
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion intercalation dynamics of electrosynthesized mesoporous WO3 thin films studied by multi-scale coupled electrogravimetric methods†
Fatemeh
Razzaghi
ab,
Catherine
Debiemme-Chouvy
ab,
Françoise
Pillier
ab,
Hubert
Perrot
ab and
Ozlem
Sel
*ab
aSorbonne Universités, UPMC Univ Paris 06, UMR 8235, Laboratoire Interfaces et Systèmes Electrochimiques, F-75005, Paris, France. E-mail: ozlem.sel@upmc.fr; Fax: +33 144274074; Tel: +33 144279615
bCNRS, UMR 8235, LISE, F-75005, Paris, France
First published on 6th May 2015
Abstract
Mesoporous WO3 thin films were prepared electrochemically by using an ionic surfactant during the synthesis, and the electrochemical properties are investigated in comparison with their dense analogues. This report specifically highlights the suitability of a time resolved coupled electrogravimetric method to follow meticulously the ion intercalation/extraction phenomena which revealed the enhanced ion intercalation/extraction behavior of electrodeposited mesoporous WO3 thin films for diverse applications in energy storage and electrochromism. This methodology (electrochemical impedance spectroscopy (EIS) and its coupling with a fast quartz crystal microbalance (QCM)) has the ability to detect the contribution of the charged or uncharged species during the electrochemical processes, and to deconvolute the global EQCM responses into the anionic, cationic, and the free solvent contributions. Our study identifies the involvement of several charged species (Li+, Li+·H2O) in the compensation of charge, and H2O molecules indirectly contribute to the process in both dense and mesoporous WO3 thin films. Even a slight contribution of ClO4− ions was detected in the case of mesoporous analogues. The results of the study indicate that the transfer resistances of Li+ and Li+·H2O are decreased when the WO3 films are mesoporous. A more significant difference is observed for the larger and partially dehydrated Li+·H2O ions, suggesting that increased surface area and pore volume created by mesoporous morphology facilitate the transfer of larger charged species. The relative concentration changes of cations are also magnified in the mesoporous films. The final concentration variations are higher in mesoporous films than that in the dense analogues; ∼4 times and ∼10 times higher for Li+ and for Li+·H2O, respectively. To the best of our knowledge, an unambiguous identification of species other than desolvated cations (e.g. Li+ ions), the information on their transfer dynamics and quantification of the transferred species have never been reported in the literature to describe the charge compensation process in WO3 based electrodes.
1. Introduction
In recent years, the use of metal oxide thin films with nanoscale porosity for diverse electrochemical applications, e.g. energy storage, electrochromic devices, etc. has become a subject of growing interest.1–4 Among these materials, tungsten(VI) oxide (WO3) is a particular metal oxide which can offer a wide range of technological applications. It is a representative metal oxide of a group of chromogenic materials due to the coloration effects associated with various processes.2–7 Aside from its potential applications including Li-ion batteries,8–10 photoelectrochemical,11–13 solar,14,15 and fuel cells;16–19 WO3 has been extensively studied as a promising electrochromic material. Meanwhile, WO3 has already been integrated in low-voltage electrochromic devices for smart windows, which emphasizes the high technological relevance of this kind of material. Nevertheless, currently available devices still suffer from several shortcomings, such as quite long electrochromic response times.20 Thus, new developments and approaches to improve such systems may have a direct technological impact.Even though not all the underlying details of the mechanism of electrochromism are fully understood, the overall process generally involves the simultaneous insertion of cations and electrons into the inorganic matrix during the reduction step (W+6/W+5), and the subsequent formation of coloring centers (W+5).7,20,21 This reversible electrochemical insertion or intercalation of electrolyte ions into an electrode material is the fundamental operation principle of many electrochemical devices; therefore it is crucial to optimize the interactions between electrolyte ions and electrode materials.
For the improvement of the performance of metal oxide based electrodes, main attention has been attributed to their nanostructuration with various morphologies due to the unique properties and functionalities that can effectively be exploited in electrochemical devices.2–4,20–22 Various synthesis methods such as sol–gel,2,3,20 spray pyrolysis/sputtering,23,24 and hydrothermal synthesis,25,26 are commonly employed for the elaboration of WO3 leading to different morphologies. However, electrochemical synthesis remains to be a fast, simple, low-cost and low-temperature technique which may also lead to the formation of homogenous nanoscale films with desirable qualities.6,27–30 The incorporation of surfactant molecules in the electrogenerated film results in the production of thin nanostructured films by using potential-controlled self-assembly of surfactant–inorganic aggregates at solid–liquid interfaces.6 This approach manipulates surfactant–inorganic assemblies only in the thin interfacial region by electrochemically controlling surface interactions, which allows the formation of nanostructured films from dilute surfactant solutions. After deposition, surfactants can be easily removed from the pores by washing with a suitable alcohol, leading to the porous inorganic replicas of the surfactant phases.6 These nanostructures, with small sizes, and large surface area to volume ratios, are expected to facilitate the ion intercalation/extraction process. These improvements are often attributed to (i) a facilitated transfer and short diffusion length for ions transport, (ii) a high electrode/electrolyte contact area, and (iii) a better stress/strain management of the material during ion intercalation/extraction.
The morphology dependent performance and ion intercalation behaviour of electrochromic WO3 thin films have been investigated by several in situ or ex situ characterization techniques,31–35 including electrochemical and optical methods,5,20,22 wide-angle X-ray scattering combined with electrochemical studies,20 Raman spectroscopy,21 and X-ray photoelectron spectroscopy studying electrochromic materials before and after cation intercalation.5,36 However, none of these methods alone provide the information on the exact identification of the intercalated ionic species, their dynamics of transfer at the interfaces, as well as the role of electrolyte composition and the effect of ions solvation on the intercalation/extraction phenomena. The status of inserted ions is not a quite solved problem and the characterization of their transfer dynamics is not straightforward using conventional characterization tools. The main challenge is to find an eligible technique offering mechanistic solutions in a single body that can be used to study ion intercalation phenomena in situ and in contact with an electrolyte. There have been studies using quartz crystal microbalance (QCM) as an in situ gravimetric probe to study the insertion and/or electroadsorption of ions in metal oxide or carbon based electrodes.37–42 Conducting electrodes coated on the QCM resonator surface are used as working electrodes in an electrochemical cell, and thus, the electrodeposition process can be monitored. Materials of interest deposited are studied in terms of mass changes during the electrochemical processes. Santos et al. studied the Li+ ion intercalation/extraction behaviour of dense WO3 films in acetonitrile solution containing LiClO4 by QCM measurements during cyclic voltammetry (EQCM).37 The mass-to-charge ratios  were calculated from the EQCM data in LiClO4/acetonitrile electrolytes, which can be used to estimate the molar mass of the species
were calculated from the EQCM data in LiClO4/acetonitrile electrolytes, which can be used to estimate the molar mass of the species  inserted in the material. If the intercalation process is only due to the charge compensation of the valence variations of W sites (W+6/W+5) by Li+, one can expect to obtain a value matching to the equivalent weight of the lithium ions. However, none of the values obtained from the MCR was in the order of the equivalent weight of either ions (Li+, or ClO4−) or solvent molecules (acetonitrile) of the study. The equivalent weight values, higher than that of Li+ (7 g mol−1) were simply attributed to the acetonitrile molecules, accompanying the Li+ ions.37 A similar study by Vondrak et al. investigated the electrochemical insertion of H+, Li+, Na+ ions into thin layers of WO3 by EQCM in propylene carbonate. EQCM data of their work also detected molar mass values higher than that would correspond to the cation present in the electrolyte. Therefore, it was suggested that ions do not enter the space lattice alone but may be accompanied by propylene carbonate solvent molecules.38 These studies are significant contributions to highlight the complexity of the electrochromic system, and to show that simplified electrical charge compensation processes may not be realistic to describe the ion intercalation/extraction mechanisms in metal oxide based electrodes. However, the limitations of the technique appear here clearly. EQCM gives a global response corresponding in fact to several possible pathways such that ions, ions with solvation shells and even indirectly free solvent molecules can contribute to the electrochemical process. Additionally, ions may lose a part of their solvation to access to the sites in smaller nanopores.42 These possible pathways, and kinetic or dynamic aspects of the ion intercalation/extraction processes have never been characterized in the previous EQCM studies due to the limitations of the technique.
inserted in the material. If the intercalation process is only due to the charge compensation of the valence variations of W sites (W+6/W+5) by Li+, one can expect to obtain a value matching to the equivalent weight of the lithium ions. However, none of the values obtained from the MCR was in the order of the equivalent weight of either ions (Li+, or ClO4−) or solvent molecules (acetonitrile) of the study. The equivalent weight values, higher than that of Li+ (7 g mol−1) were simply attributed to the acetonitrile molecules, accompanying the Li+ ions.37 A similar study by Vondrak et al. investigated the electrochemical insertion of H+, Li+, Na+ ions into thin layers of WO3 by EQCM in propylene carbonate. EQCM data of their work also detected molar mass values higher than that would correspond to the cation present in the electrolyte. Therefore, it was suggested that ions do not enter the space lattice alone but may be accompanied by propylene carbonate solvent molecules.38 These studies are significant contributions to highlight the complexity of the electrochromic system, and to show that simplified electrical charge compensation processes may not be realistic to describe the ion intercalation/extraction mechanisms in metal oxide based electrodes. However, the limitations of the technique appear here clearly. EQCM gives a global response corresponding in fact to several possible pathways such that ions, ions with solvation shells and even indirectly free solvent molecules can contribute to the electrochemical process. Additionally, ions may lose a part of their solvation to access to the sites in smaller nanopores.42 These possible pathways, and kinetic or dynamic aspects of the ion intercalation/extraction processes have never been characterized in the previous EQCM studies due to the limitations of the technique.
Here an alternative characterization tool was proposed to overcome the limitations of the classical EQCM to study the ion intercalation/extraction mechanisms in metal oxide based electrodes. Specifically, the electrochromic behavior related to the cation intercalation/extraction in WO3 thin films was investigated by coupled time resolved characterization methods (fast QCM/electrochemical impedance spectroscopy). This method, so-called ac-electrogravimetry consists of in situ coupling of electrochemical impedance spectroscopy (EIS) and fast quartz crystal microbalance (QCM).43–48 Although QCM exists more or less routinely in laboratories these days, this coupling has been developed in a limited number of laboratories worldwide. The ac-electrogravimetry simultaneously measures the electrochemical impedance, ΔE/ΔI(ω), and the mass/potential transfer functions (Δm/ΔE(ω)) during a sinusoidal potential perturbation with a small amplitude applied to the modified electrode.43–48 The mass/potential transfer functions allow the change in mass due to a unit charge passing through the electrode/film/electrolyte interfaces to be determined. It provides the access to the relevant information on (i) the kinetics of species transferred at the solid/solution interfaces, and their transport in the bulk of the materials, (ii) the nature of these species as well as their relative concentration within the material. This coupling dominates over the limitations of QCM technique and has the ability to deconvolute the global mass variations provided by QCM measurements. Specifically, it detects the contribution of the charged or uncharged species and to identify anionic, cationic, and the free solvent contributions during various redox processes, and recently has been applied to study pseudo-capacitive charge storage mechanisms in Li-birnessite type MnO2 electrodes.48
In the present work, this coupled methodology is exploited to study the ion intercalation/extraction behaviour of mesoporous WO3 films. For their fabrication, an electrochemical pathway was chosen, so-called surfactant-assisted electrodeposition,6,27,29 which provides a facile, low temperature and rapid alternative to the mesoporous thin film synthesis. In the literature, cation intercalation/extraction studied by UV-vis spectroscopy during cyclic voltammetry measurement revealed that both amorphous and crystalline mesoporous films significantly improve the electrochromic response times due to the shortening of the mean diffusion path lengths.20,49 The other studies discussed that the crystalline materials increase the energy barrier for lithium ions, and thus, their electrochemical capacity may be inferior than their amorphous analogues.5,21,50 Here, we particularly focus on the amorphous materials rather than crystalline ones to emphasize its role in the electrochemical performance of WO3 films. The structure, morphology and the composition of the mesoporous electrochemically synthesized films were characterized, and compared with their dense counterparts. The electrochemical performance and ion intercalation/extraction mechanisms were studied in aqueous LiClO4 electrolytes by electrogravimetric methods (EQCM, and ac-electrogravimetry). Special attention was given to the poorly understood aspects, such as the nature of the ions involved in the charge compensation, solvation and the role of the electrolytes and the dynamic information of ions transfer at the electrode/electrolyte interfaces. It has been demonstrated herein that the ac-electrogravimetry responses of the electrodeposited WO3 thin films can serve as a gravimetric probe to study the complex Li+ intercalation/extraction mechanisms and to extract subtleties unreachable with classical tools.
2. Experimental
Chemicals
The following chemicals were used to prepare peroxotungstic acid (PTA) solutions employed in the electrodeposition process and the characterization of the resulting thin films: tungsten powder (99.99%, Aldrich Chem, USA), 30% w/w hydrogen peroxide (analytical reagent, Sigma-Aldrich Germany), propan-2-ol (99.7% Sigma-Aldrich, Germany), platinum black (99.97%, Sigma-Aldrich, USA), sodium dodecyl sulfate (SDS) (ACS reagent, 99.0% Sigma-Aldrich, Japan) and LiClO4 (98% Sigma-Aldrich, Japan).WO3 thin film synthesis
The electrodeposition solutions for the synthesis of dense and mesoporous WO3 thin films were prepared as described elsewhere.6,30 Briefly, 0.9 g W powder was dissolved in 30 ml of 30% H2O2 which takes about 4 hours for complete dissolution. A small portion of Pt black was then added to remove the excess of H2O2. This removal process can be accelerated by heating the solution up to 70 °C to obtain a pale-yellow solution of PTA. The solution was diluted to 50 mM with a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water and isopropanol mixture. For the mesoporous thin film synthesis, the solution also contained 0.05 M sodium dodecyl sulfate (SDS).
:50 water and isopropanol mixture. For the mesoporous thin film synthesis, the solution also contained 0.05 M sodium dodecyl sulfate (SDS).
Gold-patterned quartz substrates of 9 MHz (Temex, France) were used as working electrodes. A platinum grid and an Ag/AgCl (3 M KCl saturated with AgCl) served as counter electrode and reference electrode, respectively. The electrochemical WO3 synthesis was performed by potentiostatic electrodeposition at −0.5 V vs. Ag/AgCl (3 M KCl saturated with AgCl). During electrodeposition, a salt-bridge junction equipped with a porous glass frit on the end was used to prevent the reference electrode from being contaminated by the media and vice versa. The electrodeposition was performed by using a potentiostat (Autolab PGSTAT100) and the deposition process was monitored by quartz crystal microbalance measurements. Film area is 0.25 cm2 (gold electrode on the quartz resonator) and the film thickness was controlled by the electrodeposition time, typically for 15, 20 or 30 minutes. The SDS surfactant in the WO3 thin films was extracted from the inorganic matrix by washing with ethanol–water mixture before the electrogravimetric studies.
For high resolution transmission electron microscopy (HR-TEM) studies, WO3 thin films were also deposited on gold substrates (gold foil, 0.05 mm thickness, 99.95% purity (Goodfellow)) by surfactant assisted electrodeposition at −0.5 V for 30 min, followed by a thermal treatment at 450 °C for 2 h (heating rate of 5 °C min−1). The samples were scraped off from the gold substrates and ultrasonically dispersed in ethanol.
Structural characterization
The film morphology and the thickness were investigated by field emission gun scanning electron microscopy (FEG-SEM) (Zeiss, Supra 55). The elemental analysis was performed with an energy dispersive X-ray (EDX) detector associated to the FEG-SEM equipment. The oxidation state of the tungsten and composition of the films were determined by X-ray photoelectron spectroscopy (XPS) (on a VG ESCALAB 250i-XL spectrometer using monochromatic Al Kα radiation as the X-ray source). High resolution transmission Electron Microscopy (HR-TEM) analysis was performed using a JEOL 2010 UHR microscope operating at 200 kV equipped with a TCD camera.Characterization with coupled electrochemical methods (EQCM and ac-electrogravimetry)
The electrochemical synthesis was monitored by a lab-made QCM device providing the electrodeposited mass to be determined by frequency variation (Δfm) of the quartz crystal resonator based on the Sauerbrey equation.51 The (Δfm) can be converted into the mass change (Δm) of the quartz crystal by using the Sauerbrey equation: | (1) |
The classical EQCM measurements are based on the coupling of cyclic voltammetry (CV) with QCM measurements, and provide the information on the mass variations (calculated from eqn (1)) of the electrode during electrochemical processes.
For ac-electrogravimetry, a four-channel frequency response analyzer (FRA, Solartron 1254) and a potentiostat (SOTELEM-PGSTAT) are used. The QCM is used under dynamic regime, the modified working electrode (0.2 cm2) is polarized at a selected potential, and a sinusoidal small amplitude potential perturbation (50 mV rms) is superimposed. The microbalance frequency change, Δfm, corresponding to the mass response, Δm, of the modified working electrode is measured simultaneously with the ac response, ΔI, of the electrochemical system. The frequency range is between 63 kHz and 10 mHz. The resulting signals are sent to the four-channel FRA, which allowed the electrogravimetric transfer function,  , and the electrochemical impedance,
, and the electrochemical impedance,  , to be simultaneously obtained at a given potential.
, to be simultaneously obtained at a given potential.
The electrochemical experiments were performed in aqueous 0.5 M LiClO4 solution. The gold-patterned quartz substrates of 9 MHz (Temex, France) were used as working electrodes. A platinum grid and an Ag/AgCl (3 M KCl saturated with AgCl) served as the counter electrode and the reference electrode, respectively. During electrochemical tests, a salt-bridge junction equipped with a porous glass frit on the end was used to prevent the reference electrode from being contaminated by the media and vice versa.
3. Theory
The ac-electrogravimetry methodology and theoretical background were previously discussed by Gabrielli et al.43,44Briefly, ac-electrogravimetry consists of coupling of electrochemical impedance spectroscopy (EIS) with a fast quartz crystal microbalance (QCM) used in ac-mode. It allows the response in current,  , electrical transfer function, and in mass,
, electrical transfer function, and in mass,  mass-potential transfer function to be simultaneously obtained owing to a sinusoidal potential perturbation with a small amplitude (ΔE). The combination of such transfer functions provides the possibility of a fair separation of different electrochemical processes, which involves concomitantly the mass and charge changes.
mass-potential transfer function to be simultaneously obtained owing to a sinusoidal potential perturbation with a small amplitude (ΔE). The combination of such transfer functions provides the possibility of a fair separation of different electrochemical processes, which involves concomitantly the mass and charge changes.
The transfer of two cations (c1 and c2), and an anion (a) in the electroactive films incorporating three different and independent sites, P1, P2, and P3 during the redox reaction of the host material 〈P〉 where a single electronic transfer takes place can be expressed as:
 | (2) |
 | (3) |
 | (4) |
Under the effect of a sinusoidal potential perturbation with low amplitude, ΔE, imposed to the electrode/film/electrolyte system, sinusoidal fluctuations of concentration, ΔCi, are observed. In the present case, for electroactive metal oxide thin films, the change of the concentration, ΔCi, of each species (cation 1 (c1), cation 2 (c2), anion (a), and free solvent (s)) with potential ΔE can be calculated using eqn (5)–(8):
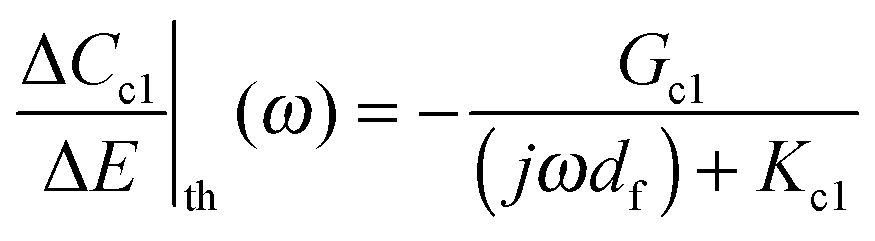 | (5) |
 | (6) |
 | (7) |
 | (8) |
 , where Ji stands for the flux of the species i crossing the film/electrolyte interface. More precisely, Ki is the kinetics rate of transfer and Gi is the inverse of the transfer resistance, Rti, of the species at the film/electrolyte interface (where i is the cation c1 or c2, the anion a, or the free solvent s). The ESI† Section B (eqn (S10)–(S14)) provides detailed information on these parameters.
, where Ji stands for the flux of the species i crossing the film/electrolyte interface. More precisely, Ki is the kinetics rate of transfer and Gi is the inverse of the transfer resistance, Rti, of the species at the film/electrolyte interface (where i is the cation c1 or c2, the anion a, or the free solvent s). The ESI† Section B (eqn (S10)–(S14)) provides detailed information on these parameters.
The concentration/potential transfer function (eqn (5)–(8)) permits us to obtain all the other theoretical transfer functions. Therefore, the following theoretical expressions are used to fit the experimental responses of electrochemical impedance, charge/potential, electrogravimetric and partial electrogravimetric transfer functions.
The charge/potential transfer function,  , is calculated for the insertion/expulsion of the two cations, c1 and c2 and an anion, a and by using the Faraday number, F, and the film thickness, df:
, is calculated for the insertion/expulsion of the two cations, c1 and c2 and an anion, a and by using the Faraday number, F, and the film thickness, df:
 | (9) |
 | (10) |
 , an interfacial capacitance must be added in parallel to the Faradaic impedance.
, an interfacial capacitance must be added in parallel to the Faradaic impedance.
The electrogravimetric transfer function,  , can be calculated theoretically, taking into account of the uncharged species contribution (solvent molecules in this case) through the two parameters Ks and Gs:
, can be calculated theoretically, taking into account of the uncharged species contribution (solvent molecules in this case) through the two parameters Ks and Gs:
 | (11) |
Partial mass/potential TFs are also estimated either by removing the c2 contribution, calculating  ; or the c1 contribution, calculating
; or the c1 contribution, calculating  ; or the anion contribution, calculating
; or the anion contribution, calculating  (ESI,† eqn (S23)–(S25)).
(ESI,† eqn (S23)–(S25)).
These theoretical expressions were used to fit the experimental responses of the electrochemical impedance,  , the charge/potential transfer function
, the charge/potential transfer function  , and the electrogravimetric transfer functions,
, and the electrogravimetric transfer functions,  . These results were compared with the experimental data which provided the key parameters (mi, Ki, Gi, and Rti) to be extracted.
. These results were compared with the experimental data which provided the key parameters (mi, Ki, Gi, and Rti) to be extracted.
4. Results and discussion
4.1 Synthesis, structure and morphology of dense and mesoporous thin films
Thin films of dense and mesoporous WO3 were synthesized on the gold electrode of a quartz resonator in a one-step potentiostatic electrodeposition method from solutions containing peroxotungstic acid (PTA) species (see Experimental section and ref. 6 and 30 for details). For the electrochemical synthesis of the mesoporous WO3 thin film, surfactant molecules (sodium dodecyl sulfate (SDS)) were added in the electrodeposition solution as templating agents. Typical current and microbalance frequency profiles, simultaneously measured on the quartz crystal during electrodeposition are presented in Fig. S1 (ESI†). The frequency variations (Δf) are converted to the corresponding mass variations (Δm) by using the Sauerbrey equation (eqn (1)). The mass variation profiles during potentiostatic electrodeposition for both dense and mesoporous films are presented in Fig. 1. The initial deposition current is higher for mesoporous thin films than that of dense WO3 thin films (Fig. 1). The deposition current density depends on both the sheet resistance of the working electrode and the electrical conductivity of the PTA electrolyte solution.53 Since identical substrates (gold coated quartz resonators) are used as working electrodes, the higher initial deposition current for the mesoporous WO3 deposit (Fig. 1B) is mainly due to the higher conductivity of the deposition solution resulted from the addition of the ionic surfactant molecules (SDS). In both cases, the deposition current initially decreases, finally approaching a steady value (Fig. 1), which is consistent with previous reports.53 The gold electrode is a conductor while the WO3 is a semiconductor and the contributions of the two components determine the relative amount of the deposition current decrease. The time for stabilization of the deposition current is higher for the films synthesized in the presence of SDS surfactant (Fig. 1B) than that of a dense film electrodeposition. In the absence of SDS templates, impingement/percolation probably takes place at relatively lower thickness of the WO3 deposits since larger grain formation may easily occur. But in the presence of SDS, the grain growth is limited due to the presence of SDS micellar structures on the gold electrode/solution interface. This may result in a slight delay in approaching a steady current value (Fig. 1B). Therefore, the differences in the deposition current profiles in Fig. 1 already give indications of the formation of different morphologies of WO3 thin films in the presence and absence of SDS templating agents. In other words, the mechanism of electrogeneration appears strongly dependent on the bath composition. | ||
| Fig. 1 Synthesis of WO3 thin films: typical electropotentiostatic deposition data and simultaneous QCM characterization (mass variation of the quartz crystal during electrodeposition) of (A) dense and (B) mesoporous WO3 thin films (deposited at −0.5 V vs. Ag/AgCl during 15 min). | ||
The characteristic frequency of the quartz resonator decreases as a function of the electrodeposition time (Fig. S1, ESI†) corresponding to a mass increase of the gold electrode of the quartz resonator. The mass variations calculated from the frequency variations are also presented in Fig. 1. The total deposited mass is higher for the films synthesized in the presence of SDS surfactant (Fig. 1B), indicating the incorporation of the surfactant molecules. These films are considered to be inorganic–organic hybrids composed of a WO3 inorganic matrix surrounding the SDS surfactant micelles, and they are extracted from the inorganic matrix in a subsequent step following the electrodeposition.
The morphology of the electrochemically synthesized WO3 thin films was characterized by FEG-SEM and HR-TEM. In the absence of the surfactant molecules, fairly dense thin films composed of sphere-like shape nanoparticles of WO3 are obtained, which is consistent with previous reports (Fig. 2).6,53 Similar morphologies are obtained for the mesoporous counterparts on the scale of FEG-SEM observations. The average film thickness is ∼300 nm under these experimental conditions (Fig. 2B), and it can be tailored by changing the electrodeposition time.
 | ||
| Fig. 2 FEG-SEM images showing the top view (A) and the cross-section (B) of an as deposited dense WO3 thin film (electrodeposited on the Au electrode of a quartz resonator at −0.5 V vs. Ag/AgCl during 20 min). | ||
The EDX analysis coupled with FEG-SEM observations indicates the presence of W and O in as-deposited dense thin films (Fig. S2, ESI†). A small contribution of Au also appears which is due to the gold electrode of the quartz crystal substrate. The EDX spectrum of films synthesized in the presence of SDS templates shows additional peaks (Fig. S3, ESI†) corresponding to Na element, indicating the successful incorporation of the SDS templates in the inorganic–organic hybrids. The contribution of Na peak is substantially decreased after the extraction of the SDS templates, signifying the facile removal of the SDS templates (Fig. S4, ESI†).
The pore morphology of the WO3 thin films electrodeposited in the presence of SDS templates was investigated by HR-TEM (Fig. 3) which revealed spherical mesopores of around 2–5 nm. The mesopores in the present films are randomly ordered and have a certain dispersity in their pore size but fairly homogeneous domains expand within the film. Compared with the mesoporous thin film preparations based on the evaporation induced self-assembly (EISA), surfactant assisted electrodeposition method employs more dilute surfactant solutions. The main principle is based on the potential-controlled self-assembly of surfactant–inorganic aggregates at solid–liquid interfaces.6 The concentration increase at the electrode surface leads to the formation of surfactant micelles, and the electrodeposition process occurs around these template structures. The subsequent removal of surfactant micelles leads to the formation of mesoporous films which are inverse replicas of the micellar structures, as shown in Fig. 3. Elemental composition analysis of the mesoporous WO3 thin films was also obtained by EDX analysis coupled with HR-TEM observations. The main contributions in the EDX spectrum in Fig. 3D are attributed to the presence of W and O elements (Cu peaks in Fig. 3D are originated from TEM grids).
 | ||
| Fig. 3 HR-TEM images (A–C) and the EDX pattern (D) of mesoporous WO3 thin films deposited by surfactant assisted electrodeposition (using SDS) at −0.5 V vs. Ag/AgCl for 30 min, sample is thermally treated at 450 °C for 2 h. | ||
To determine the exact composition and the oxidation state of the W species, as-deposited thin films were characterized by X-ray photoelectron spectroscopy (XPS) and the results are shown in Fig. 4. All peaks can be indexed to W and O elements (except the presence of Au peaks due to the substrate) indicating the high purity of as-synthesized sample. A fitting curve of the W 4f level is presented in Fig. 4B. The peaks at binding energies of 35.6 and 37.7 eV match greatly well with the reported values for the W6+ oxidation state, indicating that the synthesized WO3 is stoichiometric.36
 | ||
| Fig. 4 X-ray Photoelectron Spectroscopy (XPS) survey spectrum (A) and the corresponding W 4f core level spectrum (B) of as-prepared dense WO3 film electrosynthesized at −0.5 V vs. Ag/AgCl for 20 min. | ||
4.2 Electrochemical studies
The optical variation of the WO3 can be controlled by the application of a reversible voltage based on the double intercalation/extraction of cations and electrons into/out from the material. The general coloration process of WO3, which is a cathodic coloring electrochromic material is often presented in the literature by the following equation:| WO3(colorless) + xe− + xM+ ⇄ MxWO3(blue) | (12) |
The EQCM results of a dense and mesoporous WO3 film tested at room temperature in aqueous 0.5 M LiClO4 electrolytes are presented in Fig. 5. The CV curves show a capacitive response in the anodic region whereas the coloration reaction is observed in the potential range of −0.15 V to −0.5 V vs. Ag/AgCl. In agreement with the literature data under similar dynamic conditions, an asymmetric voltammogram is obtained with a broad anodic loop displaying an extraction of cationic species that is slower than its cathodic insertion at this scan rate (Fig. 5). The capacitive behaviour is somehow more pronounced in the CV response of the dense films compared with that for a mesoporous film (Fig. 5). This suggests a higher contribution of surface electroadsorption process, compared with the faradaic response (shown by blue arrows in Fig. 5) in the case of dense WO3 thin films. The cathodic and anodic current density of the mesoporous films (Fig. 5B) are higher than those observed for the dense films (Fig. 5A), which may indicate faster cation intercalation/extraction kinetics, as discussed in ref. 54. The current values measured are almost four times higher than that obtained for a dense film. The mass variations are also influenced by the presence of the mesopores (Fig. 5), and the QCM response is amplified (∼4 times) in the case of a mesoporous film. This enhanced behaviour can probably be attributed to the increased specific surface area and pore volume as a result of the mesopores providing a better accommodation of the electroactive sites and have an advantageous effect on the coloration process. The mass responses shown in the reduction branches (Fig. 5) correspond to the resonance frequency drops of the modified quartz resonators due to the mass increases of the films during reduction bias (coloration). In a reverse process, during oxidation bias (bleaching), the inserted species are expelled out from the film, resulting in a decrease of the electrode mass. The molecular mass of the species (cations and/or the other ionic/non-ionic species if present) involved in the charge compensation can be obtained roughly with further calculation. To do so,  function was determined
function was determined  from the EQCM data. Fig. 6 depicts the variation of
from the EQCM data. Fig. 6 depicts the variation of  values as a function of the applied potential for a dense and a mesoporous WO3 thin film. According to this calculation, an absolute molecular mass value of ∼5–6 g mol−1 was obtained for mesoporous films measured in 0.5 M aqueous solutions of LiClO4 with a scan rate of 50 mV s−1, which is close to the molecular weight of Li+. If there is strictly one species involved, the
values as a function of the applied potential for a dense and a mesoporous WO3 thin film. According to this calculation, an absolute molecular mass value of ∼5–6 g mol−1 was obtained for mesoporous films measured in 0.5 M aqueous solutions of LiClO4 with a scan rate of 50 mV s−1, which is close to the molecular weight of Li+. If there is strictly one species involved, the  function should be equivalent to the molecular mass of the species intercalated/extracted. However, for a complex electrochemical system, these values correspond to an average molecular weight related to the various species, and it is actually an average in terms of mass and kinetics. Therefore, at this point, one can arguably discuss that at this fast scan rate (50 mV s−1), Li+ ions are exclusively involved in the charge compensation process. However, it cannot guarantee the absence of any other species which may have different time constants and cannot be detected at this scan rate (such as heavier ions including ClO4−, hydrated forms of lithium ions, or free water molecules). Indeed, these results show the limitations of the classical EQCM technique. The EQCM gives a global response, and does not provide unambiguous information on which of the possible scenarios actually takes place. To investigate the subtleties that cannot be reachable by EQCM method, an ac-electrogravimetry study was performed on mesoporous WO3 films and compared with their dense analogues.
function should be equivalent to the molecular mass of the species intercalated/extracted. However, for a complex electrochemical system, these values correspond to an average molecular weight related to the various species, and it is actually an average in terms of mass and kinetics. Therefore, at this point, one can arguably discuss that at this fast scan rate (50 mV s−1), Li+ ions are exclusively involved in the charge compensation process. However, it cannot guarantee the absence of any other species which may have different time constants and cannot be detected at this scan rate (such as heavier ions including ClO4−, hydrated forms of lithium ions, or free water molecules). Indeed, these results show the limitations of the classical EQCM technique. The EQCM gives a global response, and does not provide unambiguous information on which of the possible scenarios actually takes place. To investigate the subtleties that cannot be reachable by EQCM method, an ac-electrogravimetry study was performed on mesoporous WO3 films and compared with their dense analogues.
 | ||
| Fig. 5 Cyclic voltammograms and mass responses obtained simultaneously during the electrochromic response of (A) dense and (B) mesoporous WO3 thin films deposited on the gold electrode of the quartz resonator. Measurements are in 0.5 M aqueous solution of LiClO4 with a scan rate of 50 mV s−1. | ||
 | ||
Fig. 6
 values corresponding to the average molecular weight of the species involved in the charge compensation as a function of the potential for a dense (A) and a mesoporous (B) WO3 film cycled in 0.5 M aqueous LiClO4 solutions (obtained from the reduction branch of the EQCM results at a scan rate of 50 mV s−1). values corresponding to the average molecular weight of the species involved in the charge compensation as a function of the potential for a dense (A) and a mesoporous (B) WO3 film cycled in 0.5 M aqueous LiClO4 solutions (obtained from the reduction branch of the EQCM results at a scan rate of 50 mV s−1). | ||
A. Ac-electrogravimetry study of dense and mesoporous WO3 thin films at −0.3 V vs. Ag/AgCl. Fig. 7 exhibits the electrochemical impedance spectroscopy,
 , of a dense and mesoporous thin film, at a selected potential, specifically at −0.3 V vs. Ag/AgCl. This potential value is presented because it corresponds to a situation where the electrodeposited film is in the reduced state. The
, of a dense and mesoporous thin film, at a selected potential, specifically at −0.3 V vs. Ag/AgCl. This potential value is presented because it corresponds to a situation where the electrodeposited film is in the reduced state. The  responses (Fig. 7A and B) have the usual shape, when dealing with an ion-blocking electrode, from which it is difficult to easily extract information. The low frequency trend was related to a parasitic response which is more pronounced in the mesoporous films (Fig. 7B), and was fitted by using eqn (S21) in the ESI.† The experimental transfer function and the fitted data from the model (see theoretical part, eqn (10) and the ESI,† eqn (S21)) are reported on the same graphs; a good agreement between the two sets of data is evident in term of shape and frequency distribution (Fig. 7). It should be noted that there is no evident part with a slope equals to 45° or below in the electrochemical impedance response; therefore, the rate limiting step is not the mass transport in the films or in the solution but rather ionic transfer between the solution and the film.40 The charge/potential transfer functions (TFs),
responses (Fig. 7A and B) have the usual shape, when dealing with an ion-blocking electrode, from which it is difficult to easily extract information. The low frequency trend was related to a parasitic response which is more pronounced in the mesoporous films (Fig. 7B), and was fitted by using eqn (S21) in the ESI.† The experimental transfer function and the fitted data from the model (see theoretical part, eqn (10) and the ESI,† eqn (S21)) are reported on the same graphs; a good agreement between the two sets of data is evident in term of shape and frequency distribution (Fig. 7). It should be noted that there is no evident part with a slope equals to 45° or below in the electrochemical impedance response; therefore, the rate limiting step is not the mass transport in the films or in the solution but rather ionic transfer between the solution and the film.40 The charge/potential transfer functions (TFs),  , (Fig. 8A and B) permit to separate the ionic contribution without any possibility to identify the ionic species involved. It should be indicated here that the contribution of the parasitic reaction, occurring at low frequencies, was removed for keeping only the ionic transfer response. A large and slightly suppressed loop was obtained for both dense and mesoporous WO3 thin films without a fair separation. The theoretical functions in Fig. 8 indicated that there are more than one charged species involved in both cases (Fig. 8A and B). Indeed, the time constants corresponding to each ionic transfer appear to be very close to each other. It is important to note that the
, (Fig. 8A and B) permit to separate the ionic contribution without any possibility to identify the ionic species involved. It should be indicated here that the contribution of the parasitic reaction, occurring at low frequencies, was removed for keeping only the ionic transfer response. A large and slightly suppressed loop was obtained for both dense and mesoporous WO3 thin films without a fair separation. The theoretical functions in Fig. 8 indicated that there are more than one charged species involved in both cases (Fig. 8A and B). Indeed, the time constants corresponding to each ionic transfer appear to be very close to each other. It is important to note that the  response of the mesoporous film is magnified (∼5 times) (Fig. 8B), compared to the response of a dense film (Fig. 8A). At this stage, it is impossible to distinguish between the anion and the cation contributions and to determine the nature of the ionic species involved. Nevertheless, the Ki and Gi constants were determined for two ions (for dense) and three ions (for mesoporous) WO3 thin films (see Table 1 for the Ki and Gi values) and were used in the following fittings.
response of the mesoporous film is magnified (∼5 times) (Fig. 8B), compared to the response of a dense film (Fig. 8A). At this stage, it is impossible to distinguish between the anion and the cation contributions and to determine the nature of the ionic species involved. Nevertheless, the Ki and Gi constants were determined for two ions (for dense) and three ions (for mesoporous) WO3 thin films (see Table 1 for the Ki and Gi values) and were used in the following fittings.
 | ||
Fig. 7 The electrochemical impedance,  for a dense (A) and a mesoporous (B) WO3 thin film, measured in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). for a dense (A) and a mesoporous (B) WO3 thin film, measured in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). | ||
 | ||
Fig. 8 Charge/potential transfer functions,  for a dense (A) and mesoporous (B) WO3 thin film measured in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). for a dense (A) and mesoporous (B) WO3 thin film measured in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). | ||
| M i (g mol−1) | Species identification | K i × 10+5 (s cm−1) | G i × 10+6 (mol s−1 cm−2 V−1) |
|
|
|
|---|---|---|---|---|---|---|
| Dense WO3 | ||||||
| c2 (cation 2) | 7 + 18 | Li+·H2O | 5.2 ± 0.5 | 0.01 ± 0.001 | 1175.6 | 1.1 × 10−4 |
| s (solvent) | 18 | H2O | 57.8 ± 6 | 1.96 ± 0.2 | 5.3 | 10.7 × 10−4 |
| c1 (cation 1) | 7 | Li+ | 179.1 ± 18 | 2.69 ± 0.3 | 3.9 | 3.9 × 10−4 |
| Mesoporous WO3 | ||||||
| c2 (cation 2) | 7 + 18 | Li+·H2O | 8.0 ± 0.8 | 0.18 ± 0.02 | 57.5 | 18.6 × 10−4 |
| s (solvent) | 18 | H2O | 33.0 ± 3.3 | 1.32 ± 0.1 | 7.9 | 15.6 × 10−4 |
| c1 (cation 1) | 7 | Li+ | 67.4 ± 6.7 | 4.10 ± 0.4 | 2.5 | 12.7 × 10−4 |
| a (anion) | 96 | ClO4− | 2.8 ± 0.3 | −0.02 ± 0.002 | 592.2 | −1.6 × 10−4 |


Then, to identify and clarify the contribution of the charged and also the uncharged species (i.e. free electrolyte molecules), the mass/potential transfer functions,  are scrutinized. In the mass/potential TF
are scrutinized. In the mass/potential TF  , one big loop appears in the third quadrant at medium and low frequencies (MF and LF). This loop can be attributed to either one species or to two species where their time constants are not sufficiently different from each other. The loops in the third quadrant are characteristic for cation contributions or free solvent molecules in the same flux direction. Another contribution also appears in Fig. 9B (for the mesoporous WO3 thin film) at very low frequencies in the fourth quadrant (either anions or water molecules with opposite flux direction compared to cations) highlighting the challenge in obtaining an exact identification of these two or three loops. Therefore, several configurations were tested using theoretical functions (eqn (11)) to determine the exact contribution of each species.
, one big loop appears in the third quadrant at medium and low frequencies (MF and LF). This loop can be attributed to either one species or to two species where their time constants are not sufficiently different from each other. The loops in the third quadrant are characteristic for cation contributions or free solvent molecules in the same flux direction. Another contribution also appears in Fig. 9B (for the mesoporous WO3 thin film) at very low frequencies in the fourth quadrant (either anions or water molecules with opposite flux direction compared to cations) highlighting the challenge in obtaining an exact identification of these two or three loops. Therefore, several configurations were tested using theoretical functions (eqn (11)) to determine the exact contribution of each species.
 | ||
Fig. 9 The mass/potential transfer functions,  , for a dense (A) (involving three species, specifically Li+, Li+·H2O and H2O) and a mesoporous WO3 thin films (B) (involving four species, specifically Li+, Li+·H2O, ClO4− and H2O), respectively. The measurements are performed in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). , for a dense (A) (involving three species, specifically Li+, Li+·H2O and H2O) and a mesoporous WO3 thin films (B) (involving four species, specifically Li+, Li+·H2O, ClO4− and H2O), respectively. The measurements are performed in aqueous 0.5 M LiClO4 electrolyte at −0.3 V vs. Ag/AgCl (both experimental and theoretical curves are given). | ||
The mass response of a dense WO3 thin film was fitted by considering three species cation 1 (c1 = Li+), cation 2 (c2 = Li+·H2O) and free solvent molecules (s = H2O) (Fig. 9A). In the case of a mesoporous WO3 film, a similar response is obtained, but with an additional contribution from another charged species, the anion (a = ClO4−). They appear characteristically in the fourth quadrant. Here, they are the slowest species transferred in the mesoporous films as they are detected at lower frequency region in Fig. 8 or 9B. It is important to note that eqn (11) involves the molecular weight of the ionic and/or nonionic species, providing their identification by their molecular weight. Therefore, cations or their hydrated forms can be detected with an estimation of the hydration level (e.g. cation 1 (c1 = Li+), cation 2 (c2 = Li+·nH2O n = 1)). Since the bulk hydration numbers for Li+ ions is high, n ∼ 7–8,55 the cation 2 (c2 = Li+·nH2O n = 1) detected in our study can be considered as partially dehydrated. The contribution of various species to electrochemical modulation with different kinetics of transfer has already been observed in the earlier work of Hillman et al. on nickel hydroxide thin films by combining probe beam deflection (PBD) technique and EQCM.56–58
The partial mass/potential TFs were analyzed to validate our previous hypothesis involving three and four different species for dense and mesoporous films, respectively. Partial mass/potential TFs are estimated for dense films (not shown), for example by removing the c2 contribution, calculating  or by removing the c1 contribution, calculating
or by removing the c1 contribution, calculating  as only three species are involved in the case of a dense film. The partial TFs for mesoporous films were also analyzed, specifically
as only three species are involved in the case of a dense film. The partial TFs for mesoporous films were also analyzed, specifically  (Fig. S5, ESI†) and all exhibited a good agreement between the theoretical and experimental data. These partial mass/potential TFs provide a crosscheck for validating the hypothesis involving multiple species and for obtaining a better separation of the various contributions. The Ki and the Gi constants are determined by fitting the experimental data using the theoretical functions for
(Fig. S5, ESI†) and all exhibited a good agreement between the theoretical and experimental data. These partial mass/potential TFs provide a crosscheck for validating the hypothesis involving multiple species and for obtaining a better separation of the various contributions. The Ki and the Gi constants are determined by fitting the experimental data using the theoretical functions for  (eqn (9)–(11)) with the criteria of achieving a good agreement between the experimental and theoretical data (strictly for all of the TFs, including partial mass/potential TFs (eqn (S23)–(S25), ESI†)). The equivalent weight of the charged and uncharged species is determined by the electrogravimetric TF,
(eqn (9)–(11)) with the criteria of achieving a good agreement between the experimental and theoretical data (strictly for all of the TFs, including partial mass/potential TFs (eqn (S23)–(S25), ESI†)). The equivalent weight of the charged and uncharged species is determined by the electrogravimetric TF,  which provides the identification of the species (Table 1). The Gi parameters permit to calculate the resistance (Rti) of the transfer for each species (eqn (S13), ESI†). The kinetic constants (Ki) obtained from ac-electrogravimetry at −0.3 V vs. Ag/AgCl reveal the transfer rates of species at the electrode/electrolyte interfaces. Both for dense and mesoporous WO3 thin films: (i) Li+ species contribute at high frequencies (the fastest species), (ii) free water molecules appear at the intermediate frequencies, and (iii) Li+·H2O appears at low frequencies (the slowest species). In the case of mesoporous WO3 thin films, an additional contribution is observed at the lowest frequency range corresponding to the anions transfer (ClO4−) and their transfer kinetics is slower than cationic species and free water molecules (see values in Table 1). The Gi values determined from the ac-electrogravimetry study are related to the transfer resistance,
which provides the identification of the species (Table 1). The Gi parameters permit to calculate the resistance (Rti) of the transfer for each species (eqn (S13), ESI†). The kinetic constants (Ki) obtained from ac-electrogravimetry at −0.3 V vs. Ag/AgCl reveal the transfer rates of species at the electrode/electrolyte interfaces. Both for dense and mesoporous WO3 thin films: (i) Li+ species contribute at high frequencies (the fastest species), (ii) free water molecules appear at the intermediate frequencies, and (iii) Li+·H2O appears at low frequencies (the slowest species). In the case of mesoporous WO3 thin films, an additional contribution is observed at the lowest frequency range corresponding to the anions transfer (ClO4−) and their transfer kinetics is slower than cationic species and free water molecules (see values in Table 1). The Gi values determined from the ac-electrogravimetry study are related to the transfer resistance,  which can explain the ease or the difficulty of the transfer of the ionic or nonionic species at the electrode/electrolyte interface. The transfer resistance Rti values follow the order which is the inverse of that observed for the kinetics parameters (Ki) (Table 1). Specifically, for dense films, it pursues the order: Rti(Li+·H2O) > Rti(H2O) > Rti(Li+), and for mesoporous films the same order persists: Rti(ClO4−) > Rti(Li+·H2O) > Rti(H2O) > Rti(Li+), except the presence of additional ClO4− anions exhibiting the highest transfer resistance.
which can explain the ease or the difficulty of the transfer of the ionic or nonionic species at the electrode/electrolyte interface. The transfer resistance Rti values follow the order which is the inverse of that observed for the kinetics parameters (Ki) (Table 1). Specifically, for dense films, it pursues the order: Rti(Li+·H2O) > Rti(H2O) > Rti(Li+), and for mesoporous films the same order persists: Rti(ClO4−) > Rti(Li+·H2O) > Rti(H2O) > Rti(Li+), except the presence of additional ClO4− anions exhibiting the highest transfer resistance.
There are two cationic species (Li+ and Li+·H2O) detected in both dense and mesoporous thin films. The kinetic parameters and resistance values for their interfacial transfer can be compared in Table 1. One of the most remarkable differences is that the Rti(Li+·H2O) value for dense films is much higher than that for a mesoporous film. This strongly suggests that the transfer of hydrated lithium species (Li+·H2O) at the interfaces is much easier in mesoporous WO3 thin films than in dense counterparts. Thus, the transfer of larger hydrated lithium species (Li+·H2O) is favored when WO3 thin films are mesoporous. Based on the Ki values, the Li+ species are transferred slightly slower in the case of mesoporous films but their transfer resistance is in the same order of magnitude, or even slightly lower than that in dense films. These results indicate that mesoporous films facilitate both Li+ and Li+·H2O transfer compared to dense WO3 thin films. The ClO4− anions contribution was detected in mesoporous films, but their transfer is slow and more difficult compared to other species.
Another important parameter to compare is the  value which can be considered as the quantity of the species transferred per potential unit at low frequencies of modulation. In other words, it is related to the capability to store charged or uncharged species in a particular material. It is evident that the quantity of the cationic species transferred per potential unit is much higher in mesoporous WO3 thin films than in dense films. For the dense films, the higher capacitance related to the cations is obtained with Li+. On the contrary, for mesoporous films hydrated Li+ gives the highest values (Table 1). The contribution of anions which is only detected for mesoporous films, is rather small based on the quantity transferred per potential unit (Table 1). The presence of large ClO4− anions, although in small quantity, is likely related to the mesoporosity of the WO3 thin films. Probably due to its large ionic radius, ClO4− anions cannot be involved in the charge compensation process of a dense film.
value which can be considered as the quantity of the species transferred per potential unit at low frequencies of modulation. In other words, it is related to the capability to store charged or uncharged species in a particular material. It is evident that the quantity of the cationic species transferred per potential unit is much higher in mesoporous WO3 thin films than in dense films. For the dense films, the higher capacitance related to the cations is obtained with Li+. On the contrary, for mesoporous films hydrated Li+ gives the highest values (Table 1). The contribution of anions which is only detected for mesoporous films, is rather small based on the quantity transferred per potential unit (Table 1). The presence of large ClO4− anions, although in small quantity, is likely related to the mesoporosity of the WO3 thin films. Probably due to its large ionic radius, ClO4− anions cannot be involved in the charge compensation process of a dense film.
The transfer kinetics and the resistance of free water molecules are somehow similar to those of Li+ species in both dense and mesoporous films, suggesting that these are the water molecules accompanying the transfer of Li+ species most likely due to the electrodragging (Table 1). The quantity of the water molecules transferred per potential unit is slightly higher for mesoporous films, which is in agreement with the increased pore volume.
The results obtained from the measurements at E = −0.3 V vs. Ag/AgCl, first of all indicate that there are other species than only Li+ ions; such as hydrated lithium ions (Li+·H2O), free electrolyte molecules (water in the present case); or even anionic species present in the electrolyte may contribute to the charge compensation in the electrochromic process of WO3 thin films. In the following, the kinetic parameters (Ki) and the transfer resistance (Rti) of the species are investigated as a function of the applied potential.
B. Ac-electrogravimetry study of dense and mesoporous WO3 thin films as a function of potential. Fig. 10 shows the evolution of the kinetic parameters (Ki) and the transfer resistance (Rti) of the species as a function of the potential applied. This potential range corresponds to the conditions where the intercalation/extraction process of the WO3 thin films is observed. Based on the Ki values presented in Fig. 10A and B, Li+ ions are persistently the fastest species transferred both in dense and mesoporous WO3 thin films.
 | ||
| Fig. 10 Evolution of kinetic parameters, Ki, for a dense (A) and mesoporous (B) WO3 thin films, and the comparative curve of the transfer resistance values (Rti = 1/FGi (Ω cm2)) for cationic species in dense and mesoporous WO3 thin films, Li+ ions (C), and Li+·H2O ions (D), presented as a function of applied potential. | ||
It is obvious that there are two different cation populations (Li+ and Li+·H2O) for both dense and mesoporous films at all the potentials studied. Compared to the dense films, the kinetics of transfer for Li+ ions is slower in the mesoporous films and this could be related to the morphology of the films. The hydrated lithium ions Li+·H2O in the electrolyte lose their hydration shell and get transferred at the interface, (bulk electrolyte/dense WO3 film interface). But if this transfer happens at an electrode/electrolyte interface of a confined pore wall (as in the case of a mesoporous WO3 film), the dehydration of the hydrated lithium ions in a small confined space may occur slower than that may happen in the bulk of the electrolyte. Therefore, one can arguably discuss that the different dehydration kinetics may be responsible for the differences in the Ki(Li+)meso and Ki(Li+)dense values shown in Fig. 10. The transfer kinetics of the larger, partially dehydrated Li+·H2O is in the same order of magnitude in both mesoporous and dense films (Fig. 10A and B). The transfer kinetics of free water molecules are somehow close to the values of the Li+ ions in Fig. 10A and B. Additionally, these water molecules have the same flux directions with the cations, suggesting that they might be the water molecules accompanying the transfer of Li+ species most likely due to the electrodragging. At all potential values studied, independent from the film morphology there are two types of cations, specifically fully dehydrated and partially dehydrated lithium species. However, the transfer resistance values reveal significant differences between mesoporous and dense WO3 thin films (Fig. 10C and D). Both Li+ and Li+·H2O have lower transfer resistance, indicating the ease of their transfer at the electrode/electrolyte interfaces when the WO3 films are mesoporous. A more significant difference is observed for the larger and partially dehydrated Li+·H2O ions, suggesting that increased surface area and pore volume, created by the mesoporous morphology, favor the larger charged species transfer.
To quantify the role of each species,  has been estimated as a function of the applied potential. The integration of the
has been estimated as a function of the applied potential. The integration of the  against potential gives the insertion isotherm. Fig. 11 presents the relative concentration change, (Ci − C0) for Li+, Li+·H2O, H2O and ClO4− species for dense (Fig. 11A) and mesoporous WO3 (Fig. 11B) thin films. A significant difference is observed in the relative concentration change of the species when WO3 thin films are mesoporous. Both the (Ci − C0) values for Li+ and Li+·H2O cations are magnified for mesoporous films. It is important to note that the kinetics of transfer values for Li+ species were higher (faster kinetics) in dense films compared to that in mesoporous films, and their ease of transfer was in the same order of magnitude (Fig. 10A–C). This point underlines the differences between kinetics and thermodynamics related to the transferred species. However, the final concentration variation is ∼4 times higher in mesoporous films than that in the dense films. Despite of the differences in their dynamics of transfer, the final concentration variations are magnified in the mesoporous WO3 thin films. It should be noticed that these subtleties are underlined for the very first time for both dense and mesoporous WO3 films.
against potential gives the insertion isotherm. Fig. 11 presents the relative concentration change, (Ci − C0) for Li+, Li+·H2O, H2O and ClO4− species for dense (Fig. 11A) and mesoporous WO3 (Fig. 11B) thin films. A significant difference is observed in the relative concentration change of the species when WO3 thin films are mesoporous. Both the (Ci − C0) values for Li+ and Li+·H2O cations are magnified for mesoporous films. It is important to note that the kinetics of transfer values for Li+ species were higher (faster kinetics) in dense films compared to that in mesoporous films, and their ease of transfer was in the same order of magnitude (Fig. 10A–C). This point underlines the differences between kinetics and thermodynamics related to the transferred species. However, the final concentration variation is ∼4 times higher in mesoporous films than that in the dense films. Despite of the differences in their dynamics of transfer, the final concentration variations are magnified in the mesoporous WO3 thin films. It should be noticed that these subtleties are underlined for the very first time for both dense and mesoporous WO3 films.
 | ||
| Fig. 11 Evolution of the relative concentration, Ci − C0, of each species over the potential applied, for a dense (A) and a mesoporous (B) WO3 thin film, measured in 0.5 M LiClO4 aqueous electrolyte. | ||
The (Ci − C0) values for Li+·H2O present a drastic difference between the two films, and it is ∼10 times higher in mesoporous films compared to the dense WO3. Their transfer resistance at the interfaces was also significantly smaller in mesoporous films (Fig. 10D), indicating that these large and partially dehydrated Li+·H2O species are much easier to be transferred. Thus, Li+·H2O ions are easily transferred in higher quantities when WO3 films are mesoporous. The (Ci − C0) values for water molecules are in the same order of magnitude, or no significant difference is observed. The (Ci − C0) value for ClO4− anions is low compared with the cationic species, thus only a small quantity is transferred. Their kinetics of transfer was also slow and difficult as it was shown in Table 1.
Overall, the contribution of the charged species is certainly magnified in mesoporous WO3 films compared to the dense films. This qualitative and quantitative study of ionic and nonionic species contribution in the charge compensation process, together with dynamic information of their interfacial transfer further proves the advantageous nature of nanostructuration of WO3 films for potential applications. For most of these application (e.g. in electrochromism), it is important to have a fast switching during the coloration/decoloration process which is related to the charge compensation of the redox reaction between W6+/W5+. The beneficial aspect of mesoporous WO3 films is more likely related to the better accommodation of both Li+ and Li+·H2O cations. Particularly, hydrated lithium species Li+·H2O are transferred rapidly and easily at the mesoporous electrode/electrolyte interfaces, and their concentration variations in the electrode is significantly higher with respect to that in their dense analogues.
5. Conclusions
Mesoporous WO3 thin films were synthesized by surfactant assisted electrodeposition method. The idea of using an ionic surfactant (SDS) during the electrochemical preparation of WO3 films led to the creation of ∼5 nm spherical mesopores which are templated by the corresponding spherical micelles. The dense analogues were also electrochemically synthesized under similar conditions but in the absence of SDS templates.The electrochromic behavior related to the cation intercalation/extraction in amorphous mesoporous and dense WO3 thin films was investigated by coupled time resolved characterization methods (fast QCM/electrochemical impedance spectroscopy). The chemical nature and the role of each species (ions, solvated ions, and free solvent molecules), directly or indirectly involved in the electrochemical processes were estimated for the very first time.
Our study identifies the involvement of several charged species (Li+, Li+·H2O) in the charge compensation, and H2O molecules indirectly contribute to the process in both dense and mesoporous WO3 thin films. Even a slight contribution of ClO4− ions was detected in the case of mesoporous analogues.
Compared to the dense films, the kinetics of transfer for Li+ ions is slower in the mesoporous films. This surprising result can be attributed to the transfer of ions occurring at different sites in the films. This transfer probably happens at an electrode/electrolyte interface of a confined pore wall (as in the case of a mesoporous WO3 film), the dehydration of the hydrated lithium ions in a small confined space may occur slower than that may happen in the bulk of the electrolyte. Therefore, the different dehydration kinetics may be responsible for the differences in the Ki(Li+)meso and Ki(Li+)dense. The transfer kinetics of the larger, partially dehydrated Li+·H2O is in the same order of magnitude in both mesoporous and dense films, indicating that these larger ions interact with the surface or close to surface sites of the mesoporous WO3 films.
Contrary to the kinetics of transfer, both Li+ and Li+·H2O have lower transfer resistance, indicating the ease of their transfer at the electrode/electrolyte interfaces when the WO3 films are mesoporous. A more significant difference is observed for the larger and partially dehydrated Li+·H2O ions, suggesting that increased surface area and pore volume created by mesoporous morphology facilitate the larger charged species transfer.
The relative concentration changes of Li+ and Li+·H2O cations are also magnified for the mesoporous films. The final concentration variation for Li+ species is ∼4 times higher in mesoporous films than that in the dense films. For Li+·H2O, this value is ∼10 times higher in mesoporous films compared to the dense WO3.
This qualitative and quantitative study of ionic and nonionic species contribution in the charge compensation process, together with dynamic information of their interfacial transfer further proves the advantageous nature of nanostructuration of WO3 films for potential applications. The results of the study indicate that the transfer resistances of the cations are much lower when the WO3 films are mesoporous. This observation is probably in agreement with the previous reports in the literature indicating a fast switching during the coloration/decoloration process of the mesoporous films. Another beneficial aspect of mesoporous WO3 films is related to the better accommodation of both Li+ and Li+·H2O cations. Particularly, hydrated lithium species Li+·H2O are transferred rapidly and easily at the mesoporous electrode/electrolyte interfaces, and their concentration variations in the electrode is significantly higher with respect to that occurs in their dense analogues. To the best of our knowledge, an unambiguous identification of species other than Li+ ions, the information on their transfer dynamics and quantification of the transferred species have never been reported in the literature.
This study particularly focuses on the investigation of amorphous mesoporous and dense WO3 thin films. The fact that crystalline materials increase the energy barrier for lithium ions, and thus their electrochemical capacity may be inferior than their amorphous analogues has been discussed in the literature.21,50 The aspect of crystallinity of the films can also be studied when WO3 thin films thermally treated at different temperatures, thus possessing different degree of crystallinity. However, such an electrogravimetric study requires the utilization of the resonators as substrates that have better thermal resistance than quartz crystals, and will be the subject of a further study.
Overall, these results are exciting and the combination of fast QCM with electrochemical impedance spectroscopy offer a great opportunity for better understanding of the ion intercalation/extraction dynamics in porous materials which is not possible with single EQCM measurements. Therefore, the establishment of the ac-electrogravimetry characterization in the nanostructured, ion-insertion materials is significant for designing optimized materials for the next generation electrochromic and energy storage devices.
Acknowledgements
Ms Sandra Casale is acknowledged for the HR-TEM measurements.References
- W. Gomes, D. Araujo, A. Carvalho, S. Campana-Filho and F. Huguenin, J. Phys. Chem. C, 2013, 117, 16774 CAS.
- R. Ostermann and B. Smarsly, Nanoscale, 2009, 1, 266 RSC.
- C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682 CrossRef CAS.
- W. Wang, Y. Pang and S. Hodgson, J. Mater. Chem., 2010, 20, 8591 RSC.
- D. Dalavi, R. Devan, R. Patil, R. Patil, Y. Ma, S. Sadale, I. Kim, J. Kim and P. Patil, J. Mater. Chem. C, 2013, 1, 3722 RSC.
- S. Baeck, K. Choi, T. Jaramillo, G. Stucky and E. McFarland, Adv. Mater., 2003, 15, 1269 CrossRef CAS PubMed.
- S. K. Deb, Philos. Mag., 1973, 27, 801 CrossRef CAS PubMed.
- H. Wei, X. Yan, S. Wu, Z. Luo, S. Wei and Z. Guo, J. Phys. Chem. C, 2012, 116, 25052 CAS.
- M. Sasidharan, N. Gunawardhana, M. Yoshio and K. Nakashima, Nano Energy, 2012, 1, 503 CrossRef CAS PubMed.
- W. Li and Z. Fu, Appl. Surf. Sci., 2010, 256, 2447 CrossRef CAS PubMed.
- J. C. Hill and K. Choi, J. Phys. Chem. C, 2012, 116, 7612 CAS.
- V. Cristino, S. Caramori, R. Argazzi, L. Meda, G. Marra and C. Bignozzi, Langmuir, 2011, 27, 7276 CrossRef CAS PubMed.
- S. Baeck, K. Choi, T. Jaramillo, G. Stucky and E. McFarland, Adv. Mater., 2003, 15, 1269 CrossRef CAS PubMed.
- H. Zheng, Y. Tachibana and K. Kalantar-zadeh, Langmuir, 2010, 26, 19148 CrossRef CAS PubMed.
- K. Hara, Z. Zhao, Y. Cui, M. Miyauchi, M. Miyashita and S. Mori, Langmuir, 2011, 27, 12730 CrossRef CAS PubMed.
- C. Laberty-Robert, K. Valle, F. Pereira and C. Sanchez, Chem. Soc. Rev., 2011, 40, 961 RSC.
- W. Zhao, Y. Yang and H. Zhang, Electrochim. Acta, 2013, 99, 273 CrossRef CAS PubMed.
- A. J. Martin, A. M. Chaparro and L. Daza, J. Power Sources, 2011, 196, 4187 CrossRef CAS PubMed.
- Q. Chen, J. Li, X. Li, K. Huang, B. Zhou, W. Cai and W. Shangguan, Environ. Sci. Technol., 2012, 46, 11451 CrossRef CAS PubMed.
- S. Sallard, T. Brezesinski and B. M. Smarsly, J. Phys. Chem. C, 2007, 111, 7200 CAS.
- D. Chatzikyriakou, N. Krins, B. Gilbert, P. Colson, J. Dewalque, J. Denayer, R. Cloots and C. Henrist, Electrochim. Acta, 2014, 137, 75 CrossRef CAS PubMed.
- R. S. Vemuri, K. K. Bharathi, S. K. Gullapalli and C. V. Ramana, ACS Appl. Mater. Interfaces, 2010, 2, 2623 CAS.
- L. M. Bertus and A. Duta, Ceram. Int., 2012, 38, 2873 CrossRef CAS PubMed.
- L. M. Bertus, C. Faure, A. Danine, C. Labrugere, G. Campet, A. Rougier and A. Duta, Mater. Chem. Phys., 2013, 140, 49 CrossRef CAS PubMed.
- J. Yin, H. Cao, J. Zhang, M. Qu and Z. Zhou, Cryst. Growth Des., 2013, 13, 759 CAS.
- J. Wang, E. Khoo, P. Lee and J. Ma, J. Phys. Chem. C, 2008, 112, 14306 CAS.
- S. Baeck, T. Jaramillo, G. D. Stucky and E. W. McFarland, Nano Lett., 2002, 8, 831 CrossRef.
- N. Tacconi, C. R. Chenthamarakshan, K. Rajeshwar, T. Pauporte and D. Lincot, Electrochem. Commun., 2003, 5, 220 CrossRef.
- E. A. Meulenkamp, J. Electrochem. Soc., 1997, 144, 1664 CrossRef CAS PubMed.
- W. Kwong, A. Nakaruk, P. Koshy and C. Sorrell, J. Phys. Chem. C, 2013, 117, 17766 CAS.
- L. Bobics, L. Sziraki and G. G. Lang, Electrochem. Commun., 2008, 10, 283 CrossRef CAS PubMed.
- L. Kondrachova, R. May, C. Cone, D. Vanden Bout and K. J. Stevenson, Langmuir, 2009, 25, 2508 CrossRef CAS PubMed.
- P. R. Bueno, R. C. Faria, C. O. Avellaneda, E. R. Leite and L. O. S. Bulhoes, Solid State Ionics, 2003, 158, 415 CrossRef CAS.
- N. Galiote, R. Parreira, J. Rosolen and F. Huguenin, Electrochem. Commun., 2010, 12, 733 CrossRef CAS PubMed.
- J. Kim, D. Tryk, T. Amemiya, K. Hashimoto and A. Fujishima, J. Electroanal. Chem., 1997, 435, 31 CrossRef CAS.
- X. Chang, S. Sun, L. Dong, Y. Donga and Y. Yina, RSC Adv., 2014, 4, 8994 RSC.
- V. Fernandes, M. Santos and L. Bulhões, Thin Solid Films, 2007, 515, 7155 CrossRef CAS PubMed.
- J. Vondrák, M. Sedlaříková, J. Velická, P. pičák, V. Svoboda and J. Kazelle, J. Solid State Electrochem., 2007, 11, 1459 CrossRef.
- O. Bohnke, B. Vuillemin, C. Gabrielli, M. Keddam, H. Perrot, H. Takenouti and R. Torresi, Electrochim. Acta, 1995, 40, 2755 CrossRef CAS.
- O. Bohnke, B. Vuillemin, C. Gabrielli, M. Keddam and H. Perrot, Electrochim. Acta, 1995, 40, 2765 CrossRef CAS.
- P. R. Bueno, T. R. C. Faria and L. O. S. Bulhoes, Solid State Ionics, 2005, 176, 1175 CrossRef CAS PubMed.
- W. Tsai, P. Taberna and P. Simon, J. Am. Chem. Soc., 2014, 136, 8722 CrossRef CAS PubMed.
- J. J. García-Jareño, D. Gimenez-Romero, F. Vicente, C. Gabrielli, M. Keddam and H. Perrot, J. Phys. Chem. B, 2003, 107, 11321 CrossRef.
- C. Gabrielli, J. Jareno, M. Keddam, H. Perrot and F. Vicente, J. Phys. Chem. B, 2002, 106, 3192 CrossRef CAS.
- L. To Thi Kim, O. Sel, C. Debiemme-Chouvy, C. Gabrielli, C. Laberty-Robert, H. Perrot and C. Sanchez, Electrochem. Commun., 2010, 12, 1136 CrossRef PubMed.
- O. Sel, L. To Thi Kim, C. Debiemme-Chouvy, C. Gabrielli, C. Laberty-Robert and H. Perrot, Langmuir, 2013, 29, 13655 CrossRef CAS PubMed.
- O. Sel, L. To Thi Kim, C. Debiemme-Chouvy, C. Gabrielli, C. Laberty-Robert, H. Perrot and C. Sanchez, J. Electrochem. Soc., 2010, 157, F69–F76 CrossRef CAS PubMed.
- C. Ridruejo Arias, C. Debiemme-Chouvy, C. Gabrielli, C. Laberty-Robert, A. Pailleret, H. Perrot and O. Sel, J. Phys. Chem. C, 2014, 118, 26551 Search PubMed.
- T. Brezesinski, D. Fattakhova Rohlfing, S. Sallard, M. Antonietti and B. M. Smarsly, Small, 2006, 10, 1203 CrossRef PubMed.
- J. H. Choy, Y. I. Kim, J. B. Yoon and S. H. Choy, J. Mater. Chem., 2001, 11, 1506 RSC.
- G. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
- K. Bizet, C. Gabrielli and H. Perrot, Appl. Biochem. Biotechnol., 2000, 89, 139 CrossRef CAS.
- W. Kwong, N. Savvides and C. Sorrell, Electrochim. Acta, 2012, 75, 371 CrossRef CAS PubMed.
- Z. Xie, L. Gao, B. Liang, X. Wang, G. Chen, Z. Liu, J. Chao, D. Chen and G. Shen, J. Mater. Chem., 2012, 22, 19904 RSC.
- J. Mahler and I. A. Persson, Inorg. Chem., 2012, 51, 425 CrossRef PubMed.
- H. M. French, M. J. Henderson, A. R. Hillman and E. Vieil, Solid State Ionics, 2002, 150, 27 CrossRef CAS.
- H. M. French, M. J. Henderson, A. R. Hillman and E. Vieil, J. Electroanal. Chem., 2001, 500, 192 CrossRef CAS.
- M. Gonsalves and A. R. Hillman, J. Electroanal. Chem., 1998, 454, 183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Electrochemical synthesis of WO3 thin films and the corresponding QCM profiles, structural characterization via EDX during FEG-SEM analyses, the ac-electrogravimetry methodology and the details of the theoretical model considering the transfer of two cations, an anion and free solvent molecules at the electrode/electrolyte interface, and the partial electrogravimetric transfer functions of mesoporous WO3 thin films. See DOI: 10.1039/c5cp00336a |
| This journal is © the Owner Societies 2015 |