
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix site effects on vibrational frequencies of HXeCCH, HXeBr, and HXeI: a hybrid quantum-classical simulation
Keisuke
Niimi
a,
Tetsuya
Taketsugu
b and
Akira
Nakayama
*c
aGraduate School of Chemical Sciences and Engineering, Hokkaido University, Sapporo 060-0810, Japan
bDepartment of Chemistry, Faculty of Science, Hokkaido University, Sapporo 060-0810, Japan
cCatalysis Research Center, Hokkaido University, Sapporo 001-0021, Japan. E-mail: nakayama@cat.hokudai.ac.jp
First published on 17th February 2015
Abstract
The matrix shifts of the H–Xe stretching frequency of noble-gas hydrides, HXeCCH, HXeBr, and HXeI in various noble-gas matrices (in Ne, Ar, Kr, and Xe matrices) are investigated via the hybrid quantum-classical simulations. The order of the H–Xe stretching frequencies is found to be ν(gas) < ν(Ne) < ν(Xe) < ν(Kr) < ν(Ar) for HXeCCH and HXeBr, while it is ν(gas) < ν(Ne) < ν(Xe) < ν(Ar) < ν(Kr) for HXeI. This order is anomalous with respect to the matrix dielectric constants, and the calculated results reproduce the experimentally observed shifts quite successfully. We also find that the matrix shifts from the gas-phase values are Δν(HXeCCH) ≈ Δν(HXeCl) < Δν(HXeBr) < Δν(HXeI) in the same noble-gas matrix environments, which implies that the weakly bound molecules exhibit large matrix shifts. The local trapping site is analyzed in detail, and it is shown that a realistic modeling of the surrounding matrix environments is essential to describe the unusual matrix shifts accurately.
I. Introduction
The last two decades have witnessed a rebirth of interest in the field of noble-gas chemistry mainly due to the discovery of noble-gas hydrides.1–7 These compounds have the structural formula of HNgY, where Ng is a noble gas atom and Y is an electronegative fragment. The UV photolysis of the HY precursors in the noble-gas matrix produces these HNgY compounds, and a unique electronic character and also the formation process of the HNgY compounds has been the focus of intensive research. The preparation of the noble-gas compounds in large Xe clusters by UV photolysis8–11 and also by fast electrons and X-ray irradiation in the Xe matrix12,13 have been reported in the literature as well.Since these noble-gas hydrides have a strong ion-pair character of the form (HNg)+Y−, the resulting large dipole moment and also the weak bonding of these compounds sometimes induce unique solvation effects.7,14–16 For example, the vibrational spectroscopy measurements of HXeCl in various noble-gas matrices exhibited an unusual vibrational shift of the H–Xe stretching frequency. The order of the vibrational frequency was observed to be ν(Ne) < ν(Xe) < ν(Kr) < ν(Ar), and it is a non-monotonous function of the matrix dielectric constant.1,17–19 These results are in contrast to the “normal” order observed for HCl: ν(Xe) < ν(Kr) < ν(Ar) < ν(Ne) < ν(gas).20,21 The vibrational spectroscopy measurements of HXeCCH and HXeBr in various noble-gas matrices also revealed the unusual vibrational shifts of the H–Xe stretching frequency in the order ν(Ne) < ν(Xe) < ν(Kr) < ν(Ar),1,17,22–25 which is the same as that observed for HXeCl.
There have been several attempts to explain these unique features of the matrix shifts. First, it has been reported that simple calculations of neither HXeY with the polarizable continuum model (PCM) nor single-atom complex HXeY–Ng can reproduce the order of the vibrational shifts.24–26 Very recently, Kalinowski et al. performed quantum chemical calculations by MP4(SDQ) for HXeCl embedded in a single noble-gas layer cage and successfully reproduced the order of the experimentally observed vibrational shifts.19 They discussed these calculated shifts in terms of the size available for HXeCl in the cages and its implications for the stresses acting on the embedded HXeCl compound. Curiously, the vibrational frequencies of single-layer calculations by Ne atoms were strongly red-shifted from that of the isolated HXeCl; the shifts were −50 cm−1 and −192 cm−1 for harmonic and anharmonic calculations, respectively, which are the largest among the single-layer cages of four Ng atoms (Ng = Ne, Ar, Kr, and Xe). This is in contrast to the general observations that the interaction of an embedded compound with Ne atoms is the weakest and that the vibrational frequency in a Ne matrix should be closest to the situation in the gas phase.27
At almost the same time as the work of Kalinowski et al.,19 we performed the hybrid quantum-classical simulations on the vibrational shift of HXeCl, where the interaction energy between HXeCl and surrounding Ng atoms was modeled by the pairwise-additive form and the pair-interaction energies were determined by high-level quantum chemical calculations.28 We demonstrated that the experimentally observed vibrational shifts are well reproduced by the simulations and that the local matrix morphology is an important factor affecting the vibrational shifts. The largest shifts were predicted for the Ar matrix by our simulations and at that time we were not aware of the experimental results of the Ar matrix, but the very recent experimental results are in accord with our prediction.19 It is noteworthy that the two different approaches of Kalinowski et al.19 and ours predict the same order of vibrational frequency for HXeCl embedded in four noble-gas matrices.
This paper presents an application of our simulation method to other interesting noble-gas hydrides, HXeCCH, HXeBr, and HXeI in order to provide further information about the matrix shifts. The H–Xe stretching frequencies of HXeCCH22–25 and HXeBr1,17,25 have recently been reported in four different Ng matrices, and, therefore, a comparison can be made with our simulations. The spectroscopic measurements of HXeI and its complexes with various molecules (i.e. HXeI⋯HI, HXeI⋯H2O) have been reported in the Xe matrix before,1,29,30 and very recently the vibrational frequencies of HXeI have been measured in Ar and Kr matrices.31 It has been shown that the vibrational frequency of HXeI exhibits the order ν(Xe) < ν(Ar) < ν(Kr) and it is slightly different from those observed for HXeCCH, HXeCl, and HXeBr (the experimental results are summarized in Table 1). In that paper, we performed the simulations for HXeI in Ar, Kr, and Xe matrices, and the calculated order is also in accord with the experimental data.31
| HXeCCH | HXeCl | HXeBr | HXeI | |||||
|---|---|---|---|---|---|---|---|---|
| Freq. | Ref. | Freq. | Ref. | Freq. | Ref. | Freq. | Ref. | |
| a The shifts, ν(X)–ν(Ne) (X = Ar, Kr, and Xe), were shown in the parentheses. | ||||||||
| Ne matrix | 1453 | 25 | 1612 | 17 | 1453 | 1, 25 | — | — |
| Ar matrix | 1531 (+78) | 24 | 1675 (+63) | 19 | 1541 (+88) | 25 | 1238 | 31 |
| Kr matrix | 1519 (+66) | 22 | 1664 (+52) | 1 | 1527 (+74) | 25 | 1239 | 31 |
| Xe matrix | 1486 (+33) | 23 | 1648 (+36) | 1, 18 | 1504 (+51) | 17, 25 | 1193 | 1, 31 |
II. Computational details
The vibrational spectra of HXeCCH, HXeBr, and HXeI in noble-gas (Ng) matrices are computed using the hybrid quantum-classical simulations. The computational methodology is essentially the same as that employed in our previous studies; see ref. 28 and 32 for details. In its implementation, the total potential energy is represented by the pairwise-additive form, | (2.1) |
In obtaining potential energy surfaces of VHXeY and also pair interaction energies of VHXeY–Ng and VNg–Ng, the electronic structure calculations employing the coupled-cluster singles and doubles including the perturbative contributions of connected triple excitations [CCSD(T)] method are performed. For Kr, Xe, Br, and I atoms, the relativistic pseudopotentials33 are employed. The cc-pVQZ basis sets are used for H, C, Br, and I atoms, while the aug-cc-pVQZ basis sets are employed for Ng atoms. It is known that the equilibrium distances and binding energies of Ng dimers with these basis sets yield quite reliable values (see ref. 28 for details) and these basis sets were employed in our previous study on HXeCl. All ab initio calculations are carried out using the MOLPRO2010.1 package.34
The interaction potential energies between HXeY and an Ng atom (VHXeY–Ng) are obtained at each point of the normal coordinate of q(H–Xe). The position of the Ng atom is represented by polar coordinates (r, θ), where r is the distance of Ng from the center-of-mass of HXeY and θ is the polar angle measured from the molecular axis of HXeY. The numbers of grid points are 25 and 21 for r and θ, respectively. These potential energies are tabulated for use in the subsequent Monte Carlo (MC) simulations, and in the MC runs the potential energies are evaluated by the interpolation. The number of grid points for the normal coordinate of the H–Xe stretching vibration corresponds to the number of optimized basis set for representing the vibrational wavefunction in the framework of the potential-optimized discrete variable representation (PO-DVR) method.35 In this work, the optimized basis set is constructed from the solution of the sinc-DVR method36 for HXeY in the gas phase, and the number of optimized basis sets is determined to be 8, where the convergence of the H–Xe fundamental frequency with respect to the number of optimized basis sets is confirmed from the preliminary calculations.
The MC simulations are carried out under the periodic boundary conditions, and 500 Ng atoms are first placed in a cubic box with the fcc lattice structure. Then, the HXeY compound is inserted into this fcc lattice by removing a couple of Ng atoms. There are several possible sites at which Ng atom(s) can be substituted by HXeY. Three possible orientations in the fcc structures are considered for the insertion of HXeY: 〈110〉 (nearest neighbor), 〈100〉 (four-atomic window), and 〈111〉 (three-atomic windows). The number of substituted Ng atoms (Nr) is determined by a sensible choice based on the distance between the adjacent Ng atoms along each orientation. The simulation parameters are tabulated in Table 2, and the size of the unit cell has been adjusted to yield the density of experimental values under the saturated vapor conditions.37 A cutoff distance of the half the length of the unit cell is applied to all interactions. After equilibrating the system, a total of 107 MC steps are taken to obtain statistical averages.
| Temperature | Length | Density | |
|---|---|---|---|
| Ne matrix | 5 | 21.2 | 1.75 |
| Ar matrix | 15 | 26.6 | 1.77 |
| Kr matrix | 20 | 28.2 | 3.10 |
| Xe matrix | 30 | 30.6 | 3.70 |
The stability of different sites is compared using the stabilization energy, which is defined as
 | (2.2) |
The vibrational spectra are obtained from the following simple form:
I(ω) = ∫dR![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ(E1 − E0 − ω)ρ(R) δ(E1 − E0 − ω)ρ(R) | (2.3) |
III. Results and discussion
A. Equilibrium structures and vibrational analysis of HXeCCH, HXeBr, and HXeI
The geometry optimizations were performed for HXeCCH, HXeBr, and HXeI using the CCSD(T) method, and the vibrational frequency analysis followed it. The structural parameters at equilibrium geometries and the harmonic and fundamental frequencies of H–Xe stretching vibration are summarized in Tables 3 and 4, respectively (see also Fig. 1 for equilibrium structures). The results of HXeCl are also shown in the tables for reference.28 As seen in the table, the inclusion of anharmonicity induces large red shifts of 85, 137, and 179 cm−1 for HXeCCH, HXeBr, and HXeI, respectively. The inclusion of the bending motions by DVR-3D further reduces the frequencies by 16, 28, and 44 cm−1 for HXeCCH, HXeBr, and HXeI, respectively, with respect to those by DVR-1D. The natural charge and dipole moments of these compounds are given in Table 5, where one can clearly see that the compound exhibits a strong ion-pair character of the form (HXe)+Y−, in particular, for HXeCl, HXeBr, and HXeI.| Harmonic | Fundamental (DVR-1D) | Fundamental (DVR-3D) | |
|---|---|---|---|
| a Harmonic frequencies of Xe–Y (Y = CA, Cl, Br, and I) stretching and bending vibrations are also shown. b From ref. 28. | |||
| HXeCCH |
1582 (H–Xe stretching)
308 (Xe–CA stretching) 635 (bending) |
1497 (H–Xe stretching) | 1481 (H–Xe stretching) |
| HXeClb |
1759 (H–Xe stretching)
274 (Xe–Cl stretching) 561 (bending) |
1642 (H–Xe stretching) | 1619 (H–Xe stretching) |
| HXeBr |
1625 (H–Xe stretching)
192 (Xe–Br stretching) 533 (bending) |
1488 (H–Xe stretching) | 1460 (H–Xe stretching) |
| HXeI |
1354 (H–Xe stretching)
154 (Xe–I stretching) 491 (bending) |
1175 (H–Xe stretching) | 1131 (H–Xe stretching) |
 | ||
| Fig. 1 Equilibrium structures of (a) HXeCCH, (b) HXeBr, and (c) HXeI. Bond lengths are given in units of Å. | ||
B. Equilibrium structures and vibrational analysis of HXeCCH–Ng, HXeBr–Ng, and HXeI–Ng (Ng = Ne, Ar, Kr, Xe)
In this subsection, the equilibrium structures, binding energies, and vibrational frequencies of HXeY–Ng (Y = CCH, Br, I) complexes are examined. Here, the internal coordinates of HXeY are fixed at the equilibrium values of the monomer (given in Table 3). In fact, the intramolecular structures of HXeY are negligibly perturbed upon the formation of HXeY–Ng complexes, and the changes of bond lengths of HXeY between the monomer and the complexes in the fully relaxed configurations are less than 0.003 Å for all complexes. As expected from the previous studies,28,38 three minima have been located for all complexes. Two of them are on both sides of HXeY in collinear geometry (H-side and Y-side structures) and the third one is located between and almost equidistant from Xe and Y (bent structure); see Fig. 2 for equilibrium structures of HXeCCH–Xe. The structural parameters and binding energies of an Ng atom at these minima are given in Tables 6 and 7, respectively. The strong binding site is the bent structure for all complexes, and in collinear geometry the Y-side structure exhibits a slightly stronger binding energy than the X-side structure, except for HXeCCH–Kr and HXeCCH–Xe. Note that these binding energies are several times larger than those of the Ng dimer, where they are 0.095, 0.274, 0.374, and 0.489 kcal mol−1 for Ne2, Ar2, Kr2, and Xe2, respectively.28Fig. 3 shows the two-dimensional contour plot of the potential energy surface of HXeCCH–Xe, where the internal coordinates of HXeCCH are fixed at the equilibrium values of the monomer. One can see that the potential energies at the two minima in the collinear geometry are very shallow with respect to the displacement of Xe in the direction perpendicular to the molecular axis.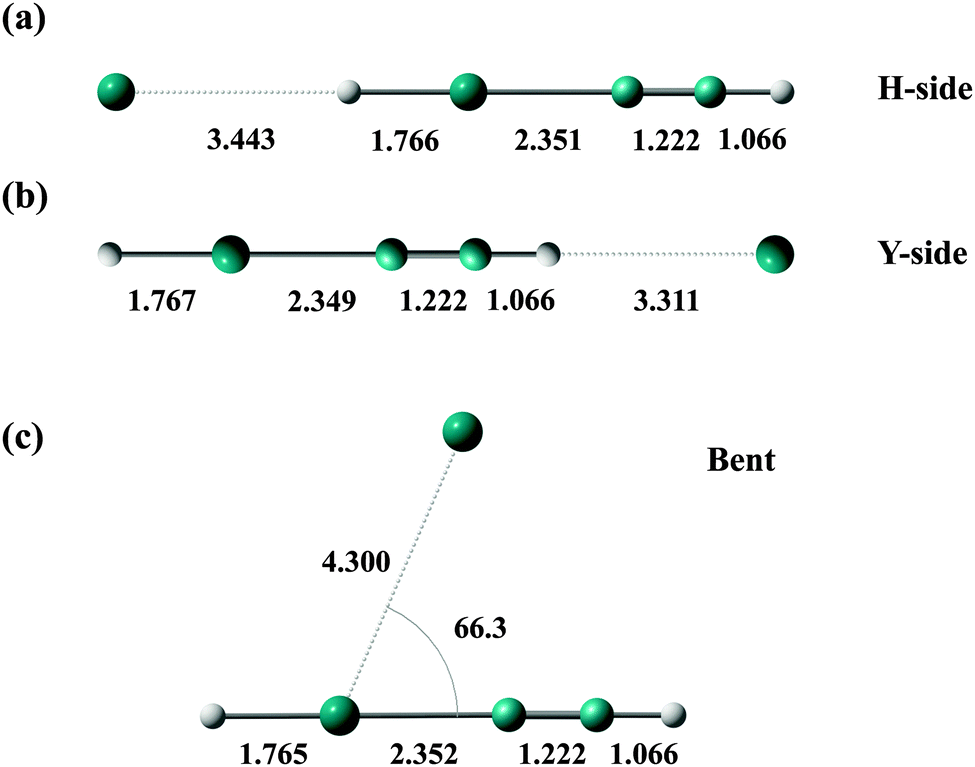 | ||
| Fig. 2 (a–c) Minimum energy structures of HXeCCH–Xe, where the internal coordinates of HXeCCH are fixed at the equilibrium values of the monomer. Bond lengths are given in units of Å and an angle is given in units of degrees. | ||
| Ng | H-side | Y-side | Bent | |
|---|---|---|---|---|
| r(Ng–H) | r(Y–Ng) | r(Ng–Xe) | θ(Ng–Xe–Y) | |
| a Distance from the nearest H atom. b From ref. 28. | ||||
| HXeCCH–Ng | ||||
| Ne | 3.176 | 2.692a | 3.734 | 69.2 |
| Ar | 3.266 | 2.995a | 3.971 | 67.9 |
| Kr | 3.314 | 3.109a | 4.088 | 67.3 |
| Xe | 3.443 | 3.311a | 4.300 | 66.3 |
| HXeCl–Ngb | ||||
| Ne | 2.970 | 3.609 | 3.715 | 70.9 |
| Ar | 3.045 | 3.742 | 3.952 | 69.9 |
| Kr | 3.091 | 3.815 | 4.066 | 69.0 |
| Xe | 3.192 | 3.952 | 4.295 | 68.0 |
| HXeBr–Ng | ||||
| Ne | 3.013 | 3.758 | 3.685 | 77.7 |
| Ar | 3.074 | 3.888 | 3.954 | 72.3 |
| Kr | 3.107 | 3.955 | 4.067 | 71.8 |
| Xe | 3.199 | 4.092 | 4.256 | 71.6 |
| HXeI–Ng | ||||
| Ne | 3.124 | 3.979 | 3.960 | 71.4 |
| Ar | 3.159 | 4.091 | 3.962 | 75.2 |
| Kr | 3.180 | 4.149 | 4.074 | 74.7 |
| Xe | 3.257 | 4.281 | 4.261 | 74.7 |
| Ng | H-side | Y-side | Bent |
|---|---|---|---|
| a From ref. 28. | |||
| HXeCCH–Ng | |||
| Ne | 0.106 | 0.176 | 0.317 |
| Ar | 0.314 | 0.355 | 0.800 |
| Kr | 0.425 | 0.422 | 1.010 |
| Xe | 0.549 | 0.478 | 1.210 |
| HXeCl–Nga | |||
| Ne | 0.149 | 0.181 | 0.327 |
| Ar | 0.447 | 0.496 | 0.828 |
| Kr | 0.618 | 0.687 | 1.052 |
| Xe | 0.823 | 0.865 | 1.265 |
| HXeBr–Ng | |||
| Ne | 0.139 | 0.167 | 0.311 |
| Ar | 0.434 | 0.493 | 0.822 |
| Kr | 0.608 | 0.659 | 1.049 |
| Xe | 0.824 | 0.837 | 1.272 |
| HXeI–Ng | |||
| Ne | 0.120 | 0.152 | 0.283 |
| Ar | 0.387 | 0.466 | 0.821 |
| Kr | 0.551 | 0.631 | 1.055 |
| Xe | 0.759 | 0.810 | 1.286 |
 | ||
| Fig. 3 Two-dimensional contour plot of the potential energy surface (in units of kcal mol−1) for HXeCCH–Xe. The internal coordinates of HXeCCH are fixed at the equilibrium values of the monomer, and the origin is set to the center-of-mass of HXeCCH. The positions of the H, Xe, C, C, and H atoms are (−2.237, 0.0), (−0.470, 0.0), (1.879, 0.0), (3.101, 0.0), and (4.167, 0.0), respectively, and they are shown in filled circles. The positions of Xe atoms in the minimum energy structures of the HXeCCH–Xe complex are given in red filled circles. | ||
In Table 7, it is seen that for each Ng the binding energies to HXeY in the bent structure are very similar to each other. Also we find that for each Ng the binding energies in the H-side and Y-side structures are very close to each other for HXeCl–Ng, HXeBr–Ng, and HXeI–Ng, and it decreases slightly as the halogen atom becomes larger. The binding energies of HXeCl–Ng, HXeBr–Ng, and HXeI–Ng are generally larger than those of HXeCCH–Ng, and this can be explained by the differences in the dipole moment of monomers (see Table 5).
The frequency analysis is made at these minimum energy structures, and the results are shown in Table 8. Note that harmonic frequencies are calculated at fully relaxed configurations and that the fundamental frequencies are obtained by fixing the position of Ng atom at the respective minima. As seen in the table, the frequencies of the H–Xe stretching vibration are most strongly affected at the H-side structures, whereas the Y-side and bent structures have a negligible effect on the H–Xe stretching frequency. For the H-side structures, the blue shifts are observed and this shift increases with the Ng size. These blue shifts were previously explained by the enhanced charge separation (HXe)+Y− upon complex formation.39 The inclusion of anharmonicity induces large red shifts with respect to the harmonic frequencies, but it is also noted that the frequency shifts from the gas-phase values become larger in the anharmonic calculations. The inclusion of the bending motion by DVR-3D further reduces the frequencies, but the frequency shifts from the gas-phase values are very similar in DVR-3D and DVR-1D calculations with the difference of ∼2 cm−1.
| Ng | H-side | Y-side | Bent | |||
|---|---|---|---|---|---|---|
| Freq. | Shift | Freq. | Shift | Freq. | Shift | |
| a Frequency shifts from monomer HXeY are also shown. b From ref. 28. | ||||||
| HXeCCH–Ng | ||||||
| Harmonic (monomer: 1582 cm−1) | ||||||
| Ne | 1585 | +3 | 1583 | +1 | 1584 | +2 |
| Ar | 1592 | +10 | 1580 | −2 | 1582 | 0 |
| Kr | 1594 | +12 | 1581 | −1 | 1586 | +5 |
| Xe | 1595 | +13 | 1581 | −1 | 1587 | +6 |
| Fundamental (DVR-1D) (monomer: 1497 cm−1) | ||||||
| Ne | 1504 | +7 | 1498 | +1 | 1497 | 0 |
| Ar | 1514 | +17 | 1496 | −1 | 1499 | +2 |
| Kr | 1518 | +21 | 1496 | −1 | 1500 | +3 |
| Xe | 1519 | +22 | 1496 | −1 | 1501 | +4 |
| Fundamental (DVR-3D) (monomer: 1481 cm−1) | ||||||
| Ne | 1488 | +7 | 1481 | 0 | 1482 | +1 |
| Ar | 1498 | +17 | 1481 | 0 | 1483 | +2 |
| Kr | 1501 | +20 | 1481 | 0 | 1484 | +3 |
| Xe | 1502 | +21 | 1481 | 0 | 1485 | +4 |
| HXeCl–Ngb | ||||||
| Harmonic (monomer: 1759 cm−1) | ||||||
| Ne | 1765 | +6 | 1759 | +0 | 1759 | +0 |
| Ar | 1777 | +18 | 1761 | +2 | 1761 | +2 |
| Kr | 1780 | +21 | 1760 | +1 | 1762 | +3 |
| Xe | 1780 | +21 | 1759 | +0 | 1768 | +9 |
| Fundamental (DVR-1D) (monomer: 1642 cm−1) | ||||||
| Ne | 1655 | +13 | 1642 | +0 | 1643 | +1 |
| Ar | 1674 | +32 | 1644 | +2 | 1645 | +3 |
| Kr | 1677 | +35 | 1644 | +2 | 1647 | +5 |
| Xe | 1678 | +36 | 1645 | +3 | 1648 | +6 |
| Fundamental (DVR-3D) (monomer: 1619 cm−1) | ||||||
| Ne | 1631 | +12 | 1619 | +0 | 1620 | +1 |
| Ar | 1648 | +29 | 1621 | +2 | 1622 | +3 |
| Kr | 1652 | +33 | 1621 | +2 | 1624 | +5 |
| Xe | 1651 | +32 | 1621 | +2 | 1625 | +6 |
| HXeBr–Ng | ||||||
| Harmonic (monomer: 1625 cm−1) | ||||||
| Ne | 1633 | +8 | 1625 | 0 | 1626 | +1 |
| Ar | 1648 | +23 | 1625 | 0 | 1629 | +4 |
| Kr | 1653 | +28 | 1626 | +1 | 1630 | +5 |
| Xe | 1656 | +31 | 1626 | +1 | 1632 | +7 |
| Fundamental (DVR-1D) (monomer: 1488 cm−1) | ||||||
| Ne | 1504 | +16 | 1488 | 0 | 1490 | +2 |
| Ar | 1529 | +41 | 1489 | +1 | 1493 | +5 |
| Kr | 1537 | +49 | 1490 | +2 | 1494 | +6 |
| Xe | 1541 | +53 | 1490 | +2 | 1497 | +9 |
| Fundamental (DVR-3D) (monomer: 1460 cm−1) | ||||||
| Ne | 1474 | +14 | 1460 | 0 | 1461 | +1 |
| Ar | 1497 | +37 | 1461 | +1 | 1464 | +4 |
| Kr | 1506 | +46 | 1461 | +1 | 1466 | +6 |
| Xe | 1510 | +50 | 1461 | +1 | 1468 | +8 |
| HXeI–Ng | ||||||
| Harmonic (monomer: 1354 cm−1) | ||||||
| Ne | 1363 | +9 | 1354 | 0 | 1353 | −1 |
| Ar | 1382 | +27 | 1354 | 0 | 1360 | +6 |
| Kr | 1391 | +37 | 1355 | 0 | 1362 | +8 |
| Xe | 1400 | +45 | 1353 | −1 | 1365 | +11 |
| Fundamental (DVR-1D) (monomer: 1175 cm−1) | ||||||
| Ne | 1194 | +19 | 1175 | 0 | 1176 | +1 |
| Ar | 1231 | +56 | 1177 | +2 | 1182 | +7 |
| Kr | 1247 | +72 | 1177 | +2 | 1184 | +9 |
| Xe | 1262 | +87 | 1176 | +1 | 1187 | +12 |
| Fundamental (DVR-3D) (monomer: 1131 cm−1) | ||||||
| Ne | 1148 | +17 | 1131 | 0 | 1132 | +1 |
| Ar | 1185 | +54 | 1133 | +2 | 1138 | +7 |
| Kr | 1202 | +71 | 1133 | +2 | 1140 | +9 |
| Xe | 1217 | +86 | 1132 | +1 | 1144 | +13 |
The order of the H–Xe stretching frequency is ν(Ne) < ν(Ar) < ν(Kr) < ν(Xe) for HXeY–Ng complexes, which is a monotonous function of the matrix dielectric constants, and it is in disagreement with the experimental results in the matrices. The PCM calculations25 for HXeCCH and HXeBr also predicted the same order of ν(gas) < ν(Ne) < ν(Ar) < ν(Kr) < ν(Xe), simply indicating that the H–Xe stretching frequency becomes higher in matrices with larger dielectric constants, but again, this PCM model cannot explain the experimental observations. As already demonstrated in our previous work,28 these results indicate that a more realistic modeling of the matrix environments is also necessary for HXeCCH, HXeBr, and HXeI.
C. Hybrid quantum-classical simulations in the solid matrices
Table 9 shows a summary of the MC simulations, where the peak positions of the vibrational spectra of H–Xe stretching motion are listed. As noted above, the DVR-1D method is employed for solving the vibrational Schrödinger equation in the MC runs. For each orientation of the substitution site, the MC simulations were initiated with different Nr, and we show only the results for the case of lower stabilization energy. As seen in the table, the 〈110〉 site exhibits a lower stabilization energy in all cases, and therefore the 〈110〉 site would be energetically favored. For the simulations in the Ne matrix, we found that the configurations of Ne atoms around HXeY become rather structureless after equilibration, and therefore the classification by the orientation in the fcc structure is somewhat vague. On the other hand, the configurations of Ar, Kr, and Xe atoms in the matrices are highly structured. For the MC simulations starting from the 〈110〉 or 〈100〉 site, the HXeY compound does not change its orientation, and the lattice structure of Ar, Kr, and Xe atoms surrounding HXeY is almost maintained. However, when the simulations are initiated from the 〈111〉 site, in most cases the compound changes its orientation and the configurations of Ar, Kr, and Xe atoms become almost identical to those of the 〈100〉 site after equilibration.| 〈110〉 | 〈100〉 | 〈111〉 | Exp.c | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N r | Freq. | Shift | ΔE | N r | Freq. | Shift | ΔE | N r | Freq. | Shift | ΔE | Freq. | |
| a N r is the number of the noble-gas atom(s) substituted with a single HXeY. Frequency shifts from the isolated gas-phase value are also shown. The cells shown as “〈100〉” indicate that after equilibration the configurations of Ng atoms become almost identical to those of the 〈100〉 site. b From ref. 28. c See Table 1 for references of the experimental data. | |||||||||||||
| HXeCCH (gas: 1497 cm−1) | |||||||||||||
| Ne matrix | 3 | 1509 | +12 | −0.8 | 3 | 1571 | +74 | +0.6 | 2 | 1557 | +60 | +0.4 | 1453 |
| Ar matrix | 2 | 1605 | +108 | −9.0 | 2 | 1554 | +57 | −5.9 | 2 | 〈100〉 | 1531 | ||
| Kr matrix | 2 | 1603 | +106 | −10.3 | 2 | 1550 | +53 | −5.2 | 2 | 〈100〉 | 1519 | ||
| Xe matrix | 2 | 1564 | +67 | −9.5 | 1 | 1590 | +93 | −4.7 | 2 | 〈100〉 | 1486 | ||
| HXeClb (gas: 1642 cm−1) | |||||||||||||
| Ne matrix | 3 | 1665 | +23 | −1.1 | 3 | 1728 | +86 | +6.5 | 2 | 1690 | +48 | +1.1 | 1612 |
| Ar matrix | 2 | 1758 | +116 | −10.2 | 2 | 1715 | +73 | −5.6 | 2 | 〈100〉 | 1675 | ||
| Kr matrix | 2 | 1747 | +105 | −11.0 | 2 | 1708 | +66 | −4.5 | 2 | 1709 | +67 | −4.9 | 1664 |
| Xe matrix | 2 | 1730 | +88 | −9.7 | 1 | 1757 | +115 | −8.1 | 2 | 〈100〉 | 1648 | ||
| HXeBr (gas: 1488 cm−1) | |||||||||||||
| Ne matrix | 3 | 1508 | +20 | +0.4 | 3 | 1580 | +92 | +0.6 | 2 | 1544 | +56 | +0.7 | 1453 |
| Ar matrix | 2 | 1630 | +142 | −10.1 | 2 | 1572 | +84 | −5.4 | 2 | 〈100〉 | 1541 | ||
| Kr matrix | 2 | 1622 | +134 | −11.3 | 2 | 1569 | +81 | −4.3 | 2 | 〈100〉 | 1527 | ||
| Xe matrix | 2 | 1599 | +111 | −10.5 | 1 | 1647 | +159 | −6.4 | 2 | 〈100〉 | 1504 | ||
| HXeI (gas: 1175 cm−1) | |||||||||||||
| Ne matrix | 3 | 1208 | +33 | +2.3 | 3 | 1296 | +121 | +3.3 | 2 | 1300 | +125 | +3.7 | — |
| Ar matrix | 2 | 1388 | +213 | −8.9 | 2 | 1303 | +128 | −5.1 | 2 | 〈100〉 | 1238 | ||
| Kr matrix | 2 | 1392 | +217 | −11.1 | 2 | 1306 | +131 | −4.5 | 2 | 〈100〉 | 1239 | ||
| Xe matrix | 2 | 1354 | +179 | −11.2 | 1 | 1436 | +261 | −3.3 | 2 | 〈100〉 | 1193 | ||
In all cases shown in Table 9, blue shifts from the isolated gas-phase values are observed. The observed blue shifts are about 2–3 times larger than those estimated from the calculations of isolated HXeY–Ng complexes (see the fundamental frequencies by DVR-1D given in Table 8). The order of H–Xe stretching frequencies is ν(Ne) < ν(Xe) < ν(Kr) < ν(Ar) for HXeCCH and HXeBr in the 〈110〉 site, which is in accord with the experimental observations.1,17,22–25 The same order has been seen for HXeCl in our previous work.28 The observed order is slightly different for HXeI in the 〈110〉 site, where it is ν(Ne) < ν(Xe) < ν(Ar) < ν(Kr), and this order is also in accord with the experiment.1,31 It is noted that the order of frequencies is different between the 〈110〉 and 〈100〉 sites.
The contour plots of two-dimensional distribution functions of Ng (= Ar, Kr, Xe) atoms around HXeCCH and HXeBr are shown in Fig. 4 in the 〈110〉 site. Here the x-axis is taken as the molecular axis of HXeY and r is the radial distance from the x-axis, and the origin is set to the center-of-mass of HXeY. Due to the highly structured configurations of solid matrices, the positions of the Ng atoms are quite localized. In the same figure, the positions of Ng atoms in the three minimum energy structures of the isolated HXeY–Ng complexes (H-side, Y-side, and bent structures) are also plotted.
 | ||
| Fig. 4 Contour plots of two-dimensional distribution functions of the Ng atoms around (a–c) HXeCCH and (d–f) HXeBr in the 〈110〉 site in the Ar, Kr, and Xe matrices. The positions of HXeCCH and HXeBr in the minimum energy structure in the gas phase are plotted for reference (shown by black filled circles), where the coordinates of HXeCCH are the same as those in Fig. 3 and the positions of the H, Xe, and Br atoms of HXeBr are (−2.761, 0.0), (−1.039, 0.0), and (1.742, 0.0), respectively. The positions of the Ng atoms in the minimum energy structures of isolated HXeCCH–Ng and HXeBr–Ng complexes are also plotted in the figure by red filled circles. | ||
As already seen in the frequency analysis of the isolated HXeY–Ng complex, the blue shifts are observed in all minimum energy structures of HXeY–Ng, and since there are several Ng atoms surrounding HXeY, the calculated larger blue shifts in the matrix environments are naturally expected. Larger blue shifts are observed in Ar and Kr matrices compared with the Xe matrix, and this could partly be explained by the observations (Fig. 4) that the Ar or Kr atom in collinear geometry on the H-side is found to be slightly closer to the hydrogen atom than that found in the minimum energy structures of HXeY–Ng, in comparison to the situation in the Xe matrix. Similar observations have been made for HXeCl and HXeI in our previous studies.28,31 As shown above in the frequency analysis of HXeY–Ng, the H–Xe stretching frequency is strongly affected by Ng atoms that are close to the hydrogen atom. In this situation, a displacement of the Ng atom toward the hydrogen atom results in a blue shift because the potential energy curve is quite repulsive when the Ng atom is located closer to the hydrogen atom.
In matrix environments, we find that four Ng atoms are located in the position of the bent structure of the HXeY–Ng complex, forming a ring around the molecular axis. Since the bent structure exhibits the strongest binding energy to HXeY (see Fig. 3 and Table 7), it is plausible that HXeY adjusts its coordinate along the molecular axis to have these surrounding four Ng atoms at the position of the bent structure. As a result of this, an Ng atom in the collinear geometry on the H-side can be close to the hydrogen atom in the case of the Ar or Kr matrix, causing the large blue shifts. Of course, the Ng atom cannot be too close to the hydrogen atom since there is quite a repulsive potential between the Ng and hydrogen atoms. Therefore, the location of HXeY in matrix environments depends on the delicate balance among stabilization and destabilization arising from the interactions between HXeY and the surrounding Ng atoms. As another explanation of larger blue shifts in Ar or Kr matrix, which is somewhat related to the above arguments, the Ar cage would be too tight for HXeY, while the Xe cage would be rather relaxed due to the difference in the lattice constants. The Ne matrix exhibits the shortest lattice constant, but the Ne matrix is “soft” due to the weak interaction between Ne atoms, and therefore HXeY would be mildly surrounded by Ne atoms. This is reflected in the small stabilization energies of ca. −1 to 2 kcal mol−1 in the Ne matrix in comparison to those in the other matrices (from −9 to −11 kcal mol−1) and also in the small frequency shifts from the gas-phase values (from +10 to +30 cm−1). The stress acting on HXeY would be an important factor affecting the vibrational shifts, which was also suggested by Kalinowski et al.19 We note in passing that the small stabilization energies in the Ne matrix (sometimes positive values) can be related to the rather structureless configurations of Ne atoms around HXeY. Also, as pointed out in the previous work,31 it may be the reason for a failure to prepare HXeI in the Ne matrix experimentally.
It is also interesting to note that the frequency shifts from the gas-phase values exhibit a trend Δν(HXeCCH) ≈ Δν(HXeCl) < Δν(HXeBr) < Δν(HXeI) in the same Ng matrix environments. The binding energies of an Ng atom to HXeCl, HXeBr and HXeI are very similar to each other in the HXeY–Ng complex and also the stabilization energies are quite close to each other for these compounds in the same Ng matrix, but there are appreciable differences in the frequency shifts. This trend is in agreement with the experiments (see Table 1), and it is consistent with the chemical intuition that the matrix effects are stronger for more weakly bound molecules.
Finally, we comment on the reliability of our calculations. First, the potential energy surfaces of embedded compounds and also the pair interaction potentials are determined using the CCSD(T) method with the (aug-)cc-pVQZ basis sets, which is known to be quite accurate, but of course further improvement would be possible (e.g., taking the complete basis set limit). We speculate that discrepancies in the absolute values of frequencies between experiments and our calculations (50–150 cm−1) stem mainly from the quality of potentials. The large discrepancies were found for HXeI, and this might be related to the weakness of the chemical bond of this compound. Second, the higher-body interactions between Ng atoms should be examined carefully because it has been reported that the three-body contributions are not negligible in bulk solids ranging from 3% (in Ne) to 7% (in Xe) in cohesive energy.40 The inclusion of the three-body contributions also increases the lattice constants by ∼1%, which might shift slightly the vibrational frequencies to the red. Lastly, the present MC simulations have been performed using the DVR-1D method for practical consideration of the computational cost. The vibrational frequencies of isolated HXeY–Ng complexes show that the H–Xe stretching frequencies decrease by ∼20 cm−1 with the inclusion of the bending motions by DVR-3D, but the shifts from the gas-phase values are very similar in DVR-1D and DVR-3D calculations with the difference of at most 4 cm−1 (see Table 8). In our previous study of HXeCl,28 the MC simulations employing DVR-3D have predicted the frequencies of 1738, 1735, and 1710 cm−1 in Ar, Kr, and Xe matrices, respectively, and therefore the shifts from the gas-phase values are +119, +116, and +91 cm−1, which is close to the results obtained by DVR-1D shown above. For HXeI, the shifts from the gas-phase values in DVR-3D calculations were +226, +230, and +196 cm−1 in Ar, Kr, and Xe matrices,31 respectively, and these shifts are slightly larger than those obtained from DVR-1D calculations (see Table 9). We expect that the differences in relative shifts between DVR-1D and DVR-3D are smaller for HXeCCH and HXeBr than HXeI considering the trend in the vibrational frequencies of these compounds, and the order of the vibrational frequency will almost certainly be unchanged.
IV. Conclusions and outlook
The hybrid quantum-classical simulations are performed to investigate the unusual vibrational spectral shifts of noble-gas hydrides, HXeCCH, HXeBr, and HXeI in matrix environments (in Ne, Ar, Kr, and Xe matrices). It is demonstrated that the H–Xe stretching frequencies are blue-shifted from the isolated gas-phase values in all matrix environments and that the amounts of blue shifts are a non-monotonous function of the matrix dielectric constants. The calculated results reproduce the experimentally observed shifts quite successfully.Our calculations have clarified that the local matrix morphology is an important factor affecting the vibrational shifts and that a realistic modeling of the surrounding matrix environments is essential to reproduce the unusual vibrational shifts observed in the experiments. The orders of the H–Xe stretching frequencies are ν(Ne) < ν(Xe) < ν(Kr) < ν(Ar) for HXeCCH and HXeBr and ν(Ne) < ν(Xe) < ν(Ar) < ν(Kr) for HXeI, and therefore the order of the matrix shifts depends on the embedded compound. Also our calculations have shown that the matrix shifts from the gas-phase values exhibit the trend Δν(HXeCCH) ≈ Δν(HXeCl) < Δν(HXeBr) < Δν(HXeI) in the same Ng matrix environments. This is consistent with the experimental results and also with the speculation that the stronger matrix effects are observed for more weakly bound molecules.
The theoretical investigations of the vibrational spectra in other matrices such as N2 or CO2 will further enhance our understanding of the matrix shifts. In N2 or CO2 matrices, the large matrix shifts were observed experimentally due to the dipole–quadrupole interactions between HXeY and matrix molecules.41 The elucidation of these shifts is also an exciting future challenge.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful to Professor Leonid Khriachtchev for useful discussions. The authors acknowledge the financial support by KAKENHI (Grant-in-Aid for Scientific Research) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT, Japan). Part of the calculations was performed on supercomputers at the Research Center for Computational Science, Okazaki, Japan.References
- M. Pettersson, J. Lundell and M. Räsänen, J. Chem. Phys., 1995, 102, 6423–6431 CrossRef CAS PubMed.
- V. I. Feldman and F. F. Sukhov, Chem. Phys. Lett., 1996, 255, 425–430 CrossRef CAS.
- M. Pettersson, J. Lundell and M. Räsänen, Eur. J. Inorg. Chem., 1999, 729–737 CrossRef CAS.
- L. Khriachtchev, M. Pettersson, N. Runeberg, J. Lundell and M. Räsänen, Nature, 2000, 406, 874–876 CrossRef CAS PubMed.
- L. Khriachtchev, M. Pettersson, A. Lignell and M. Räsänen, J. Am. Chem. Soc., 2001, 123, 8610–8611 CrossRef CAS.
- R. B. Gerber, Annu. Rev. Phys. Chem., 2004, 55, 55–78 CrossRef CAS PubMed.
- L. Khriachtchev, M. Räsänen and R. B. Gerber, Acc. Chem. Res., 2009, 42, 183–191 CrossRef CAS PubMed.
- R. Baumfalk, N. H. Nahler and U. Buck, J. Chem. Phys., 2001, 114, 4755–4758 CrossRef CAS PubMed.
- N. H. Nahler, M. Fárník and U. Buck, Chem. Phys., 2004, 301, 173–182 CrossRef CAS PubMed.
- U. Buck and M. Fárník, Int. Rev. Phys. Chem., 2006, 25, 583–612 CrossRef CAS.
- V. Poterya, O. Votava, M. Fárník, M. Ončák, P. Slavíček, U. Buck and B. Friedrich, J. Chem. Phys., 2008, 128, 104313 CrossRef PubMed.
- V. I. Feldman, F. F. Sukhov, A. Y. Orlov and I. V. Tyulpina, J. Am. Chem. Soc., 2003, 125, 4698–4699 CrossRef CAS PubMed.
- V. I. Feldman, A. V. Kobzarenko, A. Y. Orlov and F. F. Sukhov, Low Temp. Phys., 2012, 38, 766–773 CrossRef CAS PubMed.
- J. Lundell, G. M. Chaban and R. B. Gerber, Chem. Phys. Lett., 2000, 331, 308–316 CrossRef CAS.
- J. Lundell, G. M. Chaban and R. B. Gerber, J. Phys. Chem. A, 2000, 104, 7944–7949 CrossRef CAS.
- A. Lignell, L. Khriachtchev, M. Pettersson and M. Räsänen, J. Chem. Phys., 2003, 118, 11120–11128 CrossRef CAS PubMed.
- M. Lorenz, M. Räsänen and V. E. Bondybey, J. Phys. Chem. A, 2000, 104, 3770–3774 CrossRef CAS.
- J. Ahokas, K. Vaskonen, J. Eloranta and H. Kunttu, J. Phys. Chem. A, 2000, 104, 9506–9511 CrossRef CAS.
- J. Kalinowski, R. B. Gerber, M. Räsänen, A. Lignell and L. Khriachtchev, J. Chem. Phys., 2014, 140, 094303 CrossRef PubMed.
- A. J. Barnes, H. E. Hallam and G. F. Scrimshaw, Trans. Faraday Soc., 1969, 65, 3159–3171 RSC.
- A. J. Barnes, in Vibrational Spectroscopy of Trapped Species, ed. H. E. Hallam, John Wiley and Sons, London, 1973, pp. 133–177 Search PubMed.
- L. Khriachtchev, H. Tanskanen, J. Lundell, M. Pettersson, H. Kiljunen and M. Räsänen, J. Am. Chem. Soc., 2003, 125, 4696–4697 CrossRef CAS PubMed.
- H. Tanskanen, L. Khriachtchev, J. Lundell and M. Räsänen, J. Chem. Phys., 2004, 121, 8291–8298 CrossRef CAS PubMed.
- H. Tanskanen, L. Khriachtchev, J. Lundell and M. Räsänen, J. Chem. Phys., 2006, 125, 074501 CrossRef PubMed.
- M. Tsuge, A. Lignell, M. Räsänen and L. Khriachtchev, J. Chem. Phys., 2013, 139, 204303 CrossRef PubMed.
- G. Liu, Y. Zhang, Z. Wang, Y. Wang, X. Zhang and W. Zhang, Comput. Theor. Chem., 2012, 993, 118–124 CrossRef CAS PubMed.
- M. E. Jacox, Chem. Soc. Rev., 2002, 31, 108–115 RSC.
- K. Niimi, A. Nakayama, Y. Ono and T. Taketsugu, J. Phys. Chem. A, 2014, 118, 380–387 CrossRef CAS PubMed.
- M. Tsuge, S. Berski, M. Räsänen, Z. Latajka and L. Khriachtchev, J. Chem. Phys., 2013, 138, 104314 CrossRef PubMed.
- M. Tsuge, S. Berski, M. Räsänen, Z. Latajka and L. Khriachtchev, J. Chem. Phys., 2014, 140, 044323 CrossRef PubMed.
- C. Zhu, K. Niimi, T. Taketsugu, M. Tsuge, A. Nakayama and L. Khriachtchev, J. Chem. Phys., 2015, 142, 054305 CrossRef PubMed.
- A. Nakayama, K. Niimi, Y. Ono and T. Taketsugu, J. Chem. Phys., 2012, 136, 054506 CrossRef PubMed.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, et al., MOLPRO, version 2010.1, a package of ab initio programs.
- J. Echave and D. C. Clary, Chem. Phys. Lett., 1992, 190, 225–230 CrossRef CAS.
- D. T. Colbert and W. H. Miller, J. Chem. Phys., 1992, 96, 1982–1991 CrossRef CAS PubMed.
- G. L. Pollack, Rev. Mod. Phys., 1964, 36, 748–791 CrossRef CAS.
- L. Khriachtchev, A. Lignell, J. Juselius, M. Räsänen and E. Savchenko, J. Chem. Phys., 2005, 122, 014510 CrossRef PubMed.
- A. Lignell and L. Khriachtchev, J. Mol. Struct., 2008, 889, 1–11 CrossRef CAS PubMed.
- K. Rościszewski, B. Paulus, P. Fulde and H. Stoll, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 7905–7910 CrossRef.
- M. Tsuge, S. Berski, R. Stachowski, M. Räsänen, Z. Latajka and L. Khriachtchev, J. Phys. Chem. A, 2012, 116, 4510–4517 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |