
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGas phase selective hydrogenation over oxide supported Ni–Au
Fernando
Cárdenas-Lizana
and
Mark A.
Keane
*
Chemical Engineering, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh, EH14 4AS, Scotland. E-mail: M.A.Keane@hw.ac.uk; Tel: +44 (0)1314514719
First published on 2nd March 2015
Abstract
The chemoselective continuous gas phase (T = 573 K; P = 1 atm) hydrogenation of nitroarenes (p-chloronitrobenzene (p-CNB) and m-dinitrobenzene (m-DNB)) has been investigated over a series of oxide (Al2O3 and TiO2) supported Au and Ni–Au (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 mol ratio; 0.1–1 mol% Au) catalysts. Monometallic supported Au with mean particle size 3–9 nm promoted exclusive formation of p-chloroaniline (p-CAN) and m-nitroaniline (m-NAN). Selective hydrogenation rate was higher over smaller Au particles and can be attributed to increased surface hydrogen (from TPD measurements) at higher metal dispersion. (S)TEM analysis has confirmed an equivalent metal particle size for the supported bimetallics at the same Au loading where TPR indicates Ni–Au interaction and EDX surface mapping established Ni in close proximity to Au on isolated nanoparticles with a composition (Au/Ni) close to the bulk value (= 10). Increased spillover hydrogen due to the incorporation of Ni in the bimetallics resulted in elevated –NO2 group reduction rate. Full selectivity to p-CAN was maintained over all the bimetallic catalysts. Conversion of m-DNB over the lower loaded Ni–Au/Al2O3 generated m-NAN as sole product. An increase in Ni content (0.01 → 0.1 mol%) or a switch from Al2O3 to TiO2 as support resulted in full –NO2 reduction (to m-phenylenediamine). Our results demonstrate the viability of Ni-promotion of Au in the continuous production of functionalised anilines.
:10 mol ratio; 0.1–1 mol% Au) catalysts. Monometallic supported Au with mean particle size 3–9 nm promoted exclusive formation of p-chloroaniline (p-CAN) and m-nitroaniline (m-NAN). Selective hydrogenation rate was higher over smaller Au particles and can be attributed to increased surface hydrogen (from TPD measurements) at higher metal dispersion. (S)TEM analysis has confirmed an equivalent metal particle size for the supported bimetallics at the same Au loading where TPR indicates Ni–Au interaction and EDX surface mapping established Ni in close proximity to Au on isolated nanoparticles with a composition (Au/Ni) close to the bulk value (= 10). Increased spillover hydrogen due to the incorporation of Ni in the bimetallics resulted in elevated –NO2 group reduction rate. Full selectivity to p-CAN was maintained over all the bimetallic catalysts. Conversion of m-DNB over the lower loaded Ni–Au/Al2O3 generated m-NAN as sole product. An increase in Ni content (0.01 → 0.1 mol%) or a switch from Al2O3 to TiO2 as support resulted in full –NO2 reduction (to m-phenylenediamine). Our results demonstrate the viability of Ni-promotion of Au in the continuous production of functionalised anilines.
1. Introduction
The combination of two metallic elements has been shown to be an effective means of improving catalytic performance in a range of hydrogen mediated reactions, notably chemoselective hydrogenation (of nitro-compounds1), partial hydrogenation (of alkynes2) and hydrogenolysis (of chlorophenols3). Renewed interest in Au as a catalytic agent has encompassed Au in bimetallic formulations, as noted in the recent review by Villa et al.4 Gold has shown unique selectivity in the hydrogenation of polyfunctional reactants but delivers low reaction rates.5 This drawback has been ascribed to a restricted capacity for H2 activation by dissociative chemisorption.6 The combination of Au with transition metals that exhibit greater H2 chemisorption capability offers a possible means of enhancing hydrogenation rate.The hydrogenation response over bimetallic catalysts is determined by the structure and physico-chemical properties of the supported nanoparticles, which are dependent on the distribution of both metals within the crystal nanostructure. In order to achieve any degree of catalytic synergy, the two metals must be in close proximity (if not in direct contact) on the support. The size, metal ratio and nature of the support are critical variables. Bimetallic particles at the nano-scale (∼3 nm) can form solid solutions (homogeneous alloy particles) regardless of the miscibility gap between the two metallic elements.7 The incorporation of a second metal with Au (X–Au, where X = Pd,8 Pt9) has been shown to increase hydrogenation activity but at low Au/X ratios the selectivity response is affected or even governed by the second metal.8,9 The redox character and acid–base properties of the (oxide) carrier can act to stabilize small Au nanoparticles as a result of electron transfer across the metal–support interface and induce geometric and electronic modifications that impact on catalysis.5
Prior research has focused on binary metals that form (ordered or random) bulk alloys with no miscibility gap in the corresponding bulk phase diagram. This is the case with Pd–Au as the most widely studied Au-containing bimetallic.10 Interest has shifted to systems such as Ni–Au that do not mix in the bulk but can form stable alloys in the outermost surface layers.11,12 The incorporation of Au onto partially oxidised Ni resulted in partial oxidation of Au atoms and the formation of Aun–O–Nim ensembles (from XPS),13 which were suggested as active sites in the isomerisation of methylstyrenes over Ni–Au/SiO2.14 Nishikawa et al.,15 studying the promotional effect of Au on Ni, demonstrated (by XRD, TEM, XAFS and 197Au Mössbauer) the formation of an Ni–Au alloy, following reduction (to 673 K in H2) of Au/NiO co-precipitates. These were active in the hydrogenolysis of benzylic alcohols with superior catalytic activity compared with RANEY® Ni. Chin et al.16 demonstrated by EXAFS/XANES analysis of Ni–Au/MgAl2O4 (prepared by reductive deposition of Au on Ni/MgAl2O4) Au → Ni electron transfer with surface alloy formation that served to inhibit carbon deposition during n-butane steam reforming. The addition of Au to Ni (1:4) resulted in the formation of nano-crystals with a Au-core and Ni-enriched shell structure (based on EXAFS, XPS and UV-vis) with increased hydrogenolysis activity in the conversion of 2-phenoxy-1-phenylethanol.17
We have previously examined the catalytic action of supported Ni–Au with Ni/Au mol/mol = 10 in promoting gas phase nitroarene hydrogenation.18,19 That work was directed at the continuous selective production of industrially important functionalised amines under mild reactions conditions. Supported Ni–Au delivered higher hydrogenation activities but was less selective than supported Au. We have refocused our attention and have considered the possible role of Ni (introduced as the minor component; Ni/Au mol/mol = 0.1) to enhance the chemoselective hydrogenation action of Au to deliver enhanced rates to the target amine product. We assess the feasibility of controlling hydrogenation rate by varying Au particle size and the nature of the oxide support.
2. Experimental
2.1 Catalyst preparation and activation
The Al2O3 (Puralox, Condea Vista Co.) and TiO2 (Degussa) supports were used as received. High (HL, 0.1 mol%) and low loaded (LL, 0.01 mol%) Ni/Al2O3 and/or Ni/TiO2 catalyst precursors were synthesised by standard impregnation, where the support (6 g) was contacted with an aqueous Ni(NO3)2 solution (Aldrich, 7 × 10−5–7 × 10−4 M, 85 cm3). The slurry was heated (2 K min−1) to 353 K and maintained under constant agitation (600 rpm) in a He purge. The solid was dried in a flow of He at 383 K for 5 h and sieved (ATM fine test sieves) to mean particle diameter = 75 μm. The Au–Ni catalyst precursors were prepared by first reducing a batch of (LL and HL) Ni/Al2O3 and (LL) Ni/TiO2 in a (60 cm3 min−1) H2 stream at 2 K min−1 to 723 K, which was maintained for 1 h to ensure formation of zero valent Ni.20 The gas flow was switched to He, cooled to ambient temperature and the samples passivated in 1% v/v O2/He. This treatment served to provide a protective oxide layer over the surface Ni that prevented bulk oxidation upon exposure to the atmosphere. The passivated samples were treated with an aqueous HAuCl4 solution (Aldrich, 3 × 10−5–5 × 10−4 M, 85 cm3) to deliver a 10/1 Au/Ni mol ratio, post-treatment as above. Monometallic (HL, 1 mol% and LL, 0.1 mol%) Au/Al2O3 and (LL, 0.1 mol%) Au/TiO2 were prepared by standard impregnation with aqueous HAuCl4 (Aldrich, 7 × 10−4–7 × 10−3 M, 85 cm3). Bulk metal loading was determined by ICP-OES (Vista-PRO, Varian Inc.). The Au-containing samples were stored under He in the dark at 277 K to avoid the deleterious effects of light (photodecomposition of cationic gold to Au0)21 and temperature (aggregation of Au precursor species).22 Before reaction, the catalyst precursors were activated in 60 cm3 min−1 H2 at 2 K min−1 to 723 K (Ni/Al2O3 and Ni/TiO2) or 603 K (Au/Al2O3, Au/TiO2, Ni–Au/Al2O3, Ni–Au/TiO2), which was maintained for 1 h.2.2 Catalyst characterisation
Specific surface area (SSA) and total pore volume measurements were made on a Micromeritics Flowsorb II 2300 unit. Prior to analysis, the samples were outgassed at 423 K for 1 h in 20 cm3 min−1 N2. SSA was obtained in a 30% v/v N2/He flow (20 cm3 min−1) with at least three cycles of N2 adsorption–desorption using the standard single-point BET method. Total pore volume was obtained at a relative N2 pressure (P/P0) = 0.95. The data were reproducible to ±3% and values quoted in this paper are the mean.Temperature programmed reduction (TPR), H2 chemisorption and temperature programmed desorption (TPD) were determined using the CHEM-BET 3000 (Quantachrome) unit. The samples were loaded into a U-shaped Quartz cell (3.76 mm i.d.) and heated in 17 cm3 min−1 5% v/v H2/N2 (Brooks mass flow controlled) to 603 ± 1 K (Au/Al2O3, Au/TiO2, Ni–Au/Al2O3 and Ni–Au/TiO2) or 723 ± 1 K (Ni/Al2O3 and Ni/TiO2) at 2 K min−1. The effluent gas passed through a liquid N2 trap and H2 consumption was monitored by a thermal conductivity detector (TCD) with data acquisition/manipulation using the TPR Win™ software. The reduced samples were maintained at the final temperature in H2 until return to baseline. After the reduction step, the samples were cooled in a He flow and subjected to H2 chemisorption (at ambient temperature) using a pulse (10 μl) titration procedure, as described elsewhere.23 TPD was conducted in a N2 flow (65 cm3 min−1) at 50 K min−1 to 873 K with an isothermal hold until the signal returned to the baseline. The support alone was subjected to an equivalent TPR and subsequent TPD, which was used to correct for H2 desorption from Al2O3.24
Metal particle morphology, size distribution and surface composition were determined by (scanning) transmission electron microscopy using a JEOL JEM-2100F unit operating at an accelerating voltage of 200 kV with resolution to 0.14 nm and an EDAX Genesis XM 4 system 60. Samples were dispersed in 1-butanol by ultrasonic vibration, deposited on a lacey-carbon/Cu grid (200 Mesh) and dried at 383 K. Up to 300 individual metal particles were counted for each catalyst and the surface area-weighted metal diameter (d) was calculated from:
 | (1) |
2.3 Gas phase nitroarene hydrogenation
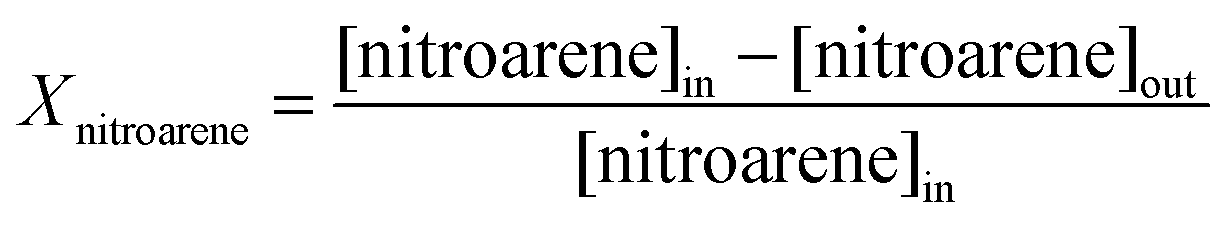 | (2) |
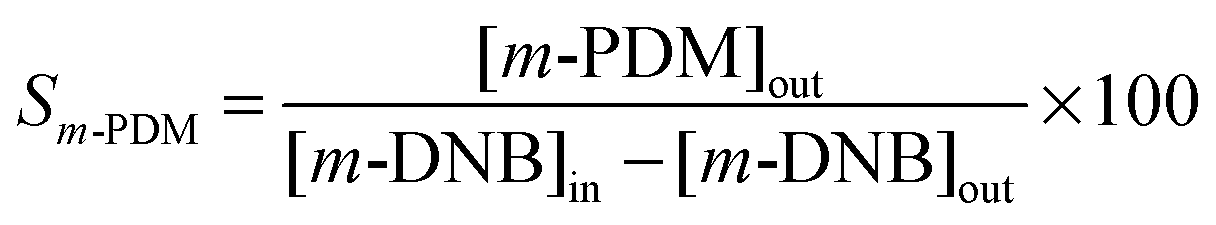 | (3) |
3. Results and discussion
3.1 Catalyst characterisation
The metal content, specific surface area (SSA), pore volume, H2 uptake/release during TPR/TPD and mean metal particle size for the (mono- and bi-metallic) catalysts used in this study are given in Table 1. The SSA and pore volume of the Al2O3 support (191 m2 g−1; 450 × 10−3 cm3 g−1) are close to values reported for commercial Puralox mesoporous γ-Al2O3 (157 m2 g−1; 420 × 10−3 cm3 g−1).25 Incorporation of Au and Ni resulted in a measurable decrease in SSA and pore volume that can be ascribed to partial pore filling by the metal(s) or Al2O3 dissolution during impregnation resulting in pore collapse.26 The TiO2 supported systems exhibited similar SSA and pore volumes to the starting support (52 m2 g−1; 120 × 10−3 cm3 g−1) that are in accord with Degussa P25 (61 m2 g−1; 120 × 10−3 cm3 g−1).27| Catalysta | SSAb (m2 g−1) | Pore volume × 10−3c (cm3 g−1) |
TPR Tmax (K) | TPR H2d,e (μmol gcatalyst−1) | H2 TPD (mmol molAu−1) | d (nm) |
|---|---|---|---|---|---|---|
|
a Au metal content (mol%): LL = 0.1, HL = 1.0, 1:10 Ni:Au molar ratio in bi-metallics.
b Specific surface area (SSA), Al2O3 = 191 m2 g−1, TiO2 = 52 m2 g−1.
c Pore volume, Al2O3 = 450 × 10−3 cm3 g−1, TiO2 = 120 × 10−3 cm3 g−1.
d H2 required for the reduction of the metal precursor.
e Experimentally determined H2 consumption.
f Surface area weighted mean metal particle size from TEM/STEM analysis.
|
||||||
| Au/Al2O3-HL | 161 | 427 | 434 | 102d/104e | 473 | 9 |
| Ni–Au/Al2O3-HL | 189 | 444 | 470 | 151d/145e | 3118 | 9 |
| Au/Al2O3-LL | 169 | 425 | 434 | 9d/16e | 1101 | 4 |
| Ni–Au/Al2O3-LL | 190 | 447 | 470 | 9d/8e | 2471 | 4 |
| Au/TiO2-LL | 49 | 117 | 383 | 11d/15e | 1233 | 3 |
| Ni–Au/TiO2-LL | 47 | 115 | 414 | 12d/15e | 1765 | 3 |
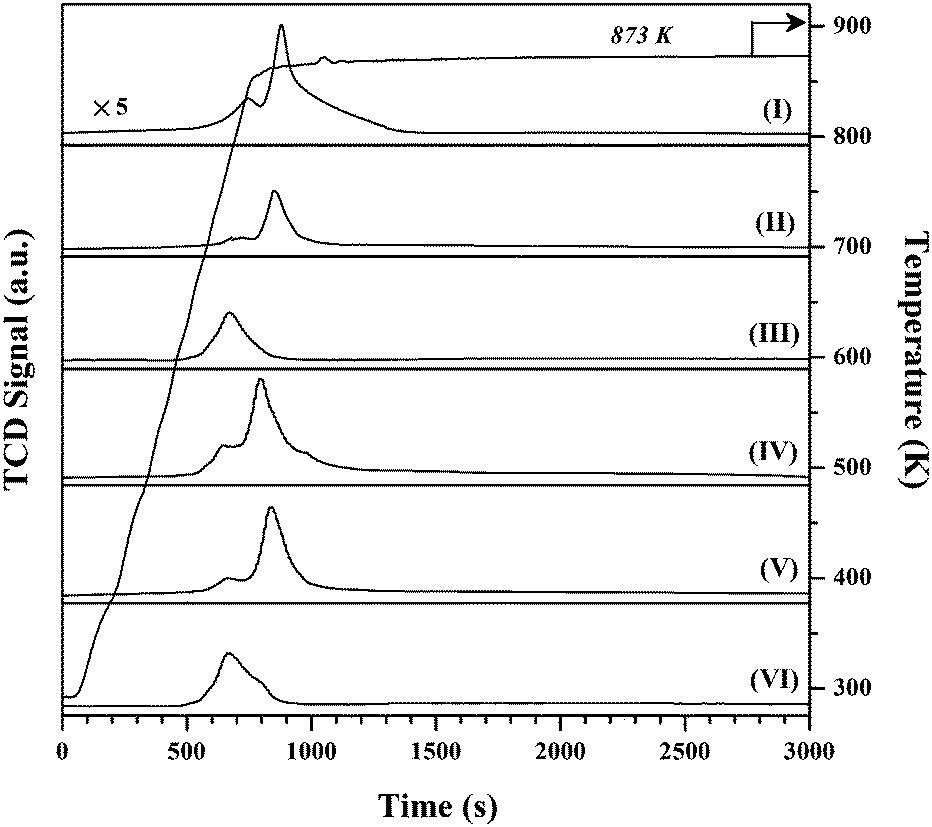
The TPR profiles generated for Au/Al2O3-HL (I), Au/Al2O3-LL (II) and Au/TiO2-LL (III) can be compared in Fig. 1. The three samples present a principal H2 consumption peak with an associated temperature maximum (Tmax) over 383–434 K, which is within the range recorded elsewhere (328–465 K) for the activation of Au/Al2O3 and Au/TiO2 and attributed to precursor reduction.28 The lower Tmax recorded for Au/TiO2-LL (relative to Au/Al2O3) is consistent with the literature.29 Moreover, Delannoy et al.30 have demonstrated (by in situ XAFS and DRIFTS) greater reducibility of cationic gold on TiO2 compared with Al2O3. Hydrogen consumption during catalyst activation was close to that required for Au3+ reduction (Table 1). Supported bimetallic catalyst preparation involved reductive deposition where Ni with a lower electrochemical potential (ECP = −0.27) acts to reduce the Au precursor (HAuCl4, ECP = 1.00).31 The TPR profiles generated for Al2O3 and TiO2 supported Ni (not shown) exhibited a single broad H2 consumption peak at the final isothermal hold (723 K) that can be associated with a combined decomposition-reduction of the precursor to metallic nickel, as noted elsewhere.1 A single stage reduction was observed for the supported Ni–Au systems at a higher Tmax (414–470 K) than that recorded for the corresponding monometallic Au catalysts. Modification to the TPR response for Au containing bimetallic catalysts with respect to monometallic counterparts has been reported and linked to interaction between both metals.32 Pu et al.33 have recently described a stabilisation of Au species (on activated carbon spheres) with the introduction of Ni by co-impregnation that inhibited reduction, displacing the temperature for AuIII → Au0 (from 583 K to 588 K) and AuI → Au0 (from 619 K to 659 K) to higher values.
 | ||
| Fig. 1 TPR profiles generated for: (I) Au/Al2O3-HL, (II) Au/Al2O3-LL, (III) Au/TiO2-LL, (IV) (1:10) Ni–Au/Al2O3-HL, (V) (1:10) Ni–Au/Al2O3-LL and (VI) (1:10) Ni–Au/TiO2-LL. | ||
Gold particle size, from TEM analysis (Fig. 2 and 3 and Table 1), is dependent on metal loading and the nature of the support. The particle size in Au/Al2O3-HL was in the range 1–20 nm (Fig. 3(I)) with a surface area weighted mean diameter of 9 nm. The lower Au loaded sample (Au/Al2O3-LL) exhibited a narrower size distribution (1–8 nm, Fig. 3(II)) and smaller mean (4 nm). This effect is consistent with literature that has shown wider size range and the formation of larger nanoparticles with increasing Au content.34 This can be linked to mobility of the chloride precursor, resulting in Au agglomeration during thermal treatment.35 Enhanced Au dispersion on TiO2 (Fig. 3(III)) is a consequence of a difference in metal/support interaction relative to Au/Al2O3.36 Loss of lattice oxygen with the formation of surface vacancies is a feature of thermal treatment in H2 for reducible oxides such as TiO2 at T ≥ 573 K.37 These surface defects act as Au nucleation sites with electronic interactions that limit particle growth, leading to enhanced dispersion.38,39
 | ||
| Fig. 2 Representative (I) medium and (II) high resolution TEM images with (III) associated diffractogram pattern of an isolated single Au nanoparticle in (A) Au/Al2O3-HL, (B) Au/Al2O3-LL and (C) Au/TiO2-LL. | ||
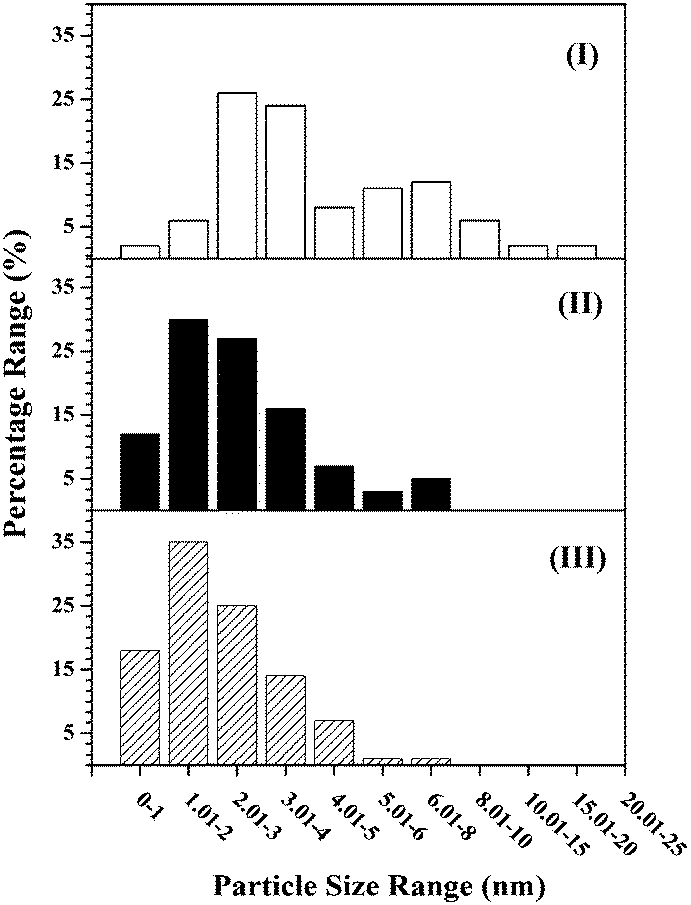 | ||
| Fig. 3 Gold particle size distributions associated with (I) Au/Al2O3-HL (open bars), (II) Au/Al2O3-LL (solid bars) and (III) Au/TiO2-LL (hatched bars). | ||
Representative (S)TEM images of Ni–Au/Al2O3-HL (I), Ni–Au/Al2O3-LL (III) and Ni–Au/TiO2-LL (IV) can be compared in Fig. 4. The metal size range in Ni–Au/Al2O3-HL (1–20 nm) coincided with Au/Al2O3-HL and there was no obvious alteration due the inclusion of Ni. This response extended to Ni–Au/Al2O3-LL and Ni–Au/TiO2-LL with an equivalent mean metal size relative to the monometallic system (see Table 1). This can be attributed to the low Ni content in the bimetallic samples. An unchanged metal particle size has been reported previously for TiO2 supported Au and Pt–Au with low Pt content (0.01–0.03 wt%).9 EDX mapping (see representative analysis for Ni–Au/Al2O3-HL in Fig. 4(II)) demonstrated that individual particles contained both Au and Ni in proportions close to the bulk value (Au/Ni = 10). EDX analysis cannot establish unequivocally the exact nature of Ni–Au interaction or rule out the occurrence of bimetallic clusters40 or surface alloy.41 Both possibilities have been suggested for Ni–Au catalysts prepared in an analogous manner and with a similar metal dispersion to that in this study.16,42,43 Deghedi and co-workers42 proposed the formation of Ni particles covered with Au adatoms (on the basis of EXAFS and TEM-EDX measurements) for Ni–Au/SiO2 synthesised by redox deposition that contained metal ensembles of 5.1 nm. Maniecki and co-workers44 reported an alloy phase (on the basis of XRD) for Ni–Au/Al2O3 prepared by co-impregnation (with Ni(NO3)2 and HAuCl4) and reduction in H2. Surface alloy formation was suggested by EXAFS/XANES for Au–Ni/MgAlO4 (10.1 nm) prepared by redox deposition.16 Molenbroek and Nørskov43 also demonstrated the occurrence of a surface alloy in Ni–Au/SiO2 (2.5–6.0 nm) and Ni–Au/MgAl2O4 (3.0–15.0 nm) by Monte Carlo simulations, which was experimentally verified by EXAFS, TEM and in situ XRD.
 | ||
| Fig. 4 Representative TEM/STEM images of (I) Ni–Au/Al2O3-HL showing (1) 12 nm and (2) 8 nm segments across individual metal particles subjected to (II) EDX mapping with the associated Ni (counts × 10) and Au distribution, (III) Ni–Au/Al2O3-LL and (IV) Ni–Au/TiO2-LL. | ||
The hydrogenation of nitroarenes is enhanced by contributions due to spillover hydrogen.19,36 The generation of spillover species involves dissociative adsorption of H2 on a (donor) supported metal site with migration of the atomic hydrogen generated to the (acceptor) support.45 Hydrogen TPD is a practical approach that can serve to quantify the degree of spillover. Ambient temperature H2 chemisorption following TPR on all the catalysts was low (≤15 mmol molAu−1) and close to the detection limits. Low uptake on supported Au has been demonstrated elsewhere and attributed to the filled d-band and high activation energy barrier for dissociative adsorption.6,46 There was no measurable difference in ambient temperature H2 uptake on the bimetallics, which may be due to the low Ni content. The H2 TPD profiles are presented in Fig. 5. In all cases, the profiles show H2 release with maxima at T ≥ 770 K where the total amount desorbed (per molAu, Table 1) was appreciably greater (by two orders of magnitude) than that taken up in the chemisorption step. This response suggests the release of spillover hydrogen generated during catalyst activation by TPR.45 The shift in the TPD peak for TiO2 systems to a lower temperature (by up to a 100 K) is indicative of a less energetically demanding desorption of H2. This is consistent with literature47,48 that has reported a down-shift (by ca. 60–70 K) in the H2-TPD peak for Ni/TiO2 and Pd–Au/Al2O3-TiO2 compared with Ni/Al2O3 and Pd–Au/Al2O3, respectively. Taking the three monometallic Au catalysts presented in Fig. 5, H2 desorbed (Table 1) increased in the order: Au/Al2O3-HL (I) < Au/Al2O3-LL (II) < Au/TiO2-LL (III) where the values obtained are close (54–932 mmol molAu−1) to those reported elsewhere for Au with similar metal size (3–5 nm) supported on Fe2O3.36 This sequence reflects that of decreasing mean Au nanoparticle size (Table 1). There is evidence in the literature23 that metal size is a crucial variable controlling spillover where a greater (donor) surface area for smaller metal nanoparticles extends the donor/acceptor interface and facilitates spillover transfer. The profiles generated for the bimetallic Ni–Au catalysts (Fig. 5, profiles IV–VI) reveals a significant increase in H2 desorbed relative to Au samples with the same loading (see Table 1). This suggests a surface Ni/Au synergism that impacts on H2 adsorption/desorption dynamics. The existing literature on Au containing bimetallics for hydrogenation applications has been from the perspective of Au as a diluent with low H2 uptake capacity and a resultant decrease in surface hydrogen associated with the bimetallic.49 As a result, there is no directly comparable H2 TPD analysis for supported bimetallic Au-containing systems where the second metal was present in small amounts. We should flag the work of Wojcieszak et al.50 who observed an increase in H2 released (0.22 → 0.38 mol g−1) during TPD of Ni–Ag/SiO2 with increasing Ni content (0.50 → 0.75 wt%). It is important to note that Ni–Au/Al2O3-HL with lower metal dispersion showed significantly greater H2 desorption (Table 1) relative to Ni–Au/Al2O3-LL. STEM-EDX analyses have demonstrated the presence of both metals on individual nanoparticles. The formation of bimetallic nano-crystals (i.e. Au decorating Ni nanoparticles) has been demonstrated (EXAFS and TEM-EDS) for Ni–Au bimetallics prepared and activated using a similar methodology.42 It is known that H2 interaction is distinct for monometallic, bimetallic and alloy nanoparticles.51,52 The greater Ni loading in Ni–Au/Al2O3-HL (0.1 mol% vs. 0.01 mol%) must facilitate H2 dissociative adsorption and spillover during TPR, which is reflected in greater H2 release during TPD. In the case of the low loading bimetallics, H2 desorption from Ni–Au/Al2O3-LL exceeded Ni–Au/TiO2-LL (Table 1). This can be tentatively linked to modifications induced by the support that impact on H2 uptake on the bimetallic phase. Likewise, Gu et al.47 observed a greater H2 TPD from Pd–Au/Al2O3 relative to Pd–Au/Al2O3-TiO2. The characterisation analysis has established the formation of nano-scale (mean = 3–9 nm) Au particles in the monometallics with greater surface hydrogen for catalysts with increasing metal dispersion. TPR measurements suggest interaction of Ni with Au in the bimetallics. STEM-EDX analysis has demonstrated the presence of Au and Ni in individual nanoparticles and the TPD results are consistent with increased spillover resulting from Au/Ni synergy that increases the available surface hydrogen.
 | ||
| Fig. 5 Hydrogen TPD profiles generated for: (I) Au/Al2O3-HL, (II) Au/Al2O3-LL, (III) Au/TiO2-LL, (IV) (1:10) Ni–Au/Al2O3-HL, (V) (1:10) Ni–Au/Al2O3-LL and (VI) (1:10) Ni–Au/TiO2-LL. | ||
3.2 Gas phase nitroarene hydrogenation
The hydrogenation of p-CNB and m-DNB over all the catalysts delivered time-invariant conversions (not shown) and exclusive nitro-group reduction with no evidence of hydrodechlorination, hydrodenitrogenation or aromatic ring reduction. This combined stability/selectivity is important in terms of the viable application of Au catalyst formulations in the production of functionalised anilines. Catalyst deactivation in hydrogenation (of naphthalene,53 1,3-butadiene54 and acetylene55,56) has been reported for Au supported on Al2O353,54,57 and TiO255,56 and ascribed to metal sintering54,55 and coking.55–57 Moreover, temporal loss of activity in the gas phase hydrogenation of nitroarenes over Pd/Al2O3 has been observed and linked to coke formation.58,59The applicability of pseudo-first order kinetics for the gas phase hydrogenation of p-CNB1,60 and m-DNB36 has been established elsewhere. The extracted specific (per molAu) pseudo-first order rate constants (k) are given in Table 2. The promoting effect of Ni is demonstrated by the significantly increased hydrogenation rate when compared with the monometallic Au catalysts with the same loading. Under the same reaction conditions, Ni/Al2O3 and Ni/TiO2 delivered negligible activity. Duan and co-workers61 using unsupported nano-crystals reported increased rate (molproduct molmetal−1) in the high pressure (40 atm) liquid phase hydrogenation of phenol (283 → 363) and benzene (164 → 261) over Ni–Rh relative to Rh, which was attributed to the bimetallic character of the nanoparticles (based on HRTEM-EDX and XRD) and possible modifications in active site electron density. Lu et al.62,63 recorded higher activity in the liquid phase hydrogenation of a series of p-substituted nitro-derivates over Ni–Pd colloids, ascribed to the formation of a Ni enriched alloy (from XRD, EXAFS, XPS). The elevated rate observed in this study can be linked to surface available reactive hydrogen as shown in Fig. 6 where H2-TPD is related to the rate constant. The increase in surface hydrogen that results from incorporation of Ni with Au was accompanied by a higher hydrogenation rate. Moreover, reaction of m-DNB uniformly generated greater rates, a result that can be attributed to the activating effect of the second –NO2 group, which is consistent with a nucleophilic mechanism, as demonstrated previously.64
:10 molar ratio) catalysts; reaction conditions: P = 1 atm, T = 573 K
| Catalyst | p-CNB | m-DNB | ||
|---|---|---|---|---|
| k | Product(s) (Sproduct (%)) | k | Product(s) (Sproduct (%)) | |
| a mol–NO2 molAu−1 h−1. | ||||
| Au/Al2O3-HL | 4 | p-CAN (100) | 6 | m-NAN (100) |
| Ni–Au/Al2O3-HL | 24 | p-CAN (100) | 40 | m-NAN (60) |
| m-PDM (40) | ||||
| Au/Al2O3-LL | 12 | p-CAN (100) | 16 | m-NAN (100) |
| Ni–Au/Al2O3-LL | 21 | p-CAN (100) | 35 | m-NAN (100) |
| Au/TiO2-LL | 18 | p-CAN (100) | 22 | m-NAN (100) |
| Ni–Au/TiO2-LL | 22 | p-CAN (100) | 25 | m-NAN (80) |
| m-PDM (20) | ||||
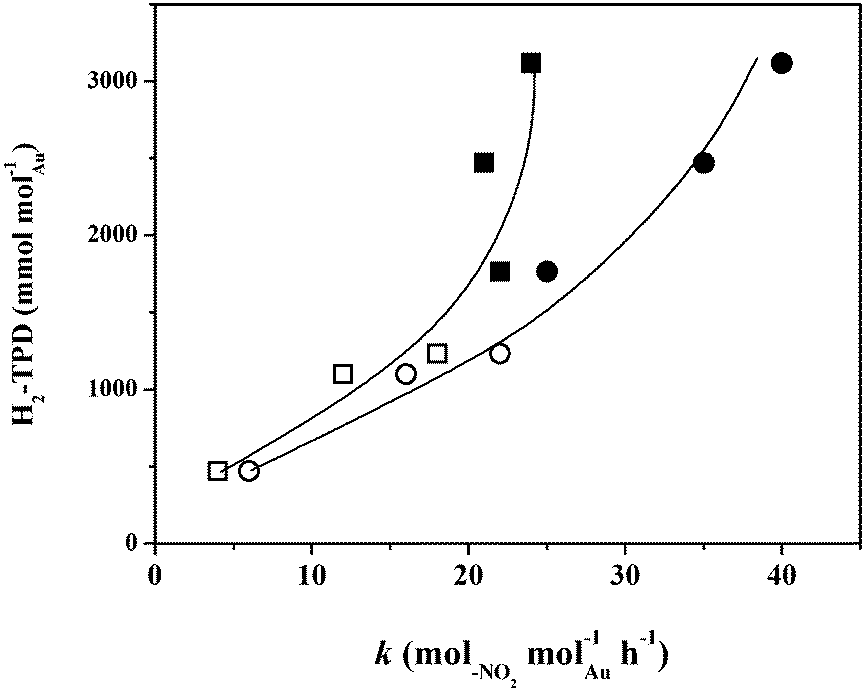 | ||
| Fig. 6 Variation in hydrogen TPD (H2-TPD) with pseudo-first order rate constant (k) in the hydrogenation of p-CNB (□, ■) and m-DNB (○, ●) over supported Au (open symbols) and Ni–Au (solid symbols) catalysts. | ||
Reaction selectivity is compared at the same degree of conversion in Table 2. p-CNB was converted solely to the target (p-chloroaniline) p-CAN over all the catalysts, which is consistent with reaction exclusivity in continuous gas phase p-CNB → p-CAN over oxide supported Au.49 In contrast, nitrobenzene,1 aniline1 and benzene65 have been observed in the gas phase hydrogenation of CNB over Ru,65 and Pd1 catalysts. Variations in product distribution are, however, evident in the hydrogenation of m-DNB and sensitive to the nature of the catalyst. Full selectivity to partially reduced (m-nitroaniline) m-NAN was obtained over monometallic Au systems. In batch liquid phase operation, high pressures (26–34 atm) have been deemed essential to achieve high selectivity (84–98%) to m-NAN.48,66 Preferential –NO2 activation on oxide supported Au in the presence of other reactive (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, carbonyl, amide and ester) functional groups has been demonstrated by FTIR analysis.67 The incorporation of Ni at low loading (Ni–Au/Al2O3-LL) served to increase rate while retaining 100% selective to m-NAN. Deghedi et al.42 reported that the combination of Au and Ni on SiO2 (Ni:Au molar ratio = 4:1) lowered the rate of CC bond and aromatic ring hydrogenation relative to Ni in the conversion of styrene, which was accounted for in terms of geometric and electronic effects. At higher loadings (Ni–Au/Al2O3-HL) complete hydrogenation to m-PDM was observed, which is a characteristic of catalysis by Ni.18,19 This may be a consequence of increased surface reactive hydrogen which facilitates reduction of both –NO2 substituents. Alternatively, m-DNB activation on Ni–Au/Al2O3-HL may be distinct from Ni–Au/Al2O3-LL with a planar adsorption on the former through the aromatic ring, resulting in the formation of a resonance structure with two positive localised charges where both –NO2 groups are activated for nucleophilic attack. It is known that substituted aromatics interact with supported Ni catalysts via the π electron-delocalised aromatic ring.68 Lonergan et al.69 have observed n-butane selectivity effects in 1,3-butadiene hydrogenation over Ni–Pt/Al2O3 with increasing Ni content (1:3 → 1:10). The formation of m-PDM was also promoted over Ni–Au/TiO2-LL, which suggests a support effect in terms of m-DNB adsorption/activation that acts in tandem with differences in available reactive hydrogen. Wang et al.70 reported a modified catalytic response in the hydrogenation of 1,3-budatiene for Pt–Ni on Al2O3 and TiO2. DFT calculations revealed stronger metal interaction with Al2O3 (binding energy ca. −50 kcal mol−1) than TiO2 (−17 kcal mol−1) with a preferential Ni–Al2O3 interaction at the metal–support interface leading to a Pt-terminated bimetallic configuration. A weaker Ni interaction in the case of the TiO2 carrier can then result in some segregation of the Ni component to the nanoparticle surface.
C, carbonyl, amide and ester) functional groups has been demonstrated by FTIR analysis.67 The incorporation of Ni at low loading (Ni–Au/Al2O3-LL) served to increase rate while retaining 100% selective to m-NAN. Deghedi et al.42 reported that the combination of Au and Ni on SiO2 (Ni:Au molar ratio = 4:1) lowered the rate of CC bond and aromatic ring hydrogenation relative to Ni in the conversion of styrene, which was accounted for in terms of geometric and electronic effects. At higher loadings (Ni–Au/Al2O3-HL) complete hydrogenation to m-PDM was observed, which is a characteristic of catalysis by Ni.18,19 This may be a consequence of increased surface reactive hydrogen which facilitates reduction of both –NO2 substituents. Alternatively, m-DNB activation on Ni–Au/Al2O3-HL may be distinct from Ni–Au/Al2O3-LL with a planar adsorption on the former through the aromatic ring, resulting in the formation of a resonance structure with two positive localised charges where both –NO2 groups are activated for nucleophilic attack. It is known that substituted aromatics interact with supported Ni catalysts via the π electron-delocalised aromatic ring.68 Lonergan et al.69 have observed n-butane selectivity effects in 1,3-butadiene hydrogenation over Ni–Pt/Al2O3 with increasing Ni content (1:3 → 1:10). The formation of m-PDM was also promoted over Ni–Au/TiO2-LL, which suggests a support effect in terms of m-DNB adsorption/activation that acts in tandem with differences in available reactive hydrogen. Wang et al.70 reported a modified catalytic response in the hydrogenation of 1,3-budatiene for Pt–Ni on Al2O3 and TiO2. DFT calculations revealed stronger metal interaction with Al2O3 (binding energy ca. −50 kcal mol−1) than TiO2 (−17 kcal mol−1) with a preferential Ni–Al2O3 interaction at the metal–support interface leading to a Pt-terminated bimetallic configuration. A weaker Ni interaction in the case of the TiO2 carrier can then result in some segregation of the Ni component to the nanoparticle surface.
4. Conclusions
Oxide (Al2O3 and TiO2) supported Au (1 and 0.1 mol%) prepared by impregnation with subsequent activation generated nano-scale metal particles of mean size 3–9 nm. A decrease in Au size was accompanied by increased surface hydrogen (from TPD analysis) with higher specific chemoselective hydrogenation rate in the conversion of p-CNB → p-CAN and m-DNB → m-NAN. Supported bimetallic synthesis by reductive deposition of Au on Ni (where Au/Ni = 10) generated an equivalent particle size at the same Au loading but elevates the available surface hydrogen with a significant increase in –NO2 group reduction rate. TPR analysis suggests Ni–Au interaction where (S)TEM-EDX mapping across isolated single particles has established Au and Ni in close proximity with a surface Au/Ni that coincided with the bulk value. Reaction of p-CNB over the bimetallics delivered full selectivity to p-CAN. Conversion of m-DNB was fully selective to m-NAN over monometallic Au and Ni–Au/Al2O3-LL. The hydrogenation of both –NO2 substituents (to m-PDM) was promoted over Ni–Au/Al2O3-HL and Ni–Au/TiO2-LL. This is attributed to the higher Ni loading that elevated the surface supply of hydrogen and possible electronic/geometric modifications induced by the support that impact on the m-DNB adsorption/activation. We have demonstrated Au/Ni synergy that serves to enhance –NO2 reduction rate.Acknowledgements
The authors are grateful to Dr Santiago Gómez-Quero, Dr Noémie Perret and Pedro Benavente Donayre for their contribution to the work.References
- F. Cárdenas-Lizana, S. Gómez-Quero, C. Amorim and M. A. Keane, Appl. Catal., A, 2014, 473, 41 CrossRef PubMed.
- A. Yarulin, I. Yuranov, F. Cárdenas-Lizana, D. T. L. Alexander and L. Kiwi-Minsker, Appl. Catal., A, 2014, 478, 186 CrossRef CAS PubMed.
- G. Yuan, C. Louis, L. Delannoy and M. A. Keane, J. Catal., 2007, 247, 256 CrossRef CAS PubMed.
- A. Villa, D. Wang, D. S. Su and L. Prati, Catal. Sci. Technol., 2015, 5, 55 CAS.
- F. Cárdenas-Lizana and M. A. Keane, in Gold Catalysis: Preparation, Characterization and Applications in the Gas and Liquid Phase, ed. L. Prati and A. Villa, Pan Stanford Publishing, Singapore, 2014 Search PubMed.
- C. Kartusch and J. A. van Bokhoven, Gold Bull., 2009, 42, 343 CrossRef CAS.
- G. C. Bond, Platinum Met. Rev., 2007, 51, 63 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, A. Hugon, L. Delannoy, C. Louis and M. A. Keane, J. Catal., 2009, 262, 235 CrossRef PubMed.
- D. P. He, X. D. Jiao, P. Jiang, J. Wang and B. Q. Xu, Green Chem., 2012, 14, 111 RSC.
- C. Louis, in Environmental Catalysis over Gold-based Materials, ed. G. Avgouropoulos and T. Tabakova, The Royal Society of Chemistry, Cambridge, 2013 Search PubMed.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Nørskov and I. Stensgaard, Science, 1998, 279, 1913 CrossRef CAS.
- L. P. Nielsen, F. Besenbacher, I. Stensgaard, E. Laegsgaard, C. Engdahl, P. Stoltze, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 1993, 71, 754 CrossRef CAS.
- A. G. Rybkin, D. Y. Usachev, A. Y. Varykhalov, A. M. Shikin and V. K. Adamchuk, Phys. Solid State, 2007, 49, 984 CrossRef CAS.
- A. V. Naumkin and A. Y. Vasil'kov, Russ. Chem. Bull., 2013, 62, 2559 CrossRef CAS.
- H. Nishikawa, D. Kawamoto, Y. Yamamoto, T. Ishida, H. Ohashi, T. Akita, T. Honma, H. Oji, Y. Kobayashi, A. Hamasaki, T. Yokoyama and M. Tokunaga, J. Catal., 2013, 307, 254 CrossRef CAS PubMed.
- Y.-H. Chin, D. L. King, H.-S. Roh, Y. Wang and S. M. Heald, J. Catal., 2006, 244, 153 CrossRef CAS PubMed.
- J. Zhang, H. Asakura, J. van Rijn, J. Yang, P. Duchesne, B. Zhang, X. Chen, P. Zhang, M. Saeys and N. Yan, Green Chem., 2014, 16, 2432 RSC.
- F. Cárdenas-Lizana, S. Gómez-Quero, G. Jacobs, Y. Ji, B. H. Davis, L. Kiwi-Minsker and M. A. Keane, J. Phys. Chem. C, 2012, 116, 11166 Search PubMed.
- F. Cárdenas-Lizana, S. Gómez-Quero, C. J. Baddeley and M. A. Keane, Appl. Catal., A, 2010, 387, 155 CrossRef PubMed.
- F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, Appl. Catal., A, 2008, 334, 199 CrossRef PubMed.
- W.-S. Lee, B.-Z. Wan, C.-N. Kuo, W.-C. Lee and S. Cheng, Catal. Commun., 2007, 8, 1604 CrossRef CAS PubMed.
- R. Zanella and C. Louis, Catal. Today, 2005, 107–108, 768 CrossRef CAS PubMed.
- C. Amorim and M. A. Keane, J. Colloid Interface Sci., 2008, 322, 196 CrossRef CAS PubMed.
- J. Chen, X. Liu and F. Zhang, Chem. Eng. J., 2015, 259, 43 CrossRef CAS PubMed.
- E. Kok, N. Cant, D. Trimm and J. Scott, Catal. Today, 2011, 178, 79 CrossRef CAS PubMed.
- J. W. Bae, Y.-J. Lee, J.-Y. Park and K.-W. Jun, Energy Fuels, 2008, 22, 2885 CrossRef CAS.
- T. R. Jagannath, PhD dissertation, Saurashtra University, 2005.
- Y.-J. Chen and C.-T. Yeh, J. Catal., 2001, 200, 59 CrossRef CAS.
- S. Y. Liu and S. M. Yang, Appl. Catal., A, 2008, 334, 92 CrossRef CAS PubMed.
- L. Delannoy, N. Weiher, N. Tsapatsaris, A. M. Beesley, L. Nchari, S. L. M. Schroeder and C. Louis, Top. Catal., 2007, 44, 263 CrossRef CAS PubMed.
- D. R. Lide, Handbook of Chemistry and Physical Properties, Taylor Francis, Boca Raton, 88th edn, 2007–2008 Search PubMed.
- F.-W. Chang, L. S. Roselin and T.-C. Ou, Appl. Catal., A, 2008, 334, 147 CrossRef CAS PubMed.
- Y. Pu, J. Zhang, X. Wang, H. Zhang, L. Yu, Y. Dong and W. Li, Catal. Sci. Technol., 2014, 4, 4426 CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, H. Idriss and M. A. Keane, J. Catal., 2009, 268, 223 CrossRef PubMed.
- A. Schulz and M. Hargittai, Chem. – Eur. J., 2001, 7, 3657 CrossRef CAS.
- F. Cárdenas-Lizana, S. Gómez-Quero, N. Perret and M. A. Keane, Catal. Sci. Technol., 2011, 1, 652 Search PubMed.
- P. Claus, A. Brückner, C. Mohr and H. Hofmeister, J. Am. Chem. Soc., 2000, 122, 11430 CrossRef CAS.
- N. Lopez, J. K. Nørskov, T. V. W. Janssens, A. Carlsson, A. Puig-Molina, B. S. Clausen and J.-D. Grunwaldt, J. Catal., 2004, 225, 86 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737 CAS.
- D. Chen, J. Li, C. Shi, X. Du, N. Zhao, J. Sheng and S. Liu, Chem. Mater., 2007, 19, 3399 CrossRef CAS.
- S.-H. Zhou, Z. Ma, H.-F. Yin, Z.-L. Wu, B. Eichhorn, S. H. Overbury and S. Dai, J. Phys. Chem. C, 2009, 113, 5758 CAS.
- L. Deghedi, J.-M. Basset, G. Bergeret, J.-P. Candy, M. C. Valero, J.-A. Dalmon, A. de Mallmann, A.-C. Dubreuil and L. Fischer, in 10th International Symposium “Scientific Bases for the Preparation of Heterogeneous Catalysts”, ed. E. M. Gaigneaux, M. Devillers, S. Hermans, P. Jacobs, J. Martens and P. Ruiz, Elsevier, Louvain-la-Newuve, 2010, vol. 175, p. 617 Search PubMed.
- A. M. Molenbroek and J. K. Nørskov, J. Phys. Chem. B, 2001, 105, 5450 CrossRef CAS.
- T. P. Maniecki, K. Bawolak, D. Gebauer, P. Mierczynski and W. K. Jozwiak, Kinet. Catal., 2009, 50, 138 CrossRef CAS.
- W. C. Conner and J. L. Falconer, Chem. Rev., 1995, 95, 759 CrossRef CAS.
- J. Harris, Surf. Sci., 1989, 221, 335 CrossRef CAS.
- Z. Gu, L. Luo and M. Teng, Indian J. Chem., 2007, 46A, 742 CAS.
- Y. X. Liu, J. X. Chen and J. Y. Zhang, Chin. J. Chem. Eng., 2007, 15, 63 CrossRef CAS PubMed.
- F. Cárdenas-Lizana and M. A. Keane, J. Mater. Sci., 2013, 48, 543 CrossRef.
- R. Wojcieszak, S. Monteverdi, J. Ghanbaja and M. M. Bettahar, J. Colloid Interface Sci., 2008, 317, 166 CrossRef CAS PubMed.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810 CrossRef CAS PubMed.
- J. Greeley and M. Mavrikakis, J. Phys. Chem. B, 2005, 109, 3460 CrossRef CAS PubMed.
- B. Pawelec, A. M. Venezia, V. La Parola, S. Thomas and J. L. G. Fierro, Appl. Catal., A, 2005, 283, 165 CrossRef CAS PubMed.
- X. Zhang, H. Shi and B.-Q. Xu, Catal. Today, 2007, 122, 330 CrossRef CAS PubMed.
- T. V. Choudhary, C. Sivadinarayana, A. K. Datye, D. Kumar and D. W. Goodman, Catal. Lett., 2003, 86, 1 CrossRef CAS.
- Y. Azizi, C. Petit and V. Pitchon, J. Catal., 2008, 256, 338 CrossRef CAS PubMed.
- F. Cárdenas-Lizana, X. Wang, D. Lamey, M. Li, M. A. Keane and L. Kiwi-Minsker, Chem. Eng. J., 2014, 255, 695 CrossRef PubMed.
- V. Vishwanathan, V. Jayasri, P. M. Basha, N. Mahata, L. M. Sikhwivhilu and N. J. Coville, Catal. Commun., 2008, 9, 453 CrossRef CAS PubMed.
- K. K. Yeong, A. Gavriilidis, R. Zapf and V. Hessel, Catal. Today, 2003, 81, 641 CrossRef CAS.
- F. Cárdenas-Lizana, B. Bridier, C. C. K. Shin, J. Pérez-Ramírez and L. Kiwi-Minsker, ChemCatChem, 2012, 4, 668 CrossRef.
- H. Duan, D. Wang, Y. Kou and Y. Li, Chem. Commun., 2013, 49, 303 RSC.
- P. Lu and N. Toshima, Bull. Chem. Soc. Jpn., 2000, 73, 751 CrossRef CAS.
- P. Lu, T. Teranishi, K. Asakura, M. Miyake and N. Toshima, J. Phys. Chem. B, 1999, 103, 9673 CrossRef CAS.
- F. Cárdenas-Lizana, Z. Martínez de Pedro, S. Gómez-Quero and M. A. Keane, J. Mol. Catal., 2010, 326, 48 CrossRef PubMed.
- C. Xiao, X. D. Wang, C. Lian, H. Q. Liu, M. H. Liang and Y. Wang, Curr. Org. Chem., 2012, 16, 280 CrossRef CAS.
- V. L. Khilnani and S. B. Chandalia, Org. Process Res. Dev., 2001, 5, 263 CrossRef CAS.
- K. Shimizu, Y. Miyamoto, T. Kawasaki, T. Tanji, Y. Tai and A. Satsuma, J. Phys. Chem. C, 2009, 113, 17803 CAS.
- E.-J. Shin and M. A. Keane, J. Catal., 1998, 173, 450 CrossRef CAS.
- W. W. Lonergan, D. G. Vlachos and J. G. Chen, J. Catal., 2010, 271, 239 CrossRef CAS PubMed.
- T. Wang, G. Mpourmpakis, W. W. Lonergan, D. G. Vlachos and J. G. Chen, Phys. Chem. Chem. Phys., 2013, 15, 12156 RSC.
| This journal is © the Owner Societies 2015 |