
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComplex transition metal hydrides incorporating ionic hydrogen: thermal decomposition pathway of Na2Mg2FeH8 and Na2Mg2RuH8
Terry D.
Humphries
*a,
Motoaki
Matsuo
b,
Guanqiao
Li
a and
Shin-ichi
Orimo
ab
aWPI-Advanced Institute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: terry_humphries81@hotmail.com; Fax: +81-22-215-2091; Tel: +81-22-215-2094
bInstitute for Materials Research, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: orimo@imr.tohoku.ac.jp; Fax: +81-22-215-2091; Tel: +81-22-215-2093
First published on 17th February 2015
Abstract
Complex transition metal hydrides have potential technological application as hydrogen storage materials, smart windows and sensors. Recent exploration of these materials has revealed that the incorporation of anionic hydrogen into these systems expands the potential number of viable complexes, while varying the countercation allows for optimisation of their thermodynamic stability. In this study, the optimised synthesis of Na2Mg2TH8 (T = Fe, Ru) has been achieved and their thermal decomposition properties studied by ex situ Powder X-ray Diffraction, Gas Chromatography and Pressure-Composition Isotherm measurements. The temperature and pathway of decomposition of these isostructural compounds differs considerably, with Na2Mg2FeH8 proceeding via NaMgH3 in a three-step process, while Na2Mg2RuH8 decomposes via Mg2RuH4 in a two-step process. The first desorption maxima of Na2Mg2FeH8 occurs at ca. 400 °C, while Na2Mg2RuH8 has its first maxima at 420 °C. The enthalpy and entropy of desorption for Na2Mg2TH8 (T = Fe, Ru) has been established by PCI measurements, with the ΔHdes for Na2Mg2FeH8 being 94.5 kJ mol−1 H2 and 125 kJ mol−1 H2 for Na2Mg2RuH8.
Introduction
Transition metals are renowned for their diverse range of valence states and structural conformations.1,2 As such, in the last five decades a swathe of homoleptic transition metal hydrides have been synthesised to determine their potential for technological applications. Mg2NiH4 was first realised for its reversible hydrogenation properties in 1968,3 and has since been investigated for a variety technological applications including smart windows and sensors.4–7 Mg2FeH6, with a gravimetric hydrogen storage content of 5.5 wt% has since been developed,8–14 along with a host of other transition metal hydride congeners and derivatives.1,2,15–22The transition metal hydrides of Group 8 often form octahedral [TH6]4− anions, of which are limited to four-fold coordination by counterions (M) in the form of M+M′+M′′+M′′′+, M2+M′+M′′+, M3+M′+, M2+M′2+M′′+. Expanding the diversity of coordination can be achieved by increasing the anionic charge of the system, for instance by the inclusion of H−. A recent DFT study by Takagi et al. established that the incorporation of anionic hydrogen into complex transition metal hydride compounds enables inclusion of a wider variety of cations, thereby allowing tuning of these materials in order to optimise their thermodynamic properties or hydrogen storage capacities.23 To date, a variety of quaternary complex hydrides have been synthesised and their structural and physical properties explored, these include LaMg2NiH7 (La3+·2Mg2+·3H−·[NiH4]4−),18,24 Na2Mg2NiH6 (2Na+·2Mg2+·2H−·[NiH4]4−),19,25 Na2Mg2TH8 (2Na+·2Mg2+·2H−·[TH6]4−) (T = Fe, Ru),20 MMg2FeH8 (M2+·2Mg2+·2H−·[TH6]4−) (M = Ba, Ca, Sr; T = Fe, Ru, Os)15,16,26 and M4Mg4Fe3H22 (4Ca2+·4Mg2+·4H−·3[FeH6]4−) (M = Ca, Yb).27,28 Thermodynamic data for these materials are scarce, although some experimental15,28 and DFT calculated23 values have been determined. SrMg2FeH8 and BaMg2FeH8 decompose at ca. 440 and 450 °C under 0.1 MPa H2,15 respectively, while Ca4Mg4Fe3H22 and Yb4Mg4Fe3H22 decompose at ca. 395 and 420 °C,1,27,28 respectively. The enthalpy of desorption of Ca4Mg4Fe3H22 and Yb4Mg4Fe3H22 to their corresponding binary hydrides has been calculated to be 122 and 137 kJ mol−1 H2, respectively.28 These values are significantly larger than those determined for the ternary Mg2FeH6 at 87 kJ mol−1 H2,29 which decomposes at ca. 300 °C,13 and indicates the increased stabilisation offered by the incorporation of anionic hydrogen and varied cations into these quaternary compounds.
The isostructural compounds of Na2Mg2FeH8 (5.1 wt% H) and Na2Mg2RuH8 (4.0 wt% H) hold potential as hydrogen storage materials.20 To make a fair assumption of their prospective application, a true understanding of the physical properties of these novel quaternary complex transition metal hydrides must be determined, unto which the data is extremely sparse. As a consequence, the influence of H− on the thermal stability and decomposition process of these materials is generally unknown and must be understood. As such, ex situ powder X-ray diffraction (PXD) and Pressure-Composition Isotherm Measurements (PCI) have been conducted. Their temperatures and pathways of decomposition have been established and the associated enthalpies and entropies of H2 desorption have been calculated and compared to literature values.
Experimental
All preparation and manipulation was performed in a Miwa glove box filled with purified argon (<1 ppm O2 and the dew point of H2O below 190 K) to avoid contamination.The synthesis of Na2Mg2FeH8 was carried out by two methods: S1 followed a four step process, which first required the synthesis of Mg2FeH6. This was achieved by mechanically milling (Fritsch Pulverisette 7) MgH2 (hydrogen storage grade, Sigma Aldrich) and Fe (99.99%, Mitsuwa) powders at a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for 2 h at 400 rpm (ball-to-powder ratio 40:1), under argon with subsequent heat treatment of the pelletised powder at 400 °C for 20 h under 3 MPa H2. The resultant olive green powder was then mechanically milled with NaH (95%, Sigma Aldrich) at a molar ratio of 1:2 for 20 h under argon (S1-BM) with subsequent heat treatment of the pelletised powder at 400 °C for 20 h under 30 MPa H2. The product was yielded as an olive green powder.
:1 for 2 h at 400 rpm (ball-to-powder ratio 40:1), under argon with subsequent heat treatment of the pelletised powder at 400 °C for 20 h under 3 MPa H2. The resultant olive green powder was then mechanically milled with NaH (95%, Sigma Aldrich) at a molar ratio of 1:2 for 20 h under argon (S1-BM) with subsequent heat treatment of the pelletised powder at 400 °C for 20 h under 30 MPa H2. The product was yielded as an olive green powder.
The synthesis of S2 (Na2Mg2FeH8) followed a two-step process. NaH, MgH2 and Fe powders at a molar ratio of 2:2:1 were mechanically milled for 20 h at 400 rpm (ball-to-powder ratio 40:1), under argon (S2-BM) with subsequent heat treatment of the pelletised powder at 400 °C for 60 h under 30 MPa H2. The product was yielded as an olive green powder.
The synthesis of Na2Mg2RuH8 followed a two-step process. MgH2, NaH and Ru (99.9%, Kojundo Chemical Laboratory) were mechanically milled (identical parameters as employed with Na2Mg2FeH8) at a molar ratio of 2:2:1 for 20 h under argon (S3-BM), before subsequent heat treatment of the pelletised powder at 500 °C for 20 h under 30 MPa H2. The product was yielded as a light grey powder.
Powder X-ray diffraction (PXD) measurements were conducted using a conventional X-ray diffractometer (Lab-PXD, PANalytical X'Pert-Pro, CuKα radiation) in flat plate mode. Data were collected using a X'Celerator X linear position sensitive detector within a 2θ range of 10–90° using 0.02° steps at 0.04°/s with X-ray generator operating conditions of 45 kV and 40 mA. The PXD samples were loaded in an Ar glovebox and the sample holder covered by Mylar film to prevent oxygen/moisture contamination during data collection. PANalytical HighScore Plus v. 3.0, DICVOL,30 CHEKCELL31 and GSAS32,33 were used for phase identification, indexing, space group identification and Rietveld refinement, respectively.
A GC323 (Gas Chromatography) (GL sciences Inc.) was used to detect the desorbed H2 by means of a TCD detector, with a column temperature of 200 °C. Samples were heated at a rate of 5 °C min−1 under an Ar flow of 40 ml min−1.
Typical Pressure-Composition Isotherm Measurements (PCI) were conducted inside custom-built manometric apparatus, where the sample cell was placed in a furnace and heated to the desired temperature at a hydrogen pressure of 6 MPa or 30 MPa. The experiment was controlled by software developed by Suzuki Shokan Co., Ltd.
Results and discussion
Synthesis of Na2Mg2TH8 (T = Fe, Ru)
The synthesis of Na2Mg2FeH8 was first reported to utilise Mg2FeH6 as a starting material, which was consequently milled for 20 h with NaH (S1-BM), followed by hydrogenation at 30 MPa H2 at 400 °C (eqn (1) and (2)).20 Overall, this method is time intensive (in excess of 100 h due to the requirement of two milling and two hydrogenation procedures), and the final product also contains Fe, NaH, and Mg2FeH6 impurities. In order to reduce the time requirements and levels of impurities, alternative synthetic routes were investigated. It was determined that successful synthesis is achievable by ball milling 2NaH + MgH2 + Fe (S2-BM), followed by hydrogenation at 30 MPa H2 at 400 °C (eqn (3)), with an associated time requirement of 80 h. The products after milling of each material differ considerably (Fig. 1), the constituents of S1-BM, identified by PXD are not altered during milling (Mg2FeH6 + NaH) despite a broadening of the peaks due to a decrease of crystallite size (or reduced crystallinity).34 The products of S2-BM were NaMgH3 + Fe.35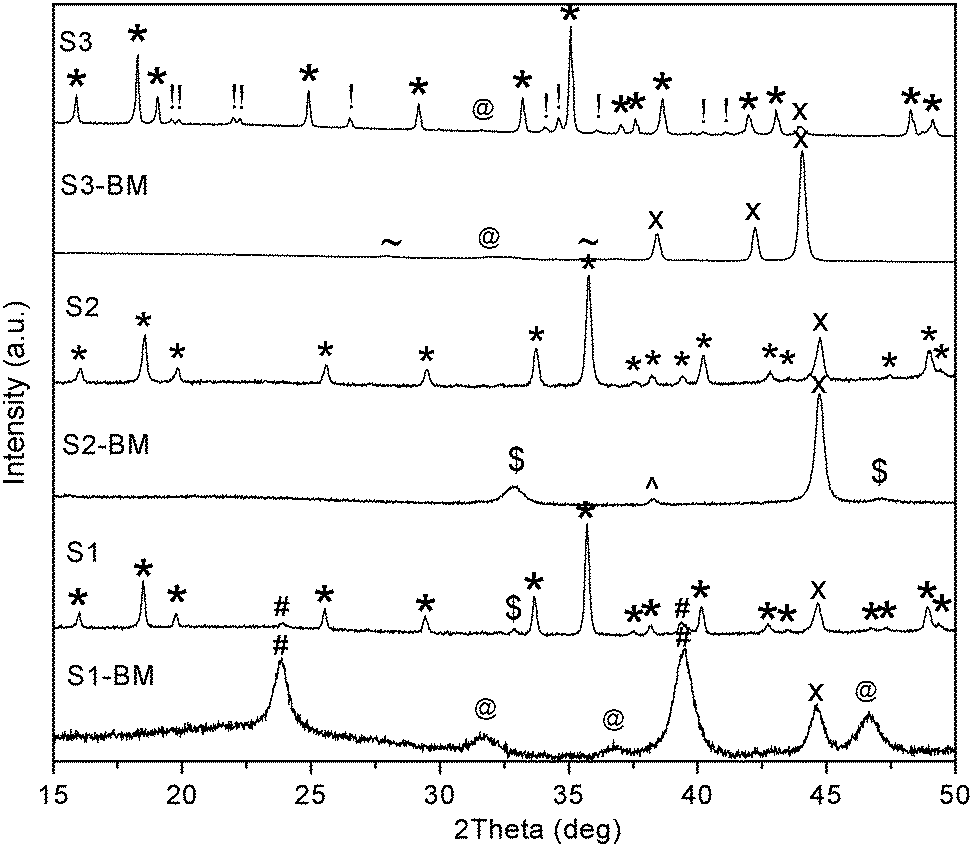 | ||
| Fig. 1 PXD of ball milled samples S1-BM (Mg2FeH6 + 2NaH), S2-BM (2NaH + 2MgH2 + Fe) and S3-BM (2MgH2 + 2NaH + Ru) and hydrogenated samples of S1-3. λ = CuKα. * = Na2Mg2TH8 (T = Fe, Ru); # = Mg2FeH6; @ = NaH; x = T (Fe, Ru); $ = NaMgH3; ^ = NaOH; + = Mg; ∼ MgH2; ! = unknown phase. | ||
Hydrogenation of the ball milled samples S1-BM and S2-BM under standard conditions (30 MPa H2 and 400 °C for 60 h), resulted in the formation of Na2Mg2FeH8 as the major phase (Fig. 1). Analysis of hydrogenated S1 by Reitveld refinement indicates that the sample is 80.3(9)% pure and as such contains residual Mg2FeH6 (8.9(4)%), Fe (7.9(2)%), NaMgH3 (2.5(3)%) and NaH (2.7(2)%) starting materials. Analysis of hydrogenated S2 indicates that the sample contains 90.6(4)% Na2Mg2FeH8 and 9.4(3)% Fe, without residual Mg2FeH6 or NaH. This indicates that the optimal method of synthesis is via the S2 method due to the elimination of the prerequisite Mg2FeH6 synthesis (eqn (1)) and overall decrease in impurities compared to S1, primarily due to the initial (complete) formation of NaMgH3 during the milling reaction. Milling initiates the breaking of the strong Na–H bonds, which is required to ensue during the annealing phase when synthesised via Mg2FeH6 (S1). This ultimately leads to the observation of unreacted NaH and Mg2FeH6 starting materials.
The synthesis of Na2Mg2RuH8 follows a two-step reaction, where stoichiometric quantities of Ru, NaH and MgH2 are milled for 5 h (S3-BM) before hydrogenation under 30 MPa H2 at 500 °C for 20 h (S3) (eqn (4)).20 The composition of the milled material is unchanged from the starting materials (Fig. 1), whereas after hydrogenation the sample appears to be mostly Na2Mg2RuH8, but also comprises of some residual Ru. An unknown material is also identifiable within the Na2Mg2RuH8 powder (S3), of which only a few reflections are discernible. The occurrence of these additional Bragg peaks were also noted previously.20
 | (1) |
 | (2) |
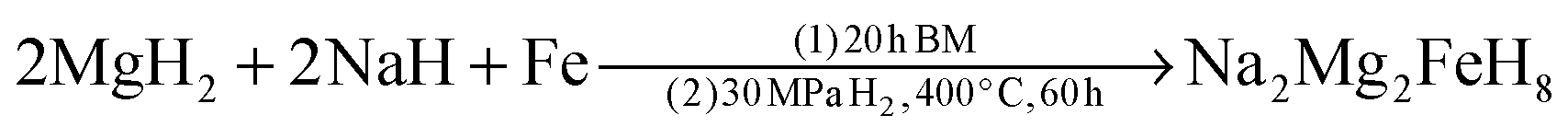 | (3) |
 | (4) |
Thermal decomposition of Na2Mg2TH8 (T = Fe, Ru)
In order to determine the thermal decomposition temperatures of Na2Mg2TH8 (T = Fe, Ru), aliquots of each material were heated at a rate of 5 °C min−1 with GC detecting the corresponding desorbed H2. The chromatograms obtained (Fig. 2a) are similar, with two stages of H2 desorption being observed for both materials (S1 and S3). The onset of decomposition for Na2Mg2FeH8 occurs at ca. 280 °C, with the first maxima being observed at ca. 400 °C, and the second at ca. 430 °C. Decomposition concludes at ca. 475 °C. Conversely, the onset of H2 desorption for Na2Mg2RuH8 occurs at ca. 325 °C, with the first maxima being observed at ca. 421 °C, and the second at ca. 460 °C. H2 is no longer detected after ca. 500 °C. | ||
| Fig. 2 (a) GC analysis of Mg2FeH6, Na2Mg2FeH8 (S1) and Na2Mg2RuH8 (S3) (ΔT = 5 °C min−1). Ex situ PXD analysis of (b) Na2Mg2FeH8 (S1) and (c) Na2Mg2RuH8 (S3) at selected temperatures (λ = CuKα). Numbers on GC plots correlate to temperatures at which samples were heated prior to PXD analysis. * = Na2Mg2TH8 (T = Fe, Ru); # = Mg2FeH6; @ = NaH; x = T (Fe, Ru); $ = NaMgH3; ^ = NaOH; + = Mg; α = Mg3Ru2; ! = unknown phase; θ = unknown phase. | ||
To ascertain the pathway of decomposition, ex situ PXD was conducted on samples heated to selected temperatures in vacuo (Fig. 2b and c). Analysis of Na2Mg2FeH8 after heating at 360 °C indicates a miniscule quantity of Na2Mg2FeH8 resides, although the majority has decomposed into NaH, Mg and Fe, while NaMgH3 is also detected. By 400 °C NaH has decomposed, while only Mg and Fe are observable by PXD. Na is not observed due to the low vapor pressure of Na at elevated temperatures. No further changes to the material are observed at higher temperatures. Therefore the decomposition of Na2Mg2FeH8 is determined to occur according to eqn (5)–(7).
| Na2Mg2FeH8 → 2NaMgH3 + Fe + H2 (1.3 wt%) | (5) |
| 2NaMgH3 + Fe → 2NaH + 2Mg + 2H2 + Fe (2.5 wt%) | (6) |
| 2NaH + 2Mg + Fe → 2Na + 2Mg + Fe + H2 (1.3 wt%) | (7) |
| Na2Mg2RuH8 → 2NaH + 2/3Mg3Ru2 + 3H2 (3.0 wt%) | (8) |
| 2NaH + Mg3Ru2 → 2Na + Mg3Ru2 + H2 (1.0 wt%) | (9) |
 | ||
| Fig. 3 PCI analysis of Na2Mg2FeH8 (a) and Na2Mg2RuH8 (b) at selected temperatures. van't Hoff plot of H2 desorption equilibrium pressures and the linear fit to the data for Na2Mg2FeH8 (c) and Na2Mg2RuH8 (d). Ex situ PXD analysis of Na2Mg2FeH8 (S1) (e) and Na2Mg2RuH8 (S3) (f) for samples collected after PCI analysis and after rehydrogenation (λ = CuKα). * = Na2Mg2TH8 (T = Fe, Ru); # = Mg2FeH6; @ = NaH; x = T (Fe, Ru); $ = NaMgH3; ^ = NaOH; + = Mg; Δ = Na; α = Mg3Ru2; δ = Mg2RuH4; ! = unknown phase; θ = unknown phase; γ = unknown phase; ε = unknown phase. | ||
 | ||
| Fig. 4 Energy diagram illustrating the experimentally determined enthalpies of formation (ΔHform kJ mol−1) of Na2Mg2FeH8, Mg2FeH6,29 and Na2Mg2RuH8. The excess Ru required to form Mg3Ru2 is acquired from the impurity Ru remaining in the Na2Mg2RuH8 starting material. | ||
The further two equilibrium plateaus below 1 MPa H2 correspond to the decomposition of NaMgH3, exhibiting mass losses of 2.5 and 1.5 wt% for eqn (6) and (7), respectively. The overall hydrogen content released was therefore determined to be 4.7 wt% at 400 °C (theoretical maximum of 5.1 wt%). ΔHdec was calculated to be 87 and 111 kJ mol−1 H2 for the latter two processes, in accord with the literature values.36 The corresponding ΔSdec also agreed with literature values with 132 and 158 J mol−1 H2/K for eqn (6) and (7), respectively. Therefore ΔHdes for the entire system is surmised to be 378 kJ mol−1 (94.5 kJ mol−1 H2). The identity of the species at each decomposition stage was determined by PXD by ending selected PCI experiments at specified pressures. Fig. 3e illustrates the final products after the PCI experiments conducted at 360 and 400 °C and also those observed after the first and during the second equilibrium step (eqn (6)). During the second equilibrium step, NaMgH3, Fe and NaH, and Mg are observed, indicating that NaMgH3 is decomposing. After the third equilibrium (final products), Na, Mg and Fe are the main constituents, although residual NaH is also observed. Therefore the decomposition process can be described according to eqn (5)–(7) and Fig. 4.
The thermal stability of Na2Mg2FeH8 is enhanced compared to that of Mg2FeH6, which exhibits a H2 desorption maxima at ca. 360 °C (Fig. 2) with an associated ΔHdes of 261 kJ mol−1.29 The additional stability achieved by the incorporation of Na+ and H− into the compound, induces a significant increase in desorption temperature maxima to 400 °C and a total ΔHdes of 378 kJ mol−1 (Fig. 4). This value correlates very well with the previous DFT calculations conducted on this compound, which determined ΔHf to be −328 kJ mol−1.23
In contrast to Na2Mg2FeH8, Na2Mg2RuH8 is stable above pressures of 0.19 MPa H2 and T >500 °C (Fig. 3a). PXD analysis of material annealed at 6 MPa H2 and 450 °C (Fig. 3f) indicates that the only modification is the disappearance of the unknown phase (observed after initial synthesis (Fig. 1)) which is replaced by another unknown phase. This material can be indexed to an orthorhombic unit cell of a = 14.5331, b = 7.9841 and c = 6.2429 and crystallises in a possible space group of Pmmn, although structural identification is inhibited by the low concentration and weak intensity of the Bragg peaks. As was observed from the GC results (Fig. 1), decomposition is also noted to follow a two-step decomposition route by PCI. At 500 °C, the first plateau is observed at an equilibrium pressure of 0.19 MPa, while the second occurs at ca. 0.07 MPa. Each step was determined to have an associated mass loss of ca. 1.9 wt%, with a total of 3.8 wt% H2 being desorbed out of a maximum theoretical capacity of 4.0 wt%. This process was also carried out at 475 and 450 °C. This allowed for ΔHdes and ΔS to be determined to be 131 kJ mol−1 H2 and 176 ΔS (J mol−1 H2/K), respectively for step 1 (R2 = 0.984) and ΔHdes = 119 kJ mol−1 H2 and ΔS = 151 ΔS (J mol−1 H2/K) for step 2 (R2 = 0.999) (Fig. 3d). Therefore ΔHdes for the entire system is surmised to be 500 kJ mol−1 (125 kJ mol−1 H2). PXD of the products at each stage allows a greater insight into those determined by ex situ heating in vacuo. After the first plateau, a substantial level of Mg2RuH4 is identifiable in the powder, along with Ru, NaOH and a small quantity of Mg3Ru2. The highly oxidisable Na (residual after evaporation) is the source of NaOH (occurring during PXD analysis), while the thermally unstable Mg2RuH4 is the source of Mg3Ru2. An unknown phase is also observed at this temperature and pressure, which due to the low intensity of the Bragg peaks associated with this material, indexing and as such, structural refinement was not possible. After the second plateau, the remaining powder consists of Mg3Ru2 and Ru. Presumably the excess Ru required to form Mg3Ru2 (without leaving excess Mg) comes from the excess Ru that remains in the starting material (Fig. 1). Therefore the decomposition process can be described according to eqn (10) and (11) and Fig. 4.
| Na2Mg2RuH8 → 2Na + Mg2RuH4 + 2H2 (2.0 wt% H2) | (10) |
| Mg2RuH4 + 2Na → Mg3Ru2 + 2Na + 2H2 (2.0 wt% H2) | (11) |
Conclusions
The optimised syntheses of Na2Mg2TH8 (T = Fe, Ru) have been reported. Ball milling of stoichiometric quantities of NaH, MgH2 and Fe followed by hydrogenation allows for a yield of >90% purity of Na2Mg2FeH8via the formation of a NaMgH3 intermediate. On the contrary, no intermediate is observed during the synthesis of Na2Mg2RuH8 using an identical procedure.The thermal decomposition of both Na2Mg2TH8 materials have been studied by ex situ PXD, GC and PCI measurements. The first desorption maxima of Na2Mg2FeH8 has been established to occur at ca. 400 °C, while Na2Mg2RuH8 has its first maxima at 420 °C. The decomposition pathways of these isostructural compounds differs considerably, with Na2Mg2FeH8 proceeding via NaMgH3 in a three-step process, while Na2Mg2RuH8 decomposes via Mg2RuH4 in a two-step process. The dissimilarity between the pathways originates from the capability of the 4d Ru metal centre to exist in a variety of [RuHx]n− complexes compared to Fe, which only exists as [FeH6]4−.
The enthalpy and entropy of desorption for Na2Mg2TH8 (T = Fe, Ru) for each stage of decomposition has been established to be by PCI measurements. The total enthalpy of desorption for Na2Mg2FeH8 is 95 kJ mol−1 H2 and 125 kJ mol−1 H2 for Na2Mg2RuH8.
Acknowledgements
We would like to acknowledge Ms. Warifune for the synthesis of the Mg2FeH6 starting material. We also appreciate the financial support from JSPS KAKENHI Grant Number 25220911.Notes and references
- K. Yvon and G. Renaudin, Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- K. Yvon, Chimia, 1998, 52, 613–619 CAS.
- J. J. Reilly and R. H. Wiswall, Inorg. Chem., 1968, 7, 2254–2256 CrossRef CAS.
- H. Blomqvist and D. Noréus, J. Appl. Phys., 2002, 91, 5141–5148 CrossRef CAS.
- M. Lelis, D. Milcius, E. Wirth, U. Halenius, L. Eriksson, K. Jansson, K. Kadir, J. Ruan, T. Sato, T. Yokosawa and D. Noréus, J. Alloys Compd., 2010, 496, 81–86 CrossRef CAS.
- R. J. Westerwaal, M. Slaman, C. P. Broedersz, D. M. Borsa, B. Dam, R. Griessen, A. Borgschulte, W. Lohstroh, B. Kooi, G. ten Brink, K. G. Tschersich and H. P. Fleischhauer, J. Appl. Phys., 2006, 100, 063518 CrossRef.
- T. J. Richardson, J. L. Slack, R. D. Armitage, R. Kostecki, B. Farangis and M. D. Rubin, Appl. Phys. Lett., 2001, 78, 3047–3049 CrossRef CAS.
- M. Polanski, T. K. Nielsen, Y. Cerenius, J. Bystrzycki and T. R. Jensen, Int. J. Hydrogen Energy, 2010, 35, 3578–3582 CrossRef CAS.
- M. Polanski, K. Witek, T. K. Nielsen, L. Jaroszewicz and J. Bystrzycki, Int. J. Hydrogen Energy, 2013, 38, 2785–2789 CrossRef CAS.
- J. J. Didisheim, P. Zolliker, K. Yvon, P. Fischer, J. Schefer, M. Gubelmann and A. F. Williams, Inorg. Chem., 1984, 23, 1953–1957 CrossRef CAS.
- G. Li, M. Matsuo, S. Deledda, R. Sato, B. C. Hauback and S. Orimo, Mater. Trans., 2013, 54, 1532–1534 CrossRef CAS.
- T. J. Richardson, J. L. Slack, B. Farangis and M. D. Rubin, Appl. Phys. Lett., 2002, 80, 1349–1351 CrossRef CAS.
- B. Bogdanović, A. Reiser, K. Schlichte, B. Spliethoff and B. Tesche, J. Alloys Compd., 2002, 345, 77–89 CrossRef.
- S. F. Parker, K. P. J. Williams, M. Bortz and K. Yvon, Inorg. Chem., 1997, 36, 5218–5221 CrossRef CAS.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1995, 227, 121–124 CrossRef CAS.
- B. Huang, F. Gingl, F. Fauth, A. Hewat and K. Yvon, J. Alloys Compd., 1997, 248, 13–17 CrossRef CAS.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1994, 204, 5–8 CrossRef.
- G. Renaudin, L. Guenee and K. Yvon, J. Alloys Compd., 2003, 350, 145–150 CrossRef CAS.
- M. Orlova, J.-P. Rapin and K. Yvon, Inorg. Chem., 2009, 48, 5052–5054 CrossRef CAS PubMed.
- T. D. Humphries, S. Takagi, G. Li, M. Matsuo, T. Sato, M. H. Sørby, S. Deledda, B. C. Hauback and S. Orimo, J. Alloys Compd., 2015 DOI:10.1016/j.jallcom.2014.12.113.
- M. Matsuo, H. Saitoh, A. Machida, R. Sato, S. Takagi, K. Miwa, T. Watanuki, Y. Katayama, K. Aoki and S. Orimo, RSC Adv., 2013, 3, 1013–1016 RSC.
- H. Saitoh, S. Takagi, M. Matsuo, Y. Iijima, N. Endo, K. Aoki and S. Orimo, APL Mater., 2014, 2, 076103 CrossRef.
- S. Takagi, T. D. Humphries, K. Miwa and S. Orimo, Appl. Phys. Lett., 2014, 104, 203901 CrossRef.
- M. Di Chio, L. Schiffini, S. Enzo, G. Cocco and M. Baricco, J. Alloys Compd., 2007, 434, 734–737 CrossRef.
- K. Kadir and D. Noréus, Inorg. Chem., 2007, 46, 3288–3289 CrossRef PubMed.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1992, 187, 227–232 CrossRef CAS.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1992, 190, 65–68 CrossRef CAS.
- B. Huang, K. Yvon and P. Fischer, J. Alloys Compd., 1993, 197, 65–68 CrossRef CAS.
- J. A. Puszkiel, P. Arneodo Larochette and F. C. Gennari, J. Alloys Compd., 2008, 463, 134–142 CrossRef CAS.
- A. Boultif and D. Louer, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- J. Laugier and B. Bochu, LMGP-Suite Suite Programs Interpret. X-ray Exp. by Jean laugier Bernard Bochu, ENSP/Laboratoire des Matériaux du Génie Phys. BP 46. 38042 Saint Martin d'Hères, Fr.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, Los Alamos National Laboratory Report, 2000, Laur 86-748 Search PubMed.
- B. E. Warren, X-ray Diffraction, Courier Dover Publications, 1969 Search PubMed.
- H. Reardon, N. Mazur and D. H. Gregory, Prog. Nat. Sci., 2013, 23, 343–350 CrossRef.
- D. A. Sheppard, M. Paskevicius and C. E. Buckley, Chem. Mater., 2011, 23, 4298–4300 CrossRef CAS.
- F. Bonhomme, K. Yvon, G. Triscone, K. Jansen, G. Auffermann, P. Müller, W. Bronger and P. Fischer, J. Alloys Compd., 1992, 178, 161–166 CrossRef CAS.
| This journal is © the Owner Societies 2015 |