
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT-based genetic algorithm search for AuCu nanoalloy electrocatalysts for CO2 reduction†
Steen
Lysgaard
,
Jón S. G.
Mýrdal
,
Heine A.
Hansen
and
Tejs
Vegge
*
Department of Energy Conversion and Storage, Technical University of Denmark, Frederiksborgvej 399, DK-4000 Roskilde, Denmark. E-mail: teve@dtu.dk; Fax: +45 4677 5858; Tel: +45 4677 5818
First published on 20th April 2015
Abstract
Using a DFT-based genetic algorithm (GA) approach, we have determined the most stable structure and stoichiometry of a 309-atom icosahedral AuCu nanoalloy, for potential use as an electrocatalyst for CO2 reduction. The identified core–shell nano-particle consists of a copper core interspersed with gold atoms having only copper neighbors and a gold surface with a few copper atoms in the terraces. We also present an adsorbate-dependent correction scheme, which enables an accurate determination of adsorption energies using a computationally fast, localized LCAO-basis set. These show that it is possible to use the LCAO mode to obtain a realistic estimate of the molecular chemisorption energy for systems where the computation in normal grid mode is not computationally feasible. These corrections are employed when calculating adsorption energies on the Cu, Au and most stable mixed particles. This shows that the mixed Cu135@Au174 core–shell nanoalloy has a similar adsorption energy, for the most favorable site, as a pure gold nano-particle. Cu, however, has the effect of stabilizing the icosahedral structure because Au particles are easily distorted when adding adsorbates.
1 Introduction
Efficient electroreduction of CO2 to fuels or chemicals is a key challenge in artificial photosynthesis from renewable energy, which has received considerable attention recently.1 Copper and gold are among the most interesting materials for CO2 reduction, because copper reduces CO2 to e.g. CO and hydrocarbons, whereas gold reduces CO2 to CO at a comparatively low overpotential.1 Alloying the two metals could be a way to break the linear scaling between the binding energies of CO and the precursor COOH.2 This is, for example, seen in CO/CO2 producing CODH enzymes.3 The electrode nanostructure has been demonstrated to be important for electrode activity and selectivity,4,5 while variations in the selectivity for CO2 reduction on Au and Cu nano-particles (NPs) with particle size have been associated with changes in the surface density of low-coordinated sites.6–8 Au3Cu bimetallic NPs show improved activity for CO evolution over Au NPs.9The theoretical prediction of structure and composition of gold–copper NPs using genetic algorithms (GAs) is not a novel idea. However, the size of the particles under investigation has often been smaller than the 2 nm particles considered here, where Au is known to display unique catalytic activity, e.g. for CO oxidation.10,11
The many-body Gupta potential has been the primary tool for structure prediction12–18 in combination with GAs, basin hopping,19,20 molecular dynamics12 or other methods for global minima discovery. The accuracy of Gupta and other empirical potentials on alloy clusters (especially gold rich) can be questioned, since the higher order contributions to the energy are non-negligible.21 The need for a computationally inexpensive potential comes from the fact that the number of homotops (distribution of A and B atoms in an AaBb cluster)22 increases combinatorially with particle size for every single composition.
Wilson and Johnston14 conducted a study of icosahedral Au–Cu particles using the Gupta potential and found that Cu in the core and Au in the surface lead to the most stable particles. Darby et al.13 found that even a single copper atom put in the center could change the lowest energy gold amorphous structure into a high-symmetrical icosahedron. Cheng et al. found Au in the core surrounded by Cu in Au43Cu12 clusters using a tight-binding approach.23 Metastable particles with Au in the core and Cu in the shell have also been observed both theoretically by simulating particle growth using molecular dynamics24 and experimentally using transmission electron microscopy methods.25,26 However, above a certain temperature, Au will segregate to the surface due to a lower surface energy. For AuCu clusters DFT studies have so far been limited to smaller particles and has been mostly used for in-depth studies of recurring structural motifs.27,28
The trend is thus that AuCu NPs are observed in high-symmetrical structures due to Cu that is mostly located in the core with Au, having a lower surface energy, being mainly in the shell. This is in good agreement with experimental observations.29,30
2 Methods
2.1 Calculation details
We employ calculations within two different levels of accuracy. The initial GA search for stable stoichiometry and composition of the AuCu icosahedral nanoparticle was performed with an semi-empirical potential based on Effective Medium Theory (EMT).31 The subsequent test of the obtained particles and calculation of adsorption energies were performed within density functional theory32,33 (DFT) using the GPAW code,34,35 a real space grid based implementation of the projector-augmented wave (PAW) method.36 The electronic wave functions were expanded using a linear combination of atomic orbitals (LCAO),37 now GPAW also has the feature of expanding the wave functions in plane waves. The LCAO mode is faster and less memory intensive than the standard finite difference (FD) mode in codes like GPAW at the cost of energetic accuracy; since we employ an incomplete basis set, we cannot expect the adsorption energies to be consistent with the results of FD grid calculations. We have therefore performed a rigorous test of adsorption energies with the LCAO mode and compared with previously published results38 of adsorption energies of all reaction intermediate adsorbates on the (111) and (211) transition metal surfaces and M13 clusters using GPAW in grid mode; these are presented in Section 3.2. The calculation of adsorption energies on all sites would not have been possible using standard DFT, i.e. the speed-up of CPU hours is on the order of 102–103 compared with similar adsorption energy calculations performed on Pt309.39,40 When reporting adsorption energies calculated using LCAO, we have included the counterpoise correction41 to avoid the basis set superposition error (BSSE). All DFT calculations were performed using the RPBE exchange–correlation functional,42 the double zeta polarized basis set and an electronic Fermi smearing of 0.1 eV. All calculations were done spin-paired except for reference calculations involving Ni. The 309 icosahedral particles were put in a cube with a side length of 32 Å (corresponding to approximately 7 Å of vacuum on each side) and a grid sampling of 184 points in each direction. The M13 particles were put in a box with 7 Å of vacuum on each side with a 96-point grid sampling in each direction. The (111) and (211) extended surfaces were sampled using a Monkhorst–Pack grid43 with k-points (6,7,1) and (5,6,1), respectively, (1 in the direction normal to the surface) and a grid spacing of 0.18 Å was employed. The presented structures and energies have all undergone geometrical optimization using the BFGS algorithm with a line search mechanism until the force on all individual atoms was less than 0.05 eV Å−1.In addition to the GA, we also performed a screening of all symmetric AuCu icosahedral particles, i.e. particles where all symmetrically equivalent sites under the point group symmetry, also called atom subshells,44,45 are occupied by the same element. All atoms in the same subshell will have the same distance to the center. In a 309-atom icosahedral particle, there are 11 symmetrically in-equivalent sites, in a bimetallic system this results in 211 different particles, which can be easily screened with the EMT potential. The symmetrical NP is represented by a string with 11 elements signifying the occupation of each subshell; e.g. the string {1Au 2Cu 3Cu 4Cu 5Cu 6Cu 7Cu 8Cu 9Cu 10Cu 11Au} represents the NP with an Au atom at the center, Au atoms at the corner sites and the rest is Cu. This approach is well known for optimizing the distribution of elements in nanoparticles.14,45,46
2.2 Genetic algorithm setup
A GA works by adapting a population of possible solutions to a problem defined using a fitness function. In this case, the challenge was to find the most stable stoichiometry and internal distribution of copper and gold atoms in an icosahedral nanoalloy containing 309 atoms in total. The fitness metric should not simply be the total energy or energy per atom since one would then not be able to compare different stoichiometries. Instead, we use the mixing energy22 defined as:14| Emix = (EAB − EANA/N − EBNB/N)/N | (1) |
We employ the GA implemented in the Atomic Simulation Environment48 (ASE). Operators previously developed for optimizing the internal distribution of atoms in clusters49 have also been employed here, with the removal of the condition that maintains stoichiometry during operations that could change the stoichiometry, e.g. the cut-splice crossover. Note that the core–shell crossover operation has not been used in this work. Two additional substitution operators have been implemented. The first operator substitutes one random atom in the cluster with a different element, directly changing the stoichiometry. In this way all stoichiometries can be examined. Thus we optimize both internal distribution and stoichiometry at the same time. One could suspect that a particle with the composition optimized would never benefit from having a random atom substituted by a different element. However, since the GA maintains a population of particles with different stoichiometries and crosses them, it is possible to approach the optimum of both parameters at the same time. The second additional operator is inspired by the screening of all symmetric AuCu particles. This operator changes all atoms in a subshell to one element type; this effectively incorporates the screening indirectly in the GA.
To sum up, the operators used in these GA runs are random permutation, permutes two atoms of a different type; center of mass to surface permutation, permutes an atom in the core region with one on the surface; rich to poor (and poor to rich) permutation, permutes two different atoms each from environments rich (poor) in their own type of atoms to environments poor (rich) of their own kind;49 random substitution, see above; symmetric subshell substitution, see above; and cut-splice crossover, cuts two particles into half through the center and joins two parts from different parents together.50 After each operation the particles undergo geometrical relaxation. We stress that by using this GA setup, the focus is on the chemical ordering of the elements rather than structural optimization. Changes in the structure can occur only during the geometrical relaxation that follows each operation. It is, however, unexpected since moving away from the icosahedral structure requires overcoming an energy barrier. This barrier can be lowered in the case of non-isolated particles.
The GA is initiated with a population of 10 particles in vacuum, each with a random Au/Cu stoichiometry. Inclusion of a support material would have been highly relevant, but it has not been taken into account in this study since it would drastically increase both computational time and complexity of the particle structure. During each generation, 10 new particles are created using the aforementioned operators, geometrically relaxed and added to the population. The particles are sorted according to the mixing energy (1) and the 10 fittest NPs form the population used to create the next generation. When no new NPs enters the population for 5 generations, it is assumed that convergence has been reached and the algorithm stops. The algorithm is thus run as a traditional generational GA, where all calculations in a generation must finish before the population is updated and the next generation can commence. It is, however, also possible to run the algorithm as a pool GA, where the population is continuously updated every time a relaxation is finished and a new structure is created.51,52
3 Results and discussion
3.1 Finding the optimum
In Fig. 1, the mixing energy as a function of Au/Cu ratio in the particles is shown. The lowest mixing energies as a function of Au/Cu ratio are connected by a red line signifying that no symmetrical particle has a lower mixing energy. In the subshell nomenclature, the {1Cu 2Cu 3Cu 4Au 5Au 6Cu 7Cu 8Au 9Au 10Au 11Au} particle has the lowest mixing energy of the structures in the screening. Two factors decide the distribution of atoms in the bimetallic icosahedral NP: (1) relieve the inherent bulk strain53 by putting the smaller element (Cu) in the core and the larger (Au) on the surface. (2) Maximize the number of stronger bonds; from the cohesive energies one can deduce individual bond strengths,14 Au–Au > Au–Cu > Cu–Cu. Here, it means that Au atoms are dispersed in the core, in subshells 4 and 5, with each Au atom having only Cu neighbors, the gain in bond energy is larger than the loss due to strain. The opposite is true for subshell 1, where a gold atom would also have 12 Cu neighbors; however, here the increase in strain energy is greater than the gain in bond energy. This is in contrast to the findings of Cheng et al.,23 who found an Au atom in the core even for stoichiometries with Cu on the surface albeit with the smaller 55-atom particle.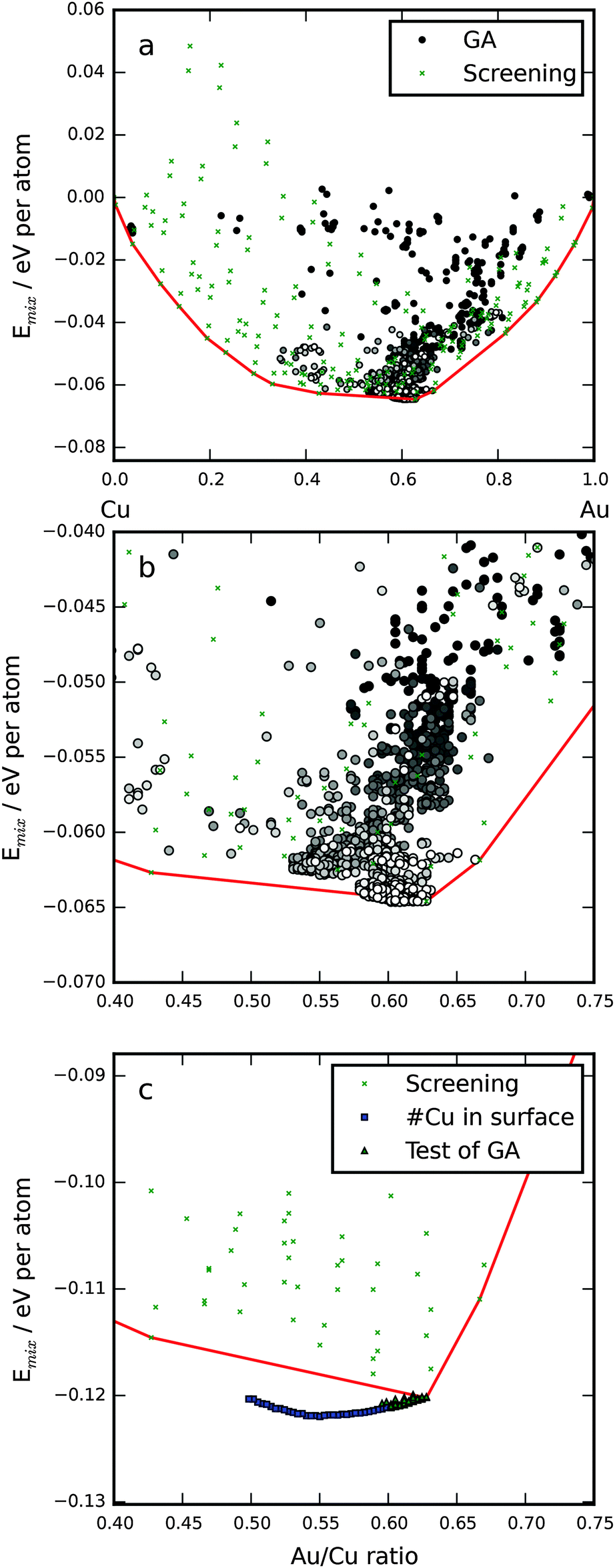 | ||
| Fig. 1 Mixing energy as a function of Au/Cu ratio in the 309-atom particles. (a) The lowest energy for each possible value of the Au/Cu ratio from the screening of 211 symmetric particles marked with x's. All the particles from a single GA run are also shown marked by circles. All are calculated using EMT. The circles are shaded corresponding to how far in the GA run the particles were investigated, black is the first generation and white is the last generation. The red line makes up the convex hull, it connects the lowest points of the screening marking the ground state line as expected from the screening. (b) A zoom-in of the minimum region of (a). (c) The minimum region now calculated using DFT. | ||
We performed a number of GA runs and in all of them a small number of, up to eight, Cu atoms were situated in the terrace sites at the surface. The only difference between the GA particles and the ones from the symmetrical screening is the surface terrace sites (the eighth subshell). Below the surface, the particles are identical to the best from the symmetrical screening. By introducing non-symmetric regions with Cu in the surface, the GA is able to predict particles with lower mixing energy, as is evident in Fig. 1b and c. We subsequently did DFT calculations on the 50 most stable particles from the screening and the 50 fittest from the GA runs, these are shown in Fig. 1c. Afterwards we tested the optimal amount of Cu in the terrace sites and discovered that EMT predicts 8 and DFT 24 (squares in Fig. 1c). The Cu atoms should be spread out as much as possible with 16 terraces only having one Cu atom and four having two. Fig. 2 shows a representation of the most stable particle. The minimum is thus at an Au/Cu ratio of 55/45, which is slightly different from previous work in the literature14 that found the lowest mixing energy for a 50/50 mixture of Cu and Au.
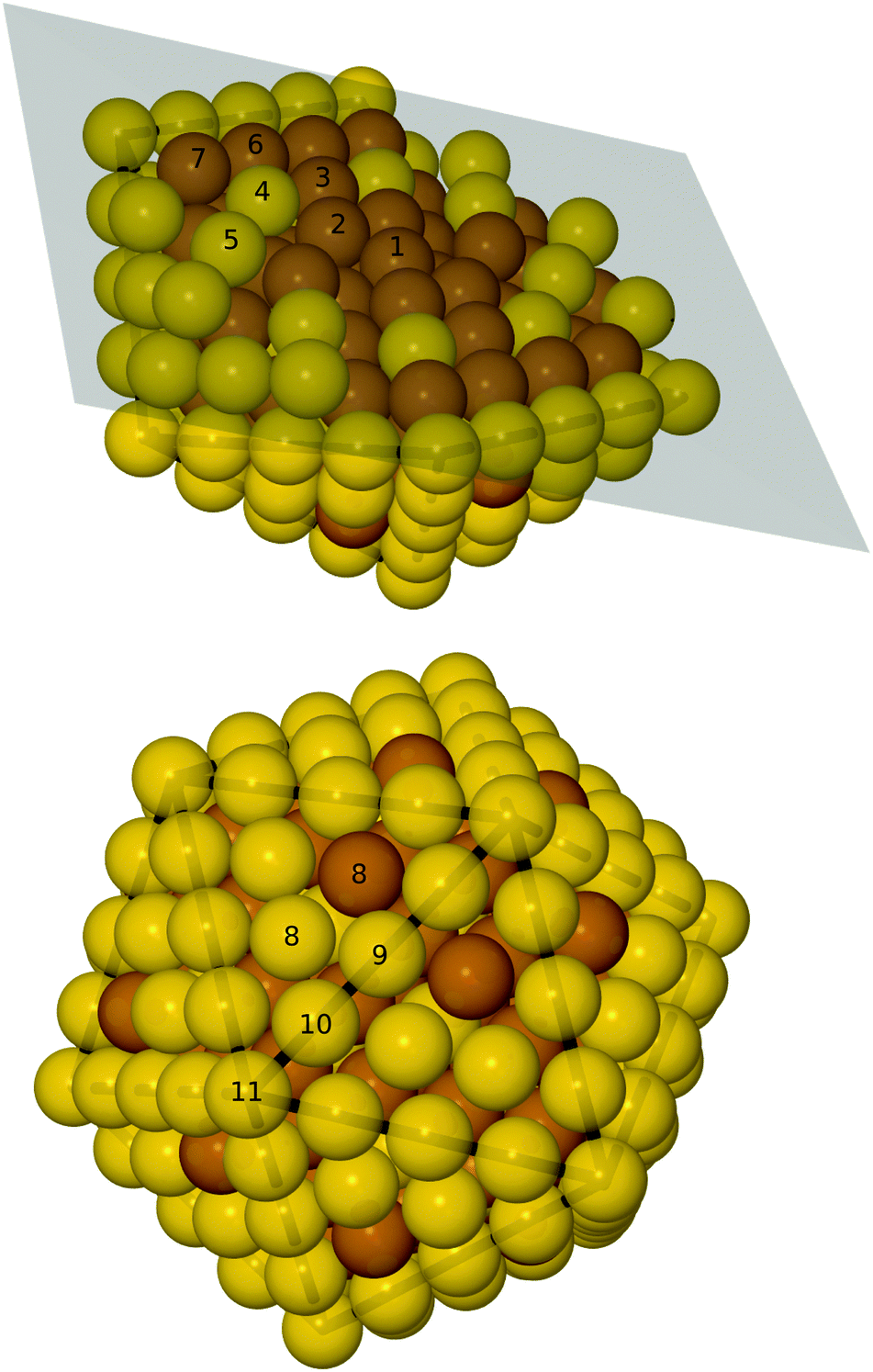 | ||
| Fig. 2 Top: cut-through of the optimal particle, the numbers on the atoms indicate the subshell number. Bottom: the whole Au174Cu135 particle. Brown: Cu, yellow: Au. | ||
For calculations of the adsorption energy, we use a particle with one Cu atom in each terrace – that is, 20 Cu atoms on the surface, in total. This leads to an Au174Cu135 particle; a pure Cu core with Au placed in atom subshells 4 and 5 (see Fig. 2), thereby the gold atoms only have copper neighbors in the core. The surface is all gold except for one Cu atom on a terrace site in each terrace.
Pure Cu particles exhibit the icosahedral structure in all sizes, whereas pure Au clusters do not exhibit highly symmetrical ground states but instead more amorphous-like low-symmetry structures.13 Au clusters are known to fluctuate in the structure meaning that there are many local minima close in energy with both low and high symmetries.54,55 We found that adding adsorbates can distort metastable icosahedral Au particles into a more energetically favorable amorphous structure. A small amount of Cu can however stabilize high-symmetry structures,13 even when adding adsorbates. Substituting the two innermost Au subshells with Cu is enough to stabilize the icosahedral 309-atom Au structure against distortion, even though the icosahedral structure is one of the least stable Au structures.56
The findings here underline that it is advantageous to employ calculations at two levels of complexity. The lighter one for fast screening and the heavier one for correct ordering of the best screening results.57–60
3.2 Accuracy of LCAO adsorption energies
In order to be able to validate the adsorption energies, we have tested the LCAO mode against values previously reported in the literature. Peterson et al.38 reported the adsorption energies of O and CO on (111) and (211) surfaces as well as M13 clusters using FD for all stable adsorption sites. We find that the error from using the faster LCAO mode depends mostly on the adsorbate. In Fig. 3, we plot the adsorption energy of CO calculated using LCAO vs. literature data on Ni, Cu, Ag, Pd, Au and Pt. It is evident that the LCAO mode generally yields too low binding energies, but the underestimation is systematic for all elements, except for Pd M13, with an acceptable standard deviation of 0.12 eV. For further discussion we have subtracted the mean error of 0.28 eV for all calculated CO adsorption energies. | ||
| Fig. 3 Adsorption energies of CO with the FD mode vs. LCAO mode in GPAW. The FD mode values are from ref. 38. The thick blue line is the no correction line. The thin red line includes the subtraction of the average error of 0.28 eV. The four lowest Pd points (the outliers) are from M13 calculations. | ||
For COOH, we do not have GPAW FD data; instead we used data obtained using the planewave code Dacapo (shown in Fig. S1, ESI†). The correction is very small and we have neglected it in the further discussion.
3.3 Adsorption energies
We calculate adsorption energies of COOH and CO intermediates at the apex site, edge sites and terrace sites of the Au, Cu and AuCu nanoalloys. The most stable adsorption geometries are displayed in Fig. 4.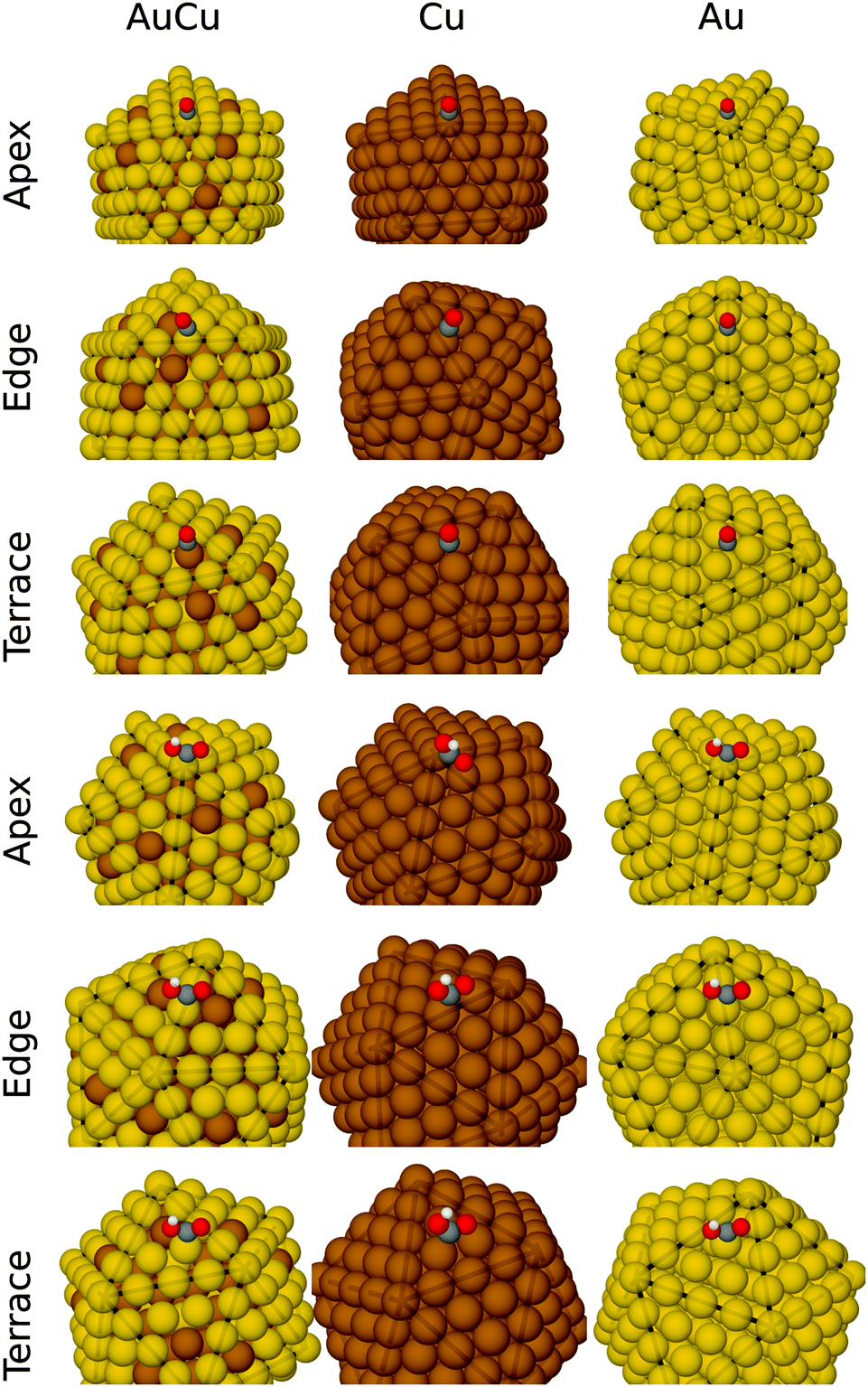 | ||
| Fig. 4 Geometries of CO and COOH adsorbed at the apex, edge and terrace sites on AuCu, Cu and Au nano-particles. Au is shown in yellow, Cu brown, C gray, O red, and H white. | ||
On the Au and Au174Cu135 NPs, adsorption of CO and COOH is the strongest on the apex sites, whereas adsorption of CO and COOH is stronger at edge sites than apex sites on Cu. On the terraces of the Cu135@Au174 core–shell nanoalloy, we find that CO binds to the Cu atom, while COOH binds with the Au atom. Because CO and COOH bind significantly stronger to Cu than Au, it is surprising that COOH binds to Au on the terrace rather than Cu. We observe no indications of the formation of oxygen bonds with the Cu atoms adjacent to the Au–C primary bond, as proposed by Kim et al.9
It has been suggested that the formation of CO on Au and Cu goes through a COOH intermediate.61–63Fig. 5 shows the CO production at 0.35 V overpotential from a kinetic model developed previously.3 In this model, the production of CO follows the reaction mechanism
| CO2(aq) + * + H+ + e− → COOH* | (R1) |
| COOH* + H+ + e− → CO* + H2O(l) | (R2) |
| CO* → CO(aq) + * | (R3) |
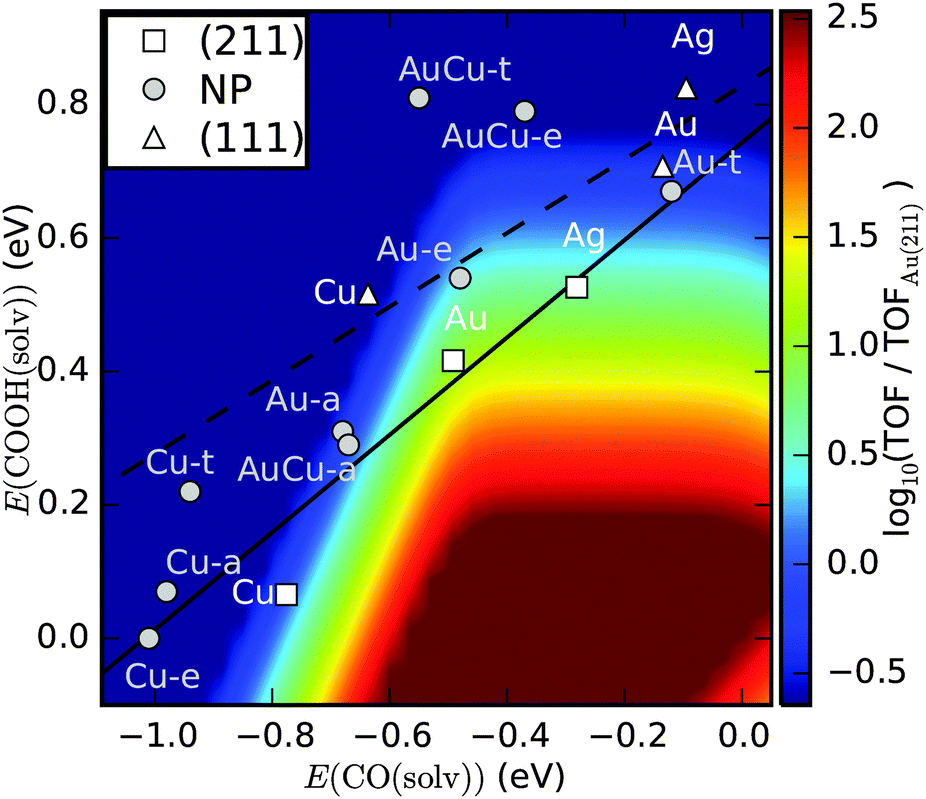 | ||
| Fig. 5 Contour plot of the turn over frequency (TOF) for CO2 to CO from a kinetic model at 0.35 V overpotential.3 The predicted activity of the apex, edge and terrace sites on Au174Cu135, Au and Cu nanoparticles from our study is shown. Activities of (111) and (211) facets are taken from ref. 64 and 2. The solid and dashed lines are trend lines for (211) and (111) surfaces, respectively, of the late transition metals. | ||
The prefactors for coupled proton–electron transfer have been fitted to experiments on Au.61 Assuming activation energies to scale with reaction energies, it is possible to describe the activity of CO evolution as a function of the COOH and CO binding energies. In Fig. 5 the activity is described as a function of the reaction energy of (R1) and (R2) as calculated from DFT and including 0.25 eV and 0.1 eV stabilization energies of COOH and CO* intermediates, due to hydrogen bonding in the solvent.63
Within the kinetic model, a good catalyst for CO production should bind COOH sufficiently strong to activate CO2 and it should not bind CO so strong that the active sites are blocked by CO.3 Activities of (111) terraces and (211) steps on Ag, Au and Cu are indicated based on previous DFT calculations.2,64
The edge sites on the Au NP are predicted to have good activity for CO production, comparable to the activity of Ag(211), but lower than Au(211). The apex sites on Au174Cu135 and Au NPs have slightly lower activity for CO evolution under the simulated conditions and are comparable to Cu(211). The CO production on these sites is limited by CO poisoning as well as by CO2 activation, so we expect that mass transport conditions are important for CO production, similar to what has been observed on Cu.65 The terrace sites on Au174Cu135 and Au NPs are predicted to be too inactive to activate CO2. The active sites on the Cu NP are found to bind too strongly for efficient CO production. It should be noted, however, that adsorbate–adsorbate interactions are not included in the model and repulsion from CO* is expected to weaken binding energies resulting in higher activity.64 It should further be noted that the model does not include H2 production, so the selectivity for CO2 reduction is not included in the model.8,66 This is a potentially critical inadequacy if accurate predictions about product selectivity are to be made, however, work is underway for including H2 production.
4 Conclusions
We have studied catalytically interesting 309-atom AuCu icosahedral nanoparticles. In order to determine the most stable stoichiometry and distribution of atoms, we have implemented GA operators that incorporate a screening of cluster symmetric subshells into a GA run. This greatly enhances the efficiency of the GA, since it now has the ability to combine highly symmetrical atomic distributions with slight but important deviations from the symmetry. This feature is present in the most stable mixed AuCu nanoalloy presented here, see Fig. 2. The most stable particle is a Cu135@Au174 core–shell nanoalloy, it is formed by a Cu core with interspersed Au that all have only Cu neighbors in the core and a surface of Au with a few Cu atoms on the terraces. During the GA runs, the energies were calculated using the EMT potential. Subsequently, the fittest particles were evaluated using DFT to obtain the correct ordering. This showed that almost half the terrace sites should be occupied by Cu (24 out of 60). It is a novel finding that a theoretical method can predict stable nanoalloys with scattered Cu and Au atoms in the shell and the core, respectively.Adsorption energies of CO and COOH were determined on the pure Au, Cu and mixed Au174Cu135 particles. This did not place any of the particles closer to the top of the activity volcano in Fig. 5. We tested the LCAO adsorption energy credibility by comparing with previously published values from the literature; the systematic errors were relatively small probably due to the systematic removal of BSSE.
The fact that the Au174Cu135 nanoalloy has approximately the same properties as Au309 regarding adsorption of molecular species on the most energetically favorable site is interesting from an economical point of view; it could be possible to reduce the amount of precious metal in a catalyst since the active metals would only be situated on the surface. This has also been previously observed for Pt–Ni particles.67
Acknowledgements
The financial support from the European Commission under the FP7 Fuel Cells and Hydrogen Joint Technology Initiative grant agreement FP7-2012-JTI-FCH-325327 for the SMARTcat project is gratefully acknowledged.Notes and references
- Y. Hori, Modern Aspects of Electrochemistry, Springer New York, New York, NY, 2008, vol. 42 Search PubMed.
- A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS.
- C. W. Li and M. W. Kanan, J. Am. Chem. Soc., 2012, 134, 7231–7234 CrossRef CAS PubMed.
- W. Tang, A. A. Peterson, A. S. Varela, Z. P. Jovanov, L. Bech, W. J. Durand, S. Dahl, J. K. Nørskov and I. Chorkendorff, Phys. Chem. Chem. Phys., 2012, 14, 76–81 RSC.
- W. Zhu, R. Michalsky, Ö. Metin, H. Lv, S. Guo, C. J. Wright, X. Sun, A. a. Peterson and S. Sun, J. Am. Chem. Soc., 2013, 135, 16833–16836 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- H. Mistry, R. Reske, Z. Zeng, Z.-J. Zhao, J. Greeley, P. Strasser and B. Roldan Cuenya, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5, 4948 CrossRef CAS PubMed.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566–575 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. J. López, P. A. Marcos and J. A. Alonso, J. Chem. Phys., 1996, 104, 1056 CrossRef PubMed.
- S. Darby, T. V. Mortimer-Jones, R. L. Johnston and C. Roberts, J. Chem. Phys., 2002, 116, 1536 CrossRef CAS PubMed.
- N. T. Wilson and R. L. Johnston, J. Mater. Chem., 2002, 12, 2913–2922 RSC.
- R. A. Lordeiro, F. F. Guimarães, J. C. Belchior and R. L. Johnston, Int. J. Quantum Chem., 2003, 95, 112–125 CrossRef CAS PubMed.
- A. Rapallo, G. Rossi, R. Ferrando, A. Fortunelli, B. C. Curley, L. D. Lloyd, G. M. Tarbuck and R. L. Johnston, J. Chem. Phys., 2005, 122, 194308 CrossRef PubMed.
- P. J. Hsu and S. K. Lai, J. Chem. Phys., 2006, 124, 044711 CrossRef CAS PubMed.
- X. Wu, W. Cai and X. Shao, J. Comput. Chem., 2009, 30, 1992–2000 CrossRef CAS PubMed.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 5639, 5111–5116 CrossRef.
- F. Calvo and E. Yurtsever, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 045423 CrossRef.
- A. Hermann, R. Krawczyk, M. Lein, P. Schwerdtfeger, I. Hamilton and J. Stewart, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 013202 CrossRef.
- J. Jellinek and E. B. Krissinel, Chem. Phys. Lett., 1996, 4, 283–292 CrossRef.
- D. Cheng, S. Huang and W. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 64117 CrossRef.
- T. J. Toai, G. Rossi and R. Ferrando, Faraday Discuss., 2008, 138, 49 RSC.
- J. A. Ascencio, H. B. Liu, U. Pal, A. Medina and Z. L. Wang, Microsc. Res. Tech., 2006, 69, 522–530 CrossRef CAS PubMed.
- F. Yin, Z. W. Wang and R. E. Palmer, J. Am. Chem. Soc., 2011, 133, 10325–10327 CrossRef CAS PubMed.
- D. D. C. Rodrigues, A. M. Nascimento, H. A. Duarte and J. C. Belchior, Chem. Phys., 2008, 349, 91–97 CrossRef CAS PubMed.
- D. T. Tran and R. L. Johnston, Phys. Chem. Chem. Phys., 2009, 11, 10340–10349 RSC.
- F. Yin, Z. Wang and R. Palmer, J. Nanopart. Res., 2012, 14, 1–8 CrossRef.
- F. Yin, Z. W. Wang and R. E. Palmer, J. Exp. Nanosci., 2012, 7, 703–710 CrossRef CAS.
- K. W. Jacobsen, P. Stoltze and J. K. Nørskov, Surf. Sci., 1996, 366, 394–402 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev. B: Solid State, 1964, 136, B864–B871 Search PubMed.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. J. Mortensen, L. B. Hansen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 35109–35111 CrossRef.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dulak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Moller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Hakkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- P. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- A. H. Larsen, M. Vanin, J. J. Mortensen, K. S. Thygesen and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 195112 CrossRef.
- A. A. Peterson, L. C. Grabow, T. P. Brennan, B. Shong, C. Ooi, D. M. Wu, C. W. Li, A. Kushwaha, A. J. Medford, F. Mbuga, L. Li and J. K. Nørskov, Top. Catal., 2012, 55, 1276–1282 CrossRef CAS.
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Chem. Lett., 2013, 4, 222–226 CrossRef CAS.
- L. Li, private communication.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- B. Hammer, L. Hansen and J. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Montejano-Carrizales and J. Moran-Lopez, Nanostruct. Mater., 1992, 1, 397–409 CrossRef CAS.
- J. E. Hearn and R. L. Johnston, J. Chem. Phys., 1997, 107, 4674 CrossRef CAS PubMed.
- G. Barcaro, L. Sementa and A. Fortunelli, Phys. Chem. Chem. Phys., 2014, 16, 24256–24265 RSC.
- L. O. Paz-Borbón, T. V. Mortimer-Jones, R. L. Johnston, A. Posada-Amarillas, G. Barcaro and A. Fortunelli, Phys. Chem. Chem. Phys., 2007, 9, 5202–5208 RSC.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- S. Lysgaard, D. D. Landis, T. Bligaard and T. Vegge, Top. Catal., 2014, 57, 33–39 CrossRef CAS PubMed.
- D. M. Deaven and K. M. Ho, Phys. Rev. Lett., 1995, 75, 288–291 CrossRef CAS.
- L. B. Vilhelmsen and B. Hammer, J. Chem. Phys., 2014, 141, 044711 CrossRef PubMed.
- A. Shayeghi, D. Gotz, J. B. A. Davis, R. Schafer and R. L. Johnston, Phys. Chem. Chem. Phys., 2015, 17, 2104–2112 RSC.
- J. P. K. Doye and D. J. Wales, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, 4859–4894 CrossRef CAS.
- I. L. Garzón, K. Michaelian, M. R. Beltrán, A. Posada-Amarillas, P. Ordejón, E. Artacho, D. Sánchez-Portal and J. M. Soler, Phys. Rev. Lett., 1998, 81, 1600–1603 CrossRef.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- S. R. Plant, L. Cao and R. E. Palmer, J. Am. Chem. Soc., 2014, 136, 7559–7562 CrossRef CAS PubMed.
- L. Vilhelmsen and B. Hammer, Phys. Rev. Lett., 2012, 108, 126101 CrossRef.
- G. Barcaro, A. Fortunelli, G. Rossi, F. Nita and R. Ferrando, J. Phys. Chem. B, 2006, 110, 23197–23203 CrossRef CAS PubMed.
- R. Ferrando, A. Fortunelli and R. L. Johnston, Phys. Chem. Chem. Phys., 2008, 10, 640–649 RSC.
- J. Rogan, G. García, C. Loyola, W. Orellana, R. Ramírez and M. Kiwi, J. Chem. Phys., 2006, 125, 214708 CrossRef PubMed.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, J. Chem. Soc., Chem. Commun., 1987, 728–729 RSC.
- H. Noda, S. Ikeda, A. Yamamoto, H. Einaga and K. Ito, Bull. Chem. Soc. Jpn., 1995, 68, 1889–1895 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- C. Shi, H. A. Hansen, A. C. Lausche and J. K. Nørskov, Phys. Chem. Chem. Phys., 2014, 16, 4720–4727 RSC.
- J. J. Kim, D. P. Summers and K. W. Frese, J. Electroanal. Chem. Interfacial Electrochem., 1988, 245, 223–244 CrossRef CAS.
- W. Zhu, Y.-J. Zhang, H. Zhang, H. Lv, Q. Li, R. Michalsky, A. A. Peterson and S. Sun, J. Am. Chem. Soc., 2014, 136, 16132–16135 CrossRef CAS PubMed.
- G. Wang, M. A. Van Hove, P. N. Ross and M. I. Baskes, J. Chem. Phys., 2005, 122, 024706 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: LCAO corrections of the adsorbed molecular species COOH, CHO and OH. See DOI: 10.1039/c5cp00298b |
| This journal is © the Owner Societies 2015 |