
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Study of structures and thermodynamics of CuNi nanoalloys using a new DFT-fitted atomistic potential
Emanuele
Panizon
a,
Jimena A.
Olmos-Asar
b,
Maria
Peressi
b and
Riccardo
Ferrando
*c
aPhysics Department, University of Genoa, Via Dodecaneso 33, 16146, Genoa, Italy
bDepartment of Physics, University of Trieste, Strada Costiera 11, 34151 Trieste, Italy
cPhysics Department, University of Genoa and CNR-IMEM, Via Dodecaneso 33, 16146, Genoa, Italy. E-mail: ferrando@fisica.unige.it
First published on 24th February 2015
Abstract
Shape, stability and chemical ordering patterns of CuNi nanoalloys are studied as a function of size, composition and temperature. A new parametrization of an atomistic potential for CuNi is developed on the basis of ab initio calculations. The potential is validated against experimental bulk properties, and ab initio results for nanoalloys of sizes up to 147 atoms and for surface alloys. The potential is used to determine the chemical ordering patterns of nanoparticles with diameters of up to 3 nm and different structural motifs (decahedra, truncated octahedra and icosahedra), both in the ground state and in a wide range of temperatures. The results show that the two elements do not intermix in the ground state, but there is a disordering towards solid–solution patterns in the core starting from room temperature. This order–disorder transition presents different characteristics in the icosahedral, decahedral and fcc nanoalloys.
1 Introduction
In recent years, nanoscale metallic alloys have become a widely studied topic at the frontier between condensed matter physics, chemistry and materials science. Such heterogeneous nanoparticles (NPs), often referred to as nanoalloys, have been shown to exhibit magnetic, catalytic and optical properties that can be different from those of bulk systems and of the elemental NPs. The size-, shape- and composition-dependence of these properties is not at all trivial but it is extremely important, since its comprehension would allow tailored design of nano-sized systems for specific applications in many technological fields, from industrial catalysis, to data storage and to medical research.1CuNi in particular is a system that has gained increasing interest for what concerns bulk surface alloys2,3 and nanoalloys,4 because of its catalytic properties. It has been shown that bulk surface CuNi alloys have a higher reactivity for carbon oxide reduction than elemental nickel or copper. They have also been used to catalyze the growth of mono- and bi-layer graphene with a fine control of thickness and uniformity.3,5 The interaction between graphene layers and CuNi nanoparticles in nanocomposites has been shown to develop electrical and photoresponse properties making them promising candidates as future photodetectors.6 Regarding biomedical applications, CuNi magnetic nanoparticles are employed in studies involving selective tissue hyperthermia.4,7 CuNi nanoalloys exhibit remarkable magnetic properties.8 Interest in small CuNi nanoalloys derived also from the possibility of producing chiral isomers.9
From a thermodynamic point of view such a system is weakly miscible, presenting a bulk miscibility gap which extends up to 630 K.10 The two elements present a small lattice mismatch and a small positive enthalpy of mixing. Such features suggest that, in clusters of small sizes, strain and pressure related driving forces, recently found to dominate segregation effects in NPs,11 should be much weaker. Moreover CuNi NPs should present smaller miscibility gaps, shifted down to and below room temperature, drastically influencing the synthesis of such clusters.12 Recent work on thermodynamic models applied to NPs in the 4–10 nm size range showed results confirming such finite-size effects on the phase diagram.13
The study of the structure and thermodynamics of nanoalloys can in principle be performed at the ab initio level. However this approach is limited either to the study of a small number of isomers with sizes of several hundred atoms,14 or to the full optimization of clusters of quite small sizes (well below 50 atoms).15 Moreover, ab initio methods are out of question when simulating growth processes, whose time scale is of several microseconds at least.16,17 On the other hand, thermodynamic modeling, which uses macroscopic concepts to study nanoscale objects, becomes less and less reliable when nanoparticle diameters decrease below ∼5 nm. For these reasons, the development of atomistic models is often necessary when studying nanoparticles and nanoalloys. Atomistic models can still provide reliable microscopic descriptions of these systems, being at the same time much less computationally expensive than ab initio calculations. Atomistic models thus allow a satisfactory exploration of the energy landscape for nanoparticles counting up to a few thousand atoms. They can be used to simulate phase transitions in such nanoparticles, and the growth on realistic time scales. The main problem with atomistic models is the validation of their accuracy, which must be performed case-by-case against experimental data, when available, and against ab initio calculations.
In this paper, a new atomistic model for the CuNi system is developed and tested, in the framework of a potential based on the second-moment approximation of the tight binding potential (SMTB),18 also known as the Gupta potential.19 SMTB potentials are nowadays used in the modeling of several nanoalloys, specifically to study problems of great complexity, such as global minimum searches,20,21 phase transitions22–24 and kinetic or growth processes,17,25 since the cost of ab initio simulations is still too high for the extensive investigation required for these studies. However computational modeling of catalytic processes is not possible at the atomistic level, hence there is a need to develop a potential as compatible as possible with ab initio methods, to select structures and compositions for further ab initio studies.
Parameter sets for elemental copper and nickel have already been published in the work of Cleri and Rosato.26 Such parameter sets were fitted to experimental bulk quantities, and developed with the aim of reproducing bulk properties as their main application. We have tried to develop a parametrization starting from Cu–Cu and Ni–Ni parameters of ref. 26, and fitting the heterogeneous Cu–Ni interactions on experimental solubility energies of impurities. The strategy of fitting on experimental values of solubility energies was successfully employed in the case of Ag–Ni, Ag–Cu and Ag–Pd heterogeneous interactions.16,17,27 However, in the case of Cu–Ni, this strategy does not work very well for two reasons: (a) the experimental values of the solubility energies of impurities present quite large uncertainties, with a large difference between the values that can be found in the literature;28,29 (b) the Ni potential of ref. 26 is not really satisfactory,30 because it produces very high surface energies and the energy difference between hcp and fcc bulk phases is too large (0.05 eV per atom against the experimental value of 0.01 eV31). This large difference between fcc and hcp phases would artificially enhance the energies of structural defects such as twin planes and stacking faults. These defects, while not important in bulk studies, can greatly influence the shape of clusters, where the crystalline symmetries are usually broken. Moreover, such a large energy difference destabilizes the hcp phase experimentally observed for Ni nanoparticles adsorbed on the MgO(100).32,33
To avoid these problems, we have adopted the strategy of fitting all parameters (for both homogeneous and heterogeneous interactions) with the aid of DFT calculations. Therefore our potential will be a DFT-based one. After fitting the potential, we checked it against further DFT data (not used in the fitting), to the available experimental results and to recent thermodynamic calculations.13 The potential is then used to study the chemical ordering in different structural motifs, both in the limit T → 0 (by global optimization searches) and at finite temperatures (by Monte Carlo simulations) with the aim of singling out phase transitions. The results shown in the following section are thus a contribution to determine the nanoscale phase diagram in the size range between 102 and 103 atoms, in which DFT calculations are too cumbersome and thermodynamic modeling is not fully reliable.
2 Models and methods
2.1 DFT calculations
Calculations were performed within the Density Functional Theory approach implemented in the Quantum-Espresso package34 with available ultrasoft pseudopotentials.35 For exchange and correlation, the spin unrestricted Generalized Gradient Approximation in the Perdew–Burke–Ernzerhof36 implementation was used. For the selection of the plane waves, energy cutoffs of 30 and 240 Ry were chosen to describe the wave function and the electronic density, respectively, and the sampling of the first Brillouin zone was performed at the Γ point.2.2 Atomistic calculations
Atomistic calculations were performed by means of the SMTB potential, whose fitting procedure is discussed in Section 3. Here we describe the calculations performed by using that potential.3 Potential fitting and validation
The SMTB potential used in the atomistic calculations has the following form; the potential energy of the system depends on the relative distances between atoms rij and it is written as the sum of single-atom contributions Ei: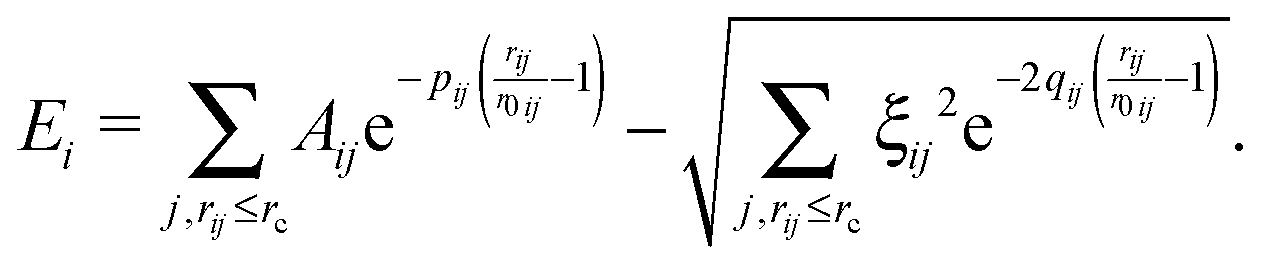 | (1) |
As stated in the Introduction, the parameters of the atomistic model are fully fitted by using DFT data. For each type of interaction (Cu–Cu, Ni–Ni, and Cu–Ni), there are four parameters (p, q, A, ξ) to be fitted.26 For the homogeneous Cu–Cu and Ni–Ni interactions, the parameters are fitted on DFT data of lattice constant a, bulk cohesive energy per atom Ec, bulk modulus B, and energy difference per atom ΔEhcp–fcc between hcp and fcc bulk phases. This fitting strategy has proved to be effective in developing atomistic potentials that are accurate for describing cluster structures, as discussed in ref. 30 and 33.
The parameters for the mixed interactions were often chosen to be the mean values of those of the elemental interactions. While this has been proven to be rather successful in systems with a tendency to form solid solutions in the bulk, such as PdPt,41 we judged that this procedure is a poor approximation for our system, which presents a miscibility gap. For this reason, only p and q have been taken as the arithmetic mean values of the pure parameters, while a and ξ have been fitted to reproduce the single impurity solubility energy Es of a Ni atom in a Cu bulk (and vice versa) as obtained in DFT simulations. The reference values are shown in Table 1. This procedure has been adopted for weakly mixing systems, such as Ag–Ni and Ag–Cu, with quite good results.14,17 To reduce the computational cost of the simulations, a cutoff rc on interatomic distances has been adopted.
| a (Å) | E c (eV) | B (GPa) | ΔEhcp–fcc (eV) | |
|---|---|---|---|---|
| Cu | 3.518 | 3.429 | 138.7 | 0.031 |
| Ni | 3.649 | 4.931 | 206.6 | 0.011 |
| p | q | A (eV) | ξ (eV) | |
|---|---|---|---|---|
| Cu–Cu | 10.653 | 2.49 | 0.092585 | 1.2437 |
| Ni–Ni | 11.7 | 2.045 | 0.096444 | 1.6111 |
| Cu–Ni | 11.1765 | 2.2675 | 0.1046 | 1.4453 |
| E s Cu impurity in Ni bulk | 0.194 eV |
| E s Ni impurity in Cu bulk | 0.113 eV |
3.1 Ground-state validation
The potential has been validated through comparison to DFT results about clusters and surfaces. In the case of bulk crystal surfaces we checked pure-metal surface energies and the vertical relaxation of Ni layers (from 1 to 4) on Cu crystals and vice versa. In the case of clusters we compared the energy ordering of different homotops for some different structures.Atomistic and DFT results about the energies of (111) and (100) bulk crystal surfaces in Cu and Ni are shown in Table 2. The agreement between the two types of calculations is quite good.
| γ(111) (mJ m−2) | γ(100) (mJ m−2) | |
|---|---|---|
| Cu (DFT) | 1135 | 1285 |
| Cu (SMTB) | 1089 | 1189 |
| Ni (DFT) | 1816 | 2147 |
| Ni (SMTB) | 1975 | 2113 |
Let us now consider the structural relaxation in the z-direction of Ni-layers on Cu crystals (and vice versa). In our DFT calculations, we fixed the position of the two lowest Cu(Ni) layers in a 5-layer slab to simulate an infinite bulk, allowing the relaxation of the other three Cu(Ni) layers. The further Ni(Cu) layers on top are all allowed to relax. For the atomistic simulations we just simulate a large slab of Cu(Ni) fcc bulk with the Ni(Cu) layers on top (2300–2600 atoms in total) and removed the periodic boundary conditions along the z axis. DFT and SMTB results are in good agreement, as can be seen in Fig. 1.
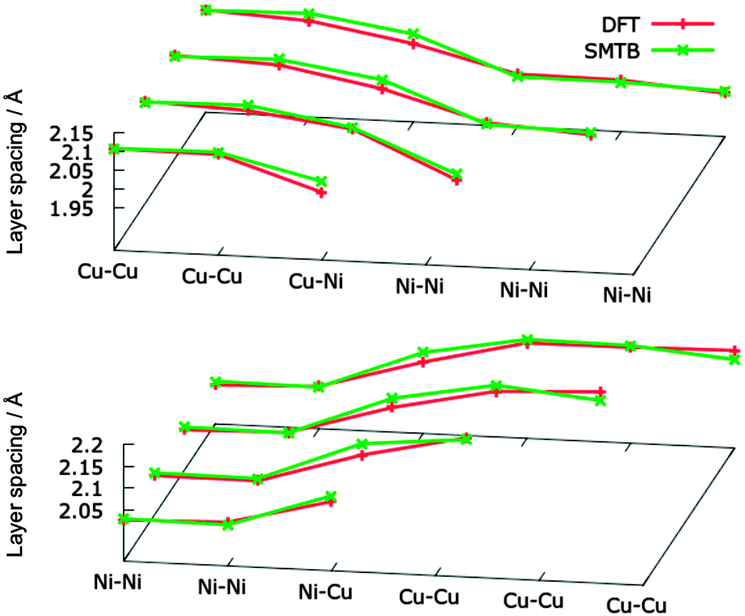 | ||
| Fig. 1 Surface relaxation in the z-direction. Top panel: 1-to-4 nickel layers above bulk copper. Bottom panel: 1-to-4 copper layers above bulk nickel. | ||
In the case of clusters, we considered three structures: the 55-atom truncated octahedron and icosahedron, and the 147-atom octahedron. For each structure, several different compositions were examined. Some of them are shown in Fig. 2. Since a thorough search of the Potential Energy Surface (PES) minima at the DFT level for these sizes is still computationally too expensive, the structures have been taken from a basin-hopping search at the atomistic level and afterwards relaxed by DFT.
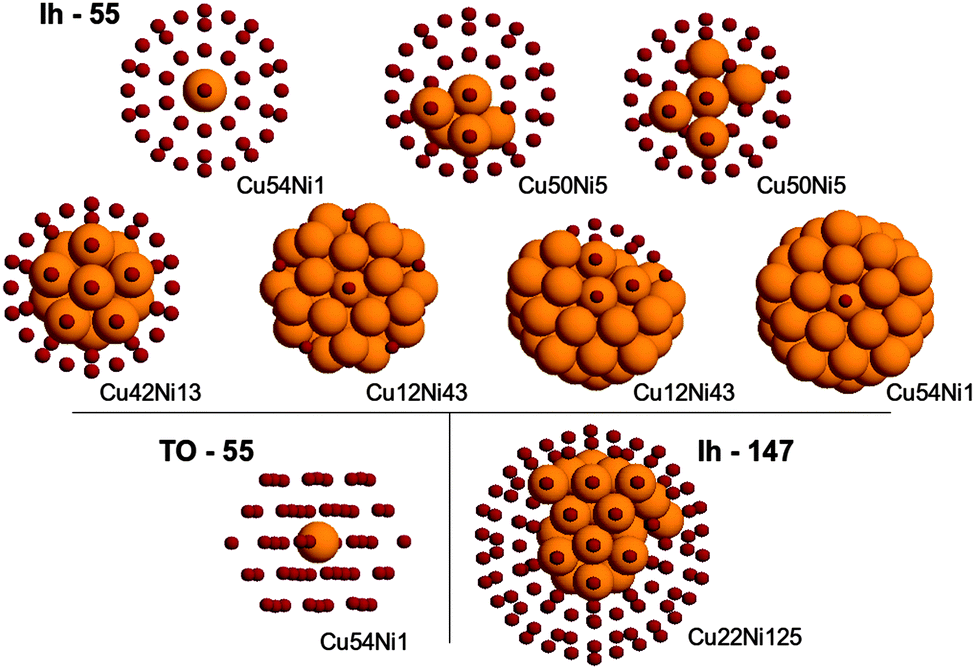 | ||
| Fig. 2 Top row: icosahedron of size 55. The left panel shows the lowest homotop of Cu54Ni1 according to both SMTB and DFT calculations. The middle and right panels show two homotops of Cu50Ni5. According to SMTB calculations, the structure in the middle panel is lower in energy, while according to DFT calculations, the structure in the right panel is lower in energy. Middle row: icosahedron of size 55. Both SMTB and DFT calculations agree on the lowest homotop of Cu42Ni13 (first panel from left) and Cu1Ni42 (fourth panel from left). In contrast, there is disagreement on the lowest homotop of Cu12Ni43, which is the one in the second panel for SMTB and in the third panel for DFT. Bottom row: best placement for a single Ni impurity inside a cuboctahedron of 55 atoms (left panel); best homotop for icosahedral Cu22Ni125. | ||
The comparison of the energy ordering shows that the preferential site for a single impurity is correctly identified in all cases, for both Ni impurity in Cu clusters and vice versa.
However, in general the agreement is not perfect, so two comments should be made. First, the energy differences between homotops in SMTB calculations are always smaller than those at the DFT level. Second, the agreement between the two methodologies, while quite good from a qualitative point of view, is subject to failure when considering the fine details: such properties as the copper segregation in the surface and the aggregation of nickel atoms in the core are very well reproduced, but the ordering of low-lying homotops is often not the same at DFT and SMTB levels.
The most evident failure is to reproduce the character of the incomplete copper capping in the case of very high nickel concentration. At the SMTB level the copper atoms occupy the less coordinated surface positions, that is all vertices first, then the edges, then the (100) facets and finally the (111) facets. This characteristic is common to all cluster shapes and sizes. There is a clear indication that this is not the case at the DFT level. For example, two representative homotops of the Cu12Ni43 icosahedron have been considered. In the first homotop, twelve Cu atoms occupy all vertices. In the second homotop, they form a compact patch on one side of the surface. While at the SMTB the potential prefers the first homotop, the homotop with the compact patch is preferred at the DFT level.
3.2 Finite temperature validation
To check whether this parametrization can be trusted for predicting the correct chemical ordering patterns at finite temperature, we validated it against a well-known experimental property, that is, the transition in the bulk phase-diagram between the segregated phase and the solid solution. For the Cu–Ni systems several phase diagrams have been proposed, but there is a good agreement that at 50% Ni concentration the miscibility gap is up to a temperature of the order of 630 K, and at 25% Ni concentration this temperature is around 500 K.10The results of the MC simulations are shown in Fig. 3. The significant percentage of heterogeneous bonds at 0 K is due to the interface between the Cu and Ni segregated phases, and is just a spurious effect of the finite size of the simulation box. The transition temperature is given by the position of the inflexion point in the order parameter curve. For the 50% Ni concentration, the transition between a well-segregated system and a quasi-random mixing can be found around 550 K. At 25% Ni concentration, the transition temperature is ∼400 K. These results are in reasonable agreement with the experimental data on bulk alloys, even though the transitions are at somewhat lower temperatures. As we will see below, our transition temperatures for nanoparticles are however higher than those given by the atomistic models used in the literature.12
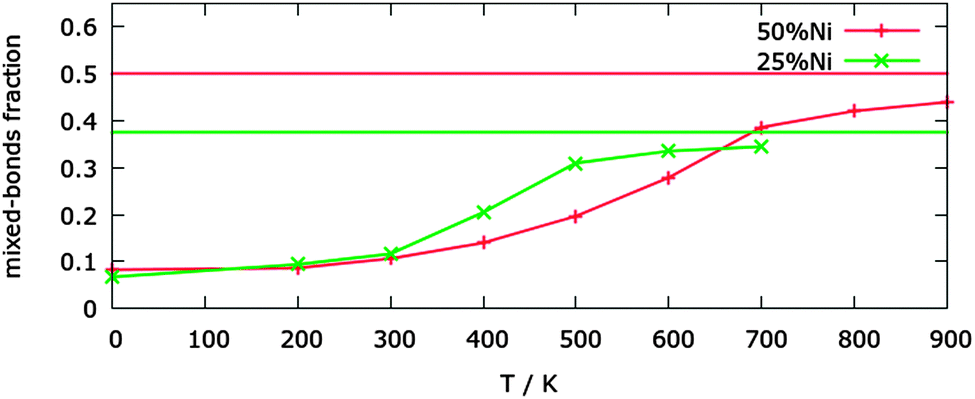 | ||
| Fig. 3 Percentage of mixed-bonds in bulk at 25% and 50% Ni concentrations as a function of the temperature. For each concentration the value for complete random mixing is shown. | ||
The mixing enthalpy of the bulk alloy has been calculated by MC simulations at different compositions at a temperature of 973 K, which is well above the miscibility gap, but still in the solid range. This temperature has been considered in the literature, both in experiments and in simulations.12,28,29 The results are shown in Fig. 4. To the best of our knowledge, there are no recent experimental data, and those present in the literature are not in complete agreement with each other.12,28,29 Our model slightly overestimates the experimental mixing enthalpy, which has a maximum value of 35 meV per atom while most previous studies show values in the range 20–25 meV. Nonetheless our results do show the correct asymmetric behaviour of the enthalpy, peaking at around 55% Ni concentration.
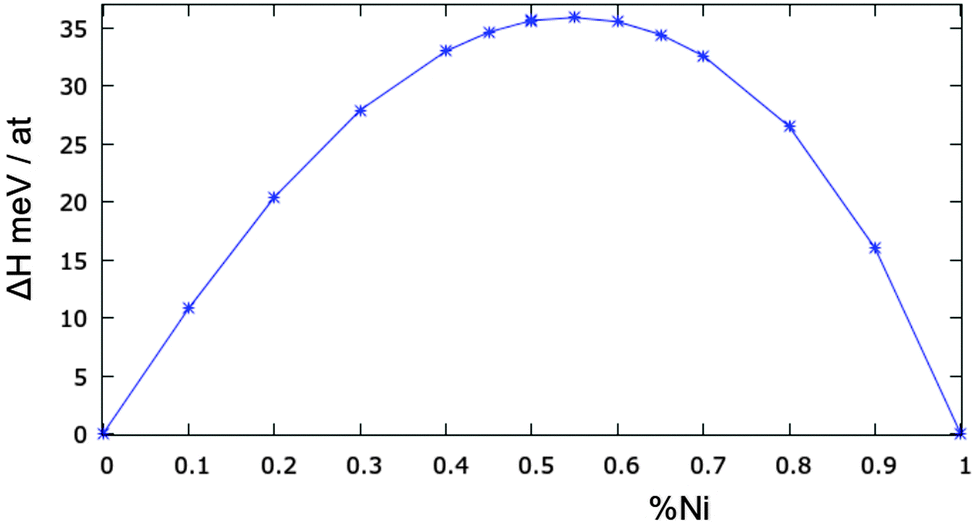 | ||
| Fig. 4 Mixing enthalpy for the CuNi bulk system at 973 K. | ||
4 Results
4.1 Optimal chemical ordering patterns
Here we determine the optimal chemical ordering according to our atomistic model for three magic-number nanoalloys: the 434-atom decahedron, the 561-atom icosahedron and the 586-atom fcc truncated octahedron. For each structure, several compositions are considered. A selection of results is shown in Fig. 5.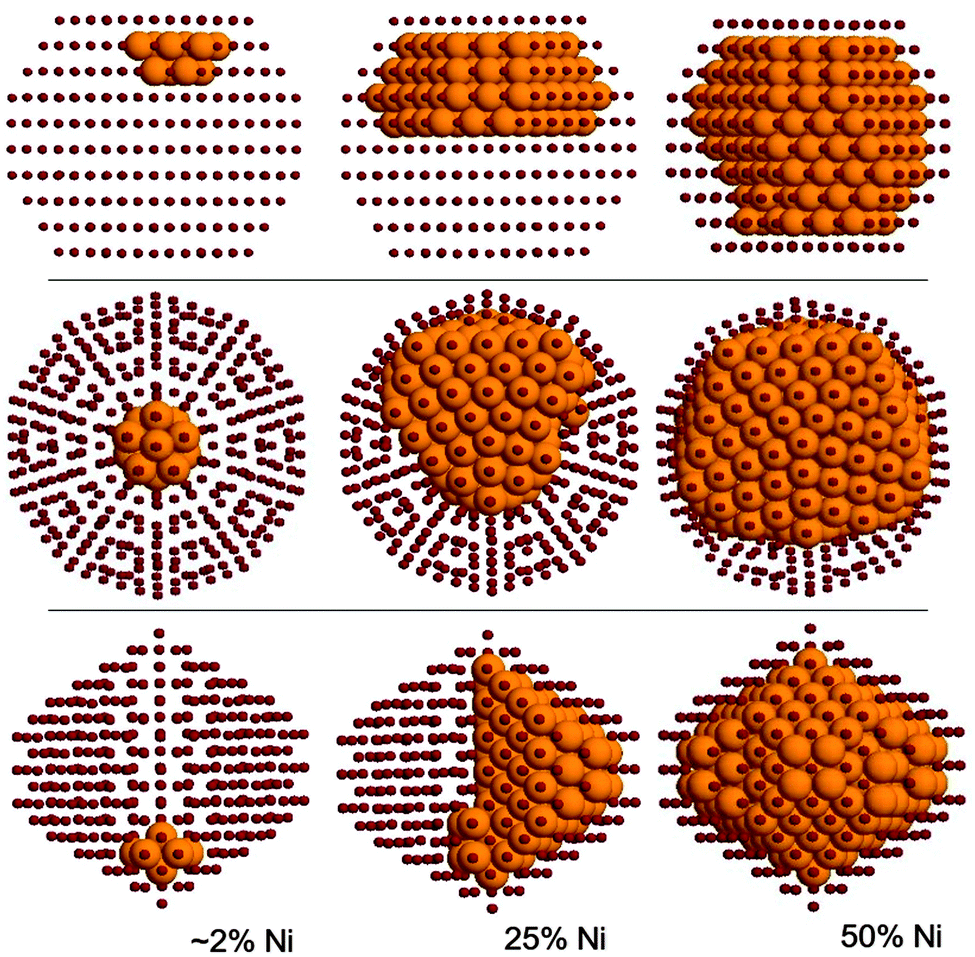 | ||
| Fig. 5 Optimal chemical ordering patterns at 0 K for different geometric structures and compositions. Top row: 586-atom TO. Middle row: 561-atom Ih. Bottom row: 434-atom Dh. Ni atoms are shown as large gold spheres, copper as small red spheres. | ||
The optimal chemical ordering patterns of these nanoalloys bear some resemblance to those of other non-mixing systems in which one of the elements has a lower surface energy and has a larger atomic radius (such as AgNi, AgCu, AgCo and AuCo11), even if the difference in the size of nickel and copper atoms is quite small. For all sizes and structures the clusters show a complete surface segregation of copper atoms, given that the number of copper atoms is sufficient to cover all nickel atoms. Moreover, for all intermediate compositions, the structures show a very clear quasi-Janus chemical pattern,42 with an off-centre nickel core with a monolayer coating of copper on one side. The main difference with AgNi, AgCu, AgCo and AuCo is that the lattice mismatch in CuNi is so small that the quasi-Janus clusters have almost no surface distortions. However, a small mismatch is sufficient to trigger the typical instabilities that lead to low-symmetry chemical ordering patterns43 such as the quasi-Janus patterns, because of strain release. Quasi-Janus patterns have been found also in simulations of another system with a small mismatch, PtIr, but the driving force in that case was not related to strain release.44
In the icosahedron, the best impurity location for a nickel atom is the central position, also for all other Ih magic-sizes, that is, 55, 147 and 309. This is expected since the Ni atom is (slightly) smaller and the centre of an icosahedron is its most compressed site. For the 561-atom cluster specifically, upon increasing the Ni concentration we first found a filling of the 55-atoms core, then a morphological instability appears (see Fig. 5, second row), similar to those already found in the literature,11 in which the core takes a low-symmetry off-centre shape.
Nickel-rich clusters show incomplete surfaces where the copper atoms occupy positions on the surface in a precise pattern from the lowest coordinated sites, i.e. vertices, to the highest coordinated sites, that is the (111) facets. This kind of decoration, though, is not in agreement with the DFT results at very small sizes, where the tendency is found to be in favour of an incomplete but segregated surface capping, so that this atomistic result should therefore be taken with some care.
The subvertex site of the decahedral structure is the preferential location for a single nickel impurity. As much as 7 atoms are found to form a small crown around the five-fold axis just under the vertex. Larger inclusions then follow a very clear and unique pattern: if we consider the decahedron as divided in five slices (the original five tetrahedra composing the structure) by its 5-fold axis, the Ni atoms occupy one slice at a time, showing magical composition numbers of 63, 104, 145, 186 and 212 nickel atoms, as shown in Fig. 6. It should be noted however that at each compositions homotops that do not completely fill these slices are very close in energy to the optimal homotop, with ΔE ∼ 0.03 eV.
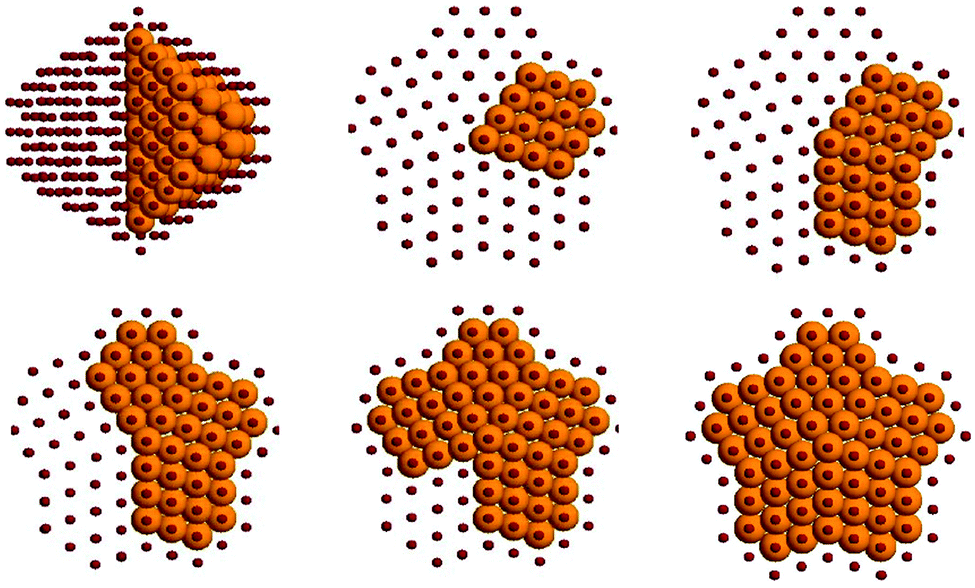 | ||
| Fig. 6 The shape of the nickel core for clusters of the 434-atom decahedral structure for five magic compositions corresponding to the filling of the five “slices”. Ni atoms are shown as large gold spheres, copper as small red spheres. | ||
In the truncated octahedra, nickel atoms always form subsurface (hence off-centre) aggregates, for all compositions. This pattern has been already found in several systems.11,45 However in CuNi the difference with respect to AgCu, AgNi or AgCo is that the surface monolayer of copper atoms has neither holes nor deformations.11
4.2 Thermodynamic stability of segregation
A complete segregation is not experimentally observed in several studies, for example in ref. 13, where some degree of intermixing is usually found. This intermixing may be caused by temperature effects. For this reason, in the following we study the temperature dependence of segregation by MC simulations. We consider again the 434-atoms Dh, the 561-atoms Ih and 586-atoms fcc TO structures. The temperature range considered is from 0 K (the ground states) up to 800 K, well above the mixing temperature of the bulk alloy, but well below the melting temperatures of the clusters (the latter will be briefly studied in Section 4.3). We decided to limit our investigation to clusters with 25% Ni composition. Since all surfaces are entirely made of copper, at this composition the core has roughly 50% nickel content and the effects of segregation and disordering are therefore more interesting.All three structures show a monotonic increase of the percentage of heterogeneous bonds with temperature, indicating a smooth transition from complete segregation to quasi-random mixing. The transition temperature is identified as the inflexion point of the curve, and it is approximately 350 K for all the motifs considered. This transition temperature is higher than those found in simulations using other atomistic models.12 It is interesting to comment that the percentage of nickel atoms in the surface is negligible even at the highest simulated temperature, meaning that the average atomic occupation for a surface atom is csurfNi < 0.005. The value of the order parameter for completely random mixing has been therefore calculated as that of a completely randomly mixed core with a pure copper surface.
Although the three structures present similar features, some differences are present. The degree of segregation in the icosahedral structure is higher than that of the other two structures even at temperatures above the transition, as can be well seen in Fig. 7, where the average occupations for clusters at 600 K are shown. In contrast, the TO core present a perfect solid solution, and for the decahedral cluster only a weak nickel preferential occupation can be seen along the 5-fold axis. These effects are clearly related to the different internal pressure profiles. While inside a TO the pressure is everywhere homogeneous (except for a slight compression of the subsurface atoms), the Ih motif has a highly compressed core and the Dh clusters present a somewhat compressed 5-fold symmetry axis. The strain release associated with the insertion of a smaller nickel atom in such location evidently has an effect on the average occupation even at temperature well above the transition.
 | ||
| Fig. 7 Top row: average local nickel occupations for Dh, Ih and TO clusters at 25% Ni composition at 600 K. Bottom panel: percentage of heterogeneous bonds for the same clusters as a function of temperature: Dh (+), TO (Â) and Ih (*) clusters. | ||
The transition temperatures between ordered and intermixed patterns can be compared to the results of the work by Guisbiers et al.,13 in which phase diagrams for 4 nm-sized nanoparticles of several shapes were calculated by thermodynamic modeling, finding transitions at 200, 400 and 400 K for Dh, Ih and fcc shapes respectively. The results for Ih and fcc nanoalloys are in good agreement with our findings, considering that our clusters have a smaller size, circa 2.5 nm, and thus are expected to present somewhat lower transition temperatures. In the case of Dh, we do not find any significant temperature difference with the other motifs.
4.3 Melting
In order to evaluate the melting range of the structures, we have performed a preliminary MD study. For each structure, several molecular dynamics simulations have been made, starting from both intermixed and segregated seeds. At these sizes pure MD simulations are often not enough to obtain precise evaluations of the melting temperature from caloric curves, as shown in ref. 46, and more sophisticated algorithms need to be implemented. Nevertheless our results are sufficient to locate the melting range with some confidence, so that they allow us to make two comments: (a) all structures melt in the range around 1000–1100 K, which is in very good agreement with the results obtained by Guisbiers et al. by means of thermodynamic calculations13 (b) all melting ranges are well above the highest temperature at which we performed MC runs at fixed structures for the study of the intermixing transition.5 Conclusions
In this paper, a new DFT-based parametrization of the SMTB atomistic potential for CuNi has been developed. The new potential is found to describe well all the properties analysed, those regarding both infinite (bulk) and semi-infinite (surface) systems and those regarding finite-size clusters. Specifically, the potential is shown to correctly reproduce structural properties, such as surface vertical relaxation in thin films and impurity locations in clusters, and thermodynamic properties, such as miscibility gap and mixing enthalpy in bulk alloys. These results make this parametrization particularly suitable to study structural properties of nanoalloys at finite temperature, but also to perform thorough explorations of the nanoalloy energy landscape which can be used as the starting point of subsequent ab initio studies.This new parametrization has then been used to study the chemical ordering pattern for several cluster shapes, sizes and compositions, both at the level of ground-state search and in finite-temperature simulations. Concerning the optimal chemical ordering patterns, the system shows qualitative trends which are similar to those of other weakly miscible systems, but with some important differences. First of all, the CuNi system present very little lattice mismatch, thus showing quasi-Janus patterns with no surface reconstruction and almost no distortion, at variance in the cases of, e.g., AgCu or AgNi. As the temperature is increased, the chemical ordering pattern changes: while the surface segregation of copper atoms is almost complete even at quite high temperatures, the internal segregation is rapidly disordered, with a transition around 350 K for an overall composition of 25% in Ni. This finite-temperature effect, which is relevant even at room temperature, has thus to be taken into account in the synthesis of such nanoalloys.
The intermixing transition has different characteristics for the different structural motifs. This is related to the intrinsic pressure profile which derives from the non-crystalline nature of icosahedra and decahedra. The more compressed sites, such as those along the five-fold axis for the Dh and especially the core sites for the Ih, retain a higher nickel occupation even at temperatures well above the order–disorder transition.
In conclusion, we have shown that atomistic modelling can be useful in reproducing the nanoscale phase diagram of CuNi nanoalloys, thus being helpful in designing appropriate synthesis methods for these nanoalloys, which present a variety of core–shell or solid–solution patterns.
Acknowledgements
The authors acknowledge support from the European Cooperation in Science and Technology through the COST Action MP0903 NANOALLOY. The authors acknowledge financial support from Italian Ministry of Foreign Affairs and International Cooperation (MAECI), Directorate General for the Country Promotion, through the Executive Programme with Argentina (PGR00190); Consortium for Physics of Trieste, Italy. Computational resources for DFT calculations have been partly obtained through Italian Super-Computing Resource Allocation (ISCRA) grants of the Consorzio Interuniversitario CINECA, partly within the agreement between the University of Trieste and CINECA.References
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- E. Vesselli, E. Monachino, M. Rizzi, S. Furlan, X. Duan, C. Dri, A. Peronio, C. Africh, P. Lacovig, A. Baldereschi, G. Comelli and M. Peressi, ACS Catal., 2013, 3, 1555–1559 CrossRef CAS.
- S. Chen, W. Cai, R. D. Piner, J. W. Suk, Y. Wu, Y. Ren, J. Kang and R. S. Ruoff, Nano Lett., 2011, 11, 3519–3525 CrossRef CAS PubMed.
- M. B. Lilly, I. A. Brezovich and W. J. Atkinson, Radiology, 1985, 154, 243–244 CrossRef CAS PubMed.
- N. Liu, L. Fu, B. Dai, K. Yan, K. Liu, R. Zhao, Y. Zhang and Z. Liu, Nano Lett., 2011, 11, 297–303 CrossRef CAS PubMed.
- A. Kumar, S. Husale, A. K. Srivastava, P. K. Dutta and A. Dhar, Nanoscale, 2014, 6, 8192–8198 RSC.
- A. A. Kuznetsov, V. G. Leontiev, V. A. Brukvin, G. N. Vorozhtsov, B. Y. Kogan, O. A. Shlyakhtin, A. M. Yunin, O. I. Tsybin and O. A. Kuznetsov, J. Magn. Magn. Mater., 2007, 311, 197–203 CrossRef CAS PubMed.
- A. V. Vinogradov, A. A. Levshanov, M. A. Kashirin, A. V. Agafonov and V. V. Vinogradov, J. Phys. Chem. C, 2015, 119, 1500–1505 CAS.
- H. Elgavi, C. Krekeler, R. Berger and D. Avnir, J. Phys. Chem. C, 2012, 116, 330–335 CAS.
- Binary Alloy Phase Diagrams, ed. T. B. Massalki, H. Okamoto and P. R. Subramanian, ASM International, Metals Park, 2nd edn, 1990 Search PubMed.
- D. Bochicchio and R. Ferrando, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165435 CrossRef.
- M. Hennes, J. Buchwald and S. G. Mayr, CrystEngComm, 2012, 14, 7633–7638 RSC.
- G. Guisbiers, S. Khanal, F. Ruiz-Zepeda, J. Roque de la Puente and M. Jose-Yacaman, Nanoscale, 2014, 6, 14630–14635 RSC.
- K. Laasonen, E. Panizon, D. Bochicchio and R. Ferrando, J. Phys. Chem. C, 2013, 117, 26405–26413 CAS.
- S. Heiles and R. L. Johnston, Int. J. Quantum Chem., 2013, 113, 2091 CrossRef CAS PubMed.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155420 CrossRef.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. Lett., 2003, 90, 135504 CrossRef CAS.
- F. Cyrot-Lackmann and F. Ducastelle, Phys. Rev. B: Solid State, 1971, 4, 2406 CrossRef.
- R. P. Gupta, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 6265 CrossRef CAS.
- L. D. Lloyd, R. L. Johnston, S. Salhi and N. T. Wilson, J. Mater. Chem., 2004, 14, 1691 RSC.
- S. Nunez and R. L. Johnston, J. Phys. Chem. C, 2010, 114, 13255–13266 CAS.
- F. Calvo, Faraday Discuss., 2008, 138, 75 RSC.
- F. Calvo, E. Cottancin and M. Broyer, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 121406 CrossRef.
- D. Palagin and J. P. K. Doye, J. Chem. Phys., 2014, 141, 214302 CrossRef PubMed.
- F. Calvo, A. Fortunelli, F. Negreiros and D. J. Wales, J. Chem. Phys., 2013, 139, 111102 CrossRef CAS PubMed.
- F. Cleri and V. Rosato, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 22 CrossRef CAS.
- C. Mottet, G. Tréglia and B. Legrand, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 16018 CrossRef CAS.
- S. Srikanth and K. T. Jacob, Mater. Sci. Technol., 1989, 5, 427–434 CrossRef CAS PubMed.
- S. An Mey, CALPHAD, 1992, 16, 255–260 CrossRef CAS.
- G. Rossi, L. Anghinolfi, R. Ferrando, F. Nita, G. Barcaro and A. Fortunelli, Phys. Chem. Chem. Phys., 2010, 12, 8564 RSC.
- A. T. Dinsdale, CALPHAD, 1991, 15, 317 CrossRef CAS.
- W. Tian, H. P. Sun, X. Q. Pan, J. H. Yu, M. Yeadon, C. B. Boothroyd, Y. P. Feng, R. A. Lukaszew and R. Clarke, Appl. Phys. Lett., 2005, 86, 131915 CrossRef PubMed.
- R. Ferrando, G. Rossi, F. Nita, G. Barcaro and A. Fortunelli, ACS Nano, 2008, 2, 1849 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892–7895 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111 CrossRef CAS.
- D. Bochicchio and R. Ferrando, Nano Lett., 2010, 10, 4211–4216 CrossRef CAS PubMed.
- J. Jellinek and E. B. Krissinel, Chem. Phys. Lett., 1996, 258, 283–292 CrossRef CAS.
- G. Rossi, R. Ferrando and C. Mottet, Faraday Discuss., 2008, 138, 193 RSC.
- L. O. Paz-Borbon, A. Gupta and R. L. Johnston, J. Mater. Chem., 2008, 18, 4154–4164 RSC.
- I. Parsina and F. Baletto, J. Phys. Chem. C, 2010, 114, 1504–1511 CAS.
- R. Ferrando, J. Phys.: Condens. Matter, 2015, 27, 013003 CrossRef PubMed.
- M. Polak and L. Rubinovich, Phys. Chem. Chem. Phys., 2014, 16, 1569 RSC.
- C. Langlois, Z. Y. Li, J. Yuan, D. Alloyeau, J. Nelayah, D. Bochicchio, R. Ferrando and C. Ricolleau, Nanoscale, 2012, 4, 3381–3388 RSC.
- A. Rapallo, J. A. Olmos-Asar, O. A. Oviedo, M. Luduena, R. Ferrando and M. M. Mariscal, J. Phys. Chem. C, 2012, 116, 17210–17218 CAS.
| This journal is © the Owner Societies 2015 |