S. Mincer and S. D. Schwartz, J. Am. Chem. Soc., 2002, 124, 3270–3276 CrossRef CAS PubMed.
- S. Nunez, D. Antoniou, V. L. Schramm and S. D. Schwartz, J. Am. Chem. Soc., 2004, 126, 15720–15729 CrossRef CAS PubMed.
- S. L. Quaytman and S. D. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12253–12258 CrossRef CAS PubMed.
- S. D. Schwartz and V. L. Schramm, Nat. Chem. Biol., 2009, 5, 551–558 CrossRef CAS PubMed.
- S. Hay, C. R. Pudney, T. A. McGrory, J. Y. Pang, M. J. Sutcliffe and N. S. Scrutton, Angew. Chem., Int. Ed., 2009, 48, 1452–1454 CrossRef CAS PubMed.
- L. O. Johannissen, N. S. Scrutton and M. J. Sutcliffe, Angew. Chem., Int. Ed., 2011, 50, 2129–2132 CrossRef CAS PubMed.
- S. Hay, M. J. Sutcliffe and N. S. Scrutton, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 507–512 CrossRef CAS PubMed.
- A. J. Adamczyk, J. Cao, S. C. L. Kamerlin and A. Warshel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14115–14120 CrossRef CAS PubMed.
- A. J. T. Smith, R. Muller, M. D. Toscano, P. Kast, H. W. Hellinga, D. Hilvert and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 15361–15373 CrossRef CAS PubMed.
- L. O. Johannissen, N. S. Scrutton and M. J. Sutcliffe, J. R. Soc., Interface, 2008, 5(suppl 3), S225–S232 CrossRef CAS PubMed.
- D. R. Glowacki, J. N. Harvey and A. J. Mulholland, Nat. Chem., 2012, 4, 169–176 CrossRef CAS PubMed.
- C. R. Pudney, S. Hay, J. Pang, C. Costello, D. Leys, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2007, 129, 13949–13956 CrossRef CAS PubMed.
- C. R. Pudney, T. McGrory, P. Lafite, J. Y. Pang, S. Hay, D. Leys, M. J. Sutcliffe and N. S. Scrutton, ChemBioChem, 2009, 10, 1379–1384 CrossRef CAS PubMed.
- D. Antoniou and S. D. Schwartz, J. Chem. Phys., 1998, 108, 3620–3625 CrossRef CAS PubMed.
- S. Hay, L. O. Johannissen, P. Hothi, M. J. Sutcliffe and N. S. Scrutton, J. Am. Chem. Soc., 2012, 134, 9749–9754 CrossRef CAS PubMed.
- S. Hay, C. R. Pudney, M. J. Sutcliffe and N. S. Scrutton, J. Phys. Org. Chem., 2010, 23, 696–701 CrossRef CAS PubMed.
- N. S. Isaacs, in Isotope Effects in Organic Chemistry, ed. E. Buncel and C. C. Lee, Elsevier, London, 1984, vol. 6, pp. 67–105 Search PubMed.
- D. B. Northrop, Philos. Trans. R. Soc., B, 2006, 361, 1341–1349 CrossRef CAS PubMed.
- N. S. Isaacs, K. Javaid and E. Rannala, Nature, 1977, 268, 372 CrossRef CAS PubMed.
- D. B. Northrop, J. Am. Chem. Soc., 1999, 121, 3521–3524 CrossRef CAS.
- S. Hay, C. R. Pudney, T. A. McGrory, J. Pang, M. J. Sutcliffe and N. S. Scrutton, Angew. Chem., Int. Ed., 2009, 48, 1452–1454 CrossRef CAS PubMed.
- S. Caratzoulas and S. D. Schwartz, J. Chem. Phys., 2001, 114, 2910–2918 CrossRef CAS PubMed.
- D. A. Ledward, High Pressure Res., 2000, 19, 391–400 CrossRef PubMed.
- R. Hoeven, D. J. Heyes, S. Hay and N. S. Scrutton, FEBS J., 2015 DOI:10.1111/febs.13193.
- J. Roche, J. A. Caro, D. R. Norberto, P. Barthe, C. Roumestand, J. L. Schlessman, A. E. Garcia, B. Garcia-Moreno and C. A. Royer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6945–6950 CrossRef CAS PubMed.
- S. M. Kandathil, M. D. Driscoll, R. V. Dunn, N. S. Scrutton and S. Hay, Phys. Chem. Chem. Phys., 2014, 16, 2256–2259 RSC.
- C. R. Pudney, A. Guerriero, N. J. Baxter, L. O. Johannissen, J. P. Waltho, S. Hay and N. S. Scrutton, J. Am. Chem. Soc., 2013, 135, 2512–2517 CrossRef CAS PubMed.
- D. Antoniou, X. X. Ge, V. L. Schramm and S. D. Schwartz, J. Phys. Chem. Lett., 2012, 3, 3538–3544 CrossRef CAS PubMed.
- L. Y. P. Luk, J. J. Ruiz-Pernia, W. M. Dawson, M. Roca, E. J. Loveridge, D. R. Glowacki, J. N. Harvey, A. J. Mulholland, I. Tunon, V. Moliner and R. K. Allemann, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16344–16349 CrossRef CAS PubMed.
- L. Y. Luk, E. J. Loveridge and R. K. Allemann, J. Am. Chem. Soc., 2014, 136, 6862–6865 CrossRef CAS PubMed.
- J. E. Masterson and S. D. Schwartz, J. Phys. Chem. A, 2013, 117, 7107–7113 CrossRef CAS PubMed.
- Z. Wang, P. Singh, C. M. Czekster, A. Kohen and V. L. Schramm, J. Am. Chem. Soc., 2014, 136, 8333–8341 CrossRef CAS PubMed.
- K. Swiderek, J. J. Ruiz-Pernia, V. Moliner and I. Tunon, Curr. Opin. Chem. Biol., 2014, 21, 11–18 CrossRef CAS PubMed.
- A. Kohen, Acc. Chem. Res., 2015 DOI:10.1021/ar5003225.
- R. B. Sessions, P. Dauber-Osguthorpe and D. J. Osguthorpe, J. Mol. Biol., 1989, 210, 617–633 CrossRef CAS.
- L. O. Johannissen, S. Hay, N. S. Scrutton and M. J. Sutcliffe, J. Phys. Chem. B, 2007, 111, 2631–2638 CrossRef CAS PubMed.
Footnote |
| † Note that these KIEs were not reported at the enzymes' optimal temperatures, but at 25, 25, & 40 °C, respectively; however, accounting for this would give the EcDHFR the largest KIE (EcDHFR has the least temperature-dependent KIE while the KIE of the other two decrease with temperature). |
| This journal is © the Owner Societies 2015 |