
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited-state intramolecular proton transfer to carbon atoms: nonadiabatic surface-hopping dynamics simulations†
Shu-Hua
Xia
a,
Bin-Bin
Xie
a,
Qiu
Fang
a,
Ganglong
Cui
*a and
Walter
Thiel
*b
aKey Laboratory of Theoretical and Computational Photochemistry, Ministry of Education, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: ganglong.cui@bnu.edu.cn
bMax-Planck-Institut für Kohlenforschung, 45470 Mülheim an der Ruhr, Germany. E-mail: thiel@mpi-muelheim.mpg.de
First published on 12th February 2015
Abstract
Excited-state intramolecular proton transfer (ESIPT) between two highly electronegative atoms, for example, oxygen and nitrogen, has been intensely studied experimentally and computationally, whereas there has been much less theoretical work on ESIPT to other atoms such as carbon. We have employed CASSCF, MS-CASPT2, RI-ADC(2), OM2/MRCI, DFT, and TDDFT methods to study the mechanistic photochemistry of 2-phenylphenol, for which such an ESIPT has been observed experimentally. According to static electronic structure calculations, irradiation of 2-phenylphenol populates the bright S1 state, which has a rather flat potential in the Franck–Condon region (with a shallow enol minimum at the CASSCF level) and may undergo an essentially barrierless ESIPT to the more stable S1 keto species. There are two S1/S0 conical intersections that mediate relaxation to the ground state, one in the enol region and one in the keto region, with the latter one substantially lower in energy. After S1 → S0 internal conversion, the transient keto species can return back to the S0 enol structure via reverse ground-state hydrogen transfer in a facile tautomerization. This mechanistic scenario is verified by OM2/MRCI-based fewest-switches surface-hopping simulations that provide detailed dynamic information. In these trajectories, ESIPT is complete within 118 fs; the corresponding S1 excited-state lifetime is computed to be 373 fs in vacuum. Most of the trajectories decay to the ground state via the S1/S0 conical intersection in the keto region (67%), and the remaining ones via the enol region (33%). The combination of static electronic structure computations and nonadiabatic dynamics simulations is expected to be generally useful for understanding the mechanistic photophysics and photochemistry of molecules with intramolecular hydrogen bonds.
Introduction
Excited-state intramolecular or intermolecular proton transfers are elementary processes occurring in many molecular and biochemical systems1–5 and electronic devices,6–9 for example, in natural and artificial photosynthesis,10,11 water-splitting photocatalysis,12 green fluorescent proteins,13,14 and photoswitches.15 Understanding these excited-state proton transfer processes is important both from fundamental and technological points of view. To this end, numerous computational studies ranging from static electronic structure calculations to nonadiabatic dynamics simulations have been performed in the past few decades.16–30 Most of these previous studies focused on excited-state proton transfer processes between two highly electronegative atoms, e.g. nitrogen, oxygen, and fluorine.17–20,22,26,27,31–37What about excited-state intramolecular proton transfer (ESIPT) in molecules without strong hydrogen bonds, for example, in alcohols or phenols? Such ESIPT processes were first investigated in the 1980s,38,39 with proton transfer to aromatic carbon atoms being first addressed at the beginning of this century.40 Since then, Wan and coworkers have systematically explored such excited-state proton transfers in many systems.41–48 They first studied photochemical deuterium incorporation at the ortho and para positions of 2-phenylphenol in various solvent mixtures49 and found that the predominant exchange at the ortho position is independent of water and methanol contents, implying an intramolecular process. They also investigated the photochemistry of o-hydroxybiaryls, which features not only an efficient excited-state proton transfer to the ortho carbon atom of the naphthyl ring, but also a novel ring-closing reaction.50 Flegel et al.51 studied the photoaddition of water and alcohols to the 9- and 10-positions of the anthracene moiety of 9-(2-hydroxyphenyl)anthracene in acetonitrile and methanol mixtures and proposed a mechanism involving water-mediated excited-state proton transfer from the phenolic OH group to the anthracene fragment. Basarić and Wan52 investigated the potential excited-state proton transfer in four derivatives of 9-(2-hydroxyphenyl)anthracene. Nayak and Wan53 explored photochemical deuterium incorporation in extended ortho-substituted biaryl systems and reported the longest solvent-assisted proton-relay chain. They proposed direct and water-assisted proton transfer mechanisms to explain photohydration at the ortho and distal positions, respectively. In these experimental studies, it was generally believed that excited-state intramolecular proton transfer to ortho positions is efficient in phenols.
The underlying photophysical and photochemical mechanisms in these systems have not yet been elucidated in detail, e.g., with regard to the relevant structures, proton transfer paths, excited-state potential energy surfaces, lifetimes, and decay channels. We are aware of only one recent theoretical study in this context,54 which employed the single-reference second-order coupled cluster (RI-CC2) method to explore direct and water-assisted excited-state proton transfer in 2-phenyl-1-naphthol. Given this situation, we decided to perform high-level multi-reference electronic structure computations and trajectory-based surface-hopping dynamics simulations to study the mechanistic photochemistry of the prototypical 2-phenylphenol molecule, with emphasis on the ESIPT process to the ortho carbon atom and the deactivation channels leading back to the ground state.
Computational details
Ab initio methods
Ground-state (S0) conformers were optimized at the B3LYP level.55–58 The resolution-of-the-identity second-order algebraic diagrammatic construction [RI-ADC(2)] method was employed to optimize excited-state minimum-energy reaction paths.59–63The state-averaged complete active space self-consistent field (SA-CASSCF) method (equal state weights) was used to optimize minima (S0 and S1) and minimum-energy conical intersections (S0/S1). In all SA-CASSCF geometric optimizations, the active space comprised 10 electrons in 8 orbitals. To obtain more accurate potential energy profiles, single-point MS-CASPT2 calculations64,65 were performed at the CASSCF optimized geometries. In these MS-CASPT2 calculations, an imaginary shift of 0.2 a.u. was applied to avoid intruder-state issues,66 and Cholesky decomposition techniques with unbiased auxiliary basis sets were used to evaluate two-electron integrals.67
Vertical excitation energies were computed at the TD-CAM-B3LYP68,69 and MS-CASPT2 levels. The 6-31G* basis set70,71 was used throughout except for the RI-ADC(2) calculations which employed the def2-SVP basis set.72 The following codes were used: TDDFT, GAUSSIAN09;73 DFT and CASSCF optimizations, GAUSSIAN03;74 MS-CASPT2, MOLCAS7.6;75 and RI-ADC(2), TURBOMOLE6.5.76
OM2/MRCI method
All semiempirical calculations were performed using the OM2/MRCI method as implemented in the MNDO99 code.77–80 During geometry optimizations, all required energies, gradients, and nonadiabatic coupling elements were computed analytically. Conical intersections were optimized using the Lagrange–Newton approach.81,82In OM2/MRCI calculations, the restricted open-shell HF formalism was applied in the self-consistent field (SCF) treatment (i.e., the orbitals were optimized for the leading configuration of the S1 state with two singly occupied orbitals). The active space in the MRCI calculations included 12 electrons in 10 orbitals (see ESI†). In terms of the SCF configuration, it comprised the five highest doubly occupied orbitals, two singly occupied orbitals, and the three lowest unoccupied orbitals. For the MRCI treatment, three configuration state functions were chosen as references, namely the SCF configuration and the two closed-shell configurations derived therefrom (i.e., all singlet configurations that can be generated from the HOMO and the LUMO of the closed-shell ground state). The MRCI wavefunction was built by allowing all single and double excitations from these three references.
The nonadiabatic dynamics was studied by performing 1 ps OM2/MRCI trajectory surface-hopping simulations. The initial atomic coordinates and velocities were randomly selected from 5 ps trajectories of ground-state molecular dynamics. The number of excited-state dynamics runs was then chosen according to the computed S0–S1 transition probability. A total of 193 surface-hopping trajectories were run, with all relevant energies, gradients, and nonadiabatic coupling vectors being computed on-the-fly as needed. For points with an S1–S0 energy gap of less than 10 kcal mol−1, the fewest-switches criterion was applied to decide whether to hop. The time step was chosen to be 0.1 fs for nuclear motion and 0.0005 fs for electronic propagation. The unitary propagator evaluated at a mid-point was used to propagate the electronic motion. The translational and rotational motions were removed in each step. The empirical decoherence correction (0.1 a.u.) proposed by Granucci et al. was employed.83 The final evaluations were done for the 148 trajectories that finished successfully and satisfied our energy continuity criterion (no changes greater than 30 kcal mol−1 between any two consecutive MD steps). Further technical details are given in our previous publications.26–29,84–86
Results
Ground-state properties and vertical excitation energies
Apart from the most stable ground-state structure of 2-phenylphenol (S0-ENOL), there is also a minimum for the keto tautomer (S0-KETO), see Fig. 1 and Table 1. For each of the two minima, OM2/MRCI and CASSCF yield similar geometries. S0-ENOL is more stable than S0-KETO by 34.7 (33.7) kcal mol−1 at the OM2/MRCI (MS-CASPT2) level. | ||
| Fig. 1 Stationary points and minimum-energy conical intersections, with selected optimized bond lengths (Å) obtained from OM2/MRCI and CASSCF (in square brackets). | ||
| Structure | C4C3C2O9 | C2C3C5C6 | C2C3C5C7 | C3C1C2O9 | ΔE |
|---|---|---|---|---|---|
| OM2/MRCI | |||||
| S0-ENOL | 179.6 | −127.7 | 52.8 | 179.7 | 0.0 |
| S0-KETO | 180.0 | 180.0 | 0.0 | 180.0 | 34.7 |
| S1S0-ENOL | −83.1 | 154.0 | −27.8 | 140.6 | 92.1 |
| S1S0-KETO | 166.8 | −107.9 | 58.2 | −171.7 | 73.7 |
| CASSCF(10,8)/6-31G* | |||||
| S0-ENOL | 179.0 | −118.1 | 62.6 | 179.7 | 0.0 |
| S0-KETO | −180.0 | 179.8 | −0.2 | −180.0 | 33.7 |
| S1-ENOL | 178.2 | −132.3 | 51.2 | 179.4 | 103.0 |
| S1-KETO | 180.0 | 180.0 | 0.0 | 180.0 | 74.4 |
| S1S0-ENOL | −96.8 | 145.9 | −35.4 | 154.8 | 98.8/100.7 |
| S1S0-KETO | 172.4 | −113.7 | 52.8 | −174.7 | 69.8/73.9 |
The computed vertical excitation energies for the first excited singlet state (S1) are collected in Table 2. The OM2/MRCI value of 4.92 eV agrees very well with the results from MS-CASPT2 (4.99 eV) and TD-CAM-B3LYP (4.93 eV). The calculations are consistent with the experimental value of 4.66 eV obtained from laser flash photolysis of 2-phenylphenol in solutions.49 A slightly lower experimental value of 4.28 eV has been reported for 2-phenyl-1-naphthol featuring more extensive conjugation.54 The S1 state at the Franck–Condon point is spectroscopically bright; its oscillator strength is computed to be 0.155 at the TD-CAM-B3LYP level. Molecular orbital analysis shows that the S0–S1 electronic transition mainly originates from the HOMO–LUMO single excitation (Fig. 2). The HOMO is mainly localized in the phenolic part, whereas the LUMO is localized in the phenyl group. Hence, the S1 state is of charge-transfer character, which sets the stage for the subsequent excited-state proton transfer. In fact, this kind of electronic structure change has been found in many similar intramolecularly hydrogen-bonded systems.18,20,22,26,27,30
| OM2/MRCI | MS-CASPT2 | TD-CAM-B3LYP | Exp. | |
|---|---|---|---|---|
| kcal mol−1 | 113.5 | 115.0 | 113.8 | 107.5 |
| eV | 4.92 | 4.99 | 4.93 | 4.66 |
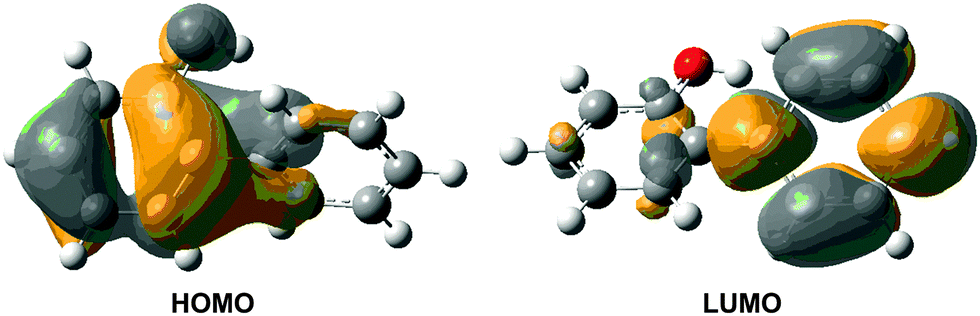 | ||
| Fig. 2 CAM-B3LYP/6-31G* computed HOMO and LUMO of S0-ENOL responsible for the S0 → S1 vertical excitation. | ||
Excited-state minima
At the CASSCF level, there is a shallow S1 minimum in the Franck–Condon region of 2-phenylphenol (S1-ENOL), which is computed to lie 103.0 kcal mol−1 above the S0-ENOL minimum in single-point MS-CASPT2 calculations. At the OM2/MRCI level, no such S1 minimum could be located since all minimizations starting from the S0-ENOL equilibrium geometry led directly to the S1 keto species (S1-KETO), see the left panel of Fig. 3. Likewise, the minimum-energy path for proton transfer computed at the RI-ADC(2)/def2-SVP level indicates an essentially barrierless excited-state enol–keto tautomerization, see the right panel of Fig. 3. This is also verified by OM2/MRCI nonadiabatic dynamics simulations (vide infra). | ||
| Fig. 3 (left) OM2/MRCI optimization path starting from the enol minimum, which leads directly to a keto species after about 50 steps; (right) RI-ADC(2)/def2-SVP computed minimum-energy reaction path with respect to the C7–H10 proton transfer reaction coordinate. See text for discussion. | ||
CASSCF optimization yields another S1 minimum in the keto region (S1-KETO, see Fig. 1). At this geometry, there is no significant charge transfer in the S1 state; the S0 → S1 transition involves mostly the central C3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 double-bond region, causing an elongation of this bond from 1.396 to 1.480 Å (S0versus S1 keto minimum, CASSCF values). Other geometric parameters change only slightly (Fig. 1). According to single-point MS-CASPT2 calculations, S1-KETO lies 74.4 kcal mol−1 above S0-ENOL and 28.6 kcal mol−1 below S1-ENOL (Table 1). Thus, the excited-state proton transfer that yields the keto species is highly exothermic; in other words, 2-phenylphenol is a strong photoacid in S1. As already mentioned, this proton transfer is computed to be essentially barrierless and is thus expected to be ultrafast. In terms of excited-state topology, our present results are consistent with recent RI-CC2 computations on a similar system, 2-phenyl-1-naphthol.54
C5 double-bond region, causing an elongation of this bond from 1.396 to 1.480 Å (S0versus S1 keto minimum, CASSCF values). Other geometric parameters change only slightly (Fig. 1). According to single-point MS-CASPT2 calculations, S1-KETO lies 74.4 kcal mol−1 above S0-ENOL and 28.6 kcal mol−1 below S1-ENOL (Table 1). Thus, the excited-state proton transfer that yields the keto species is highly exothermic; in other words, 2-phenylphenol is a strong photoacid in S1. As already mentioned, this proton transfer is computed to be essentially barrierless and is thus expected to be ultrafast. In terms of excited-state topology, our present results are consistent with recent RI-CC2 computations on a similar system, 2-phenyl-1-naphthol.54
Conical intersections
At the OM2/MRCI level, we were able to locate two S1/S0 minimum-energy conical intersections (S1S0-ENOL and S1S0-KETO). Selected bond lengths and dihedral angles are given in Fig. 1 and in Table 1, respectively. In S1S0-ENOL, the H10 atom is still attached to the O9 atom (phenol species) but the OH group is extruded out of the ring plane, with a C4C3C2O9 dihedral angle of −83° (OM2/MRCI). This strong out-of-plane deformation significantly increases the S0 energy, thus closing the S0–S1 energy gap and reaching an S1/S0 conical intersection. In S1S0-KETO, the H10 atom is already bonded to the C7 atom (keto species); the two rings are not coplanar with a C2C3C5C7 dihedral angle of 58°. Energetically, S1S0-ENOL [S1S0-KETO] is computed to lie 92.1 [73.7] kcal mol−1 above the S0-ENOL ground state, and 21.4 kcal mol−1 [39.8 kcal mol−1] below the S1 energy at the Franck–Condon point (113.5 kcal mol−1); hence, these two conical intersections are energetically accessible. Taking into consideration that S1S0-KETO is more stable than S1S0-ENOL by 18.4 kcal mol−1, the former is expected to play a more vital role in excited-state deactivation.It is worth stressing that OM2/MRCI and the ab initio methods give similar structures and energies for the two S1/S0 conical intersections (Table 1). Taking S1S0-ENOL as an example, the dihedral angles C2C3C5C7, C4C3C2O9, and C3C1C2O9 are computed to be −28°, −83°, and 141° at the OM2/MRCI level, compared with −35°, −97°, and 155° at the CASSCF level, respectively, (see Fig. 1). The relative energies from OM2/MRCI and single-point MS-CASPT2 calculations are also reasonably close to each other: the values of S1S0-ENOL [S1S0-KETO] are 92.1 [73.7] kcal mol−1 for OM2/MRCI, and 98.8/100.7 [69.8/73.9] kcal mol−1 for MS-CASPT2. In the latter case, the quoted S0 and S1 state energies differ slightly because they come from single-point MS-CASPT2 calculations at CASSCF-optimized geometries.
Excited-state decay paths
The preceding static electronic structure computations suggest the following scenario for the photoinduced processes in 2-phenylphenol. Upon irradiation, the spectroscopically bright S1 state is populated in the Franck–Condon region, from which the S1/S0 conical intersection with an intact phenol moiety is energetically accessible (with relaxation to the ground state via S1S0-ENOL). A competitive process involves an essentially barrierless excited-state proton transfer yielding an S1 keto minimum, which can decay to the ground state via the S1/S0 conical intersection in the keto region (S1S0-KETO); back in the S0 state, the keto species S0-KETO can return to the more stable tautomer S0-ENOL via reverse ground-state hydrogen transfer.To verify this mechanism and to explore the timescales of the underlying photophysical and photochemical events, we have performed trajectory-based fewest-switches surface-hopping dynamics simulations starting in the S1 state of 2-phenylphenol.
Hopping-point distribution
The S1–S0 hopping-point distribution extracted from all surface-hopping trajectories reflects the topology of the conical intersection seam.84 The two types of S1/S0 conical intersections in 2-phenylphenol, S1S0-ENOL and S1S0-KETO, clearly govern our nonadiabatic dynamics simulations. Fig. 4 depicts the distributions of the C7H10 distance and the C4C3C2O9 dihedral angle at all S1–S0 hopping points. Obviously, there are two main hopping regions, which cluster around two minimum-energy S1/S0 conical intersections S1S0-ENOL and S1S0-KETO. A closer examination of the C7H10 distance distribution at all hopping points in Fig. 4 shows that most of the trajectories (67%) hop to the S0 state via the keto conical intersection seam. This preference arises from two factors: first, the S1 proton transfer is essentially barrierless so that the S1 keto species is generated easily, and second, S1S0-KETO is thermodynamically favored over S1S0-ENOL because its potential energy is lower by 18.4 kcal mol−1 (OM2/MRCI). Hence, it is not surprising that most trajectories decay to the S0 state via S1S0-KETO in our dynamics simulations. | ||
| Fig. 4 Distribution of the C7H10 distance and the C4C3C2C9 dihedral angle at all S1–S0 hopping points. See text for detailed discussion. | ||
S1 lifetime
In our simulations, 118 of 148 (80%) trajectories have reached the S0 state at the end of the 1 ps nonadiabatic dynamics runs. As shown in the left panel of Fig. 5, most of the S1 → S0 hops happen between 100 and 400 fs (only 4 hops after 400 fs). Again, this ultrafast decay is consistent with the excited-state topological features, i.e. an almost barrierless proton transfer and two efficient deactivation channels (vide supra).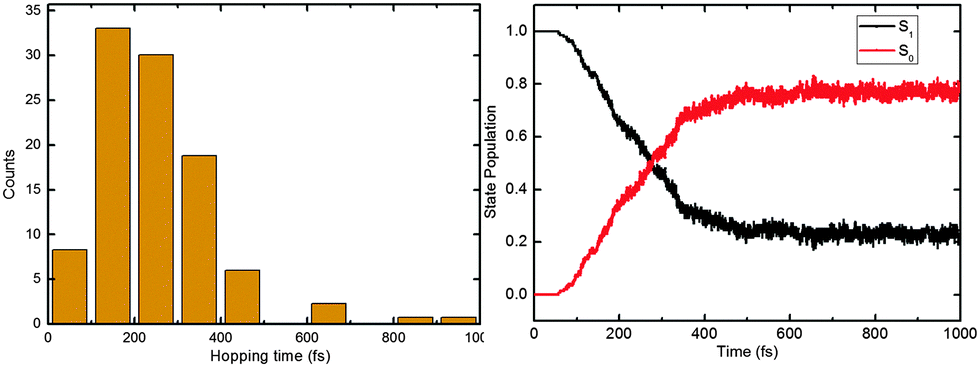 | ||
| Fig. 5 Distribution of the S1–S0 hopping times (left) and time-dependent S1 and S0 state populations (right). See text for detailed discussion. | ||
The S1 excited-state deactivation can be viewed as a first-order elementary reaction. The S1 state population is thus ruled by the following rate equation:
| p(t) = exp(−k(t − t0)) + p0 | (1) |
Product distribution
Fig. 6 shows the product distribution at the end of the 1 ps nonadiabatic simulations. Overall, there are four kinds of products, namely S1 enol (13%) and keto (5%), and S0 enol (47%) and keto (35%); the enol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :keto ratio is estimated to be 3:2. The top panel illustrates the distribution of the resulting phenol conformers. Most of the trajectories ending up in the phenol region have the H10 atom bonded to the O9 atom. However, there are also some S0 phenol products that have the H8 atom bonded to the O9 atom, not the H10 atom (see H8O9 distribution). In these trajectories, the excited-state proton transfer and the reverse ground-state hydrogen transfer involve two different hydrogen atoms (H10 and H8, respectively). The bottom panel depicts the distribution of the keto conformers after the 1 ps simulations. There are ca. 90 trajectories with the H8 atom bonded to the C7 atom, and ca. 40 trajectories with the H10 atom bonded to the C7 atom.
:keto ratio is estimated to be 3:2. The top panel illustrates the distribution of the resulting phenol conformers. Most of the trajectories ending up in the phenol region have the H10 atom bonded to the O9 atom. However, there are also some S0 phenol products that have the H8 atom bonded to the O9 atom, not the H10 atom (see H8O9 distribution). In these trajectories, the excited-state proton transfer and the reverse ground-state hydrogen transfer involve two different hydrogen atoms (H10 and H8, respectively). The bottom panel depicts the distribution of the keto conformers after the 1 ps simulations. There are ca. 90 trajectories with the H8 atom bonded to the C7 atom, and ca. 40 trajectories with the H10 atom bonded to the C7 atom.
 | ||
| Fig. 6 Distribution of the C7H10, C7H8, H8O9, and O9H10 bond lengths at the end of 1 ps simulations. See text for detailed discussion. | ||
Typical trajectories
In our nonadiabatic dynamics simulations, we see three different photocycles that start from S1-ENOL and end up at S0-ENOL: (I) the S1 state decays directly to the ground state, without excited-state intramolecular proton transfer (ESIPT); (II) the S1 state first evolves towards the S1 keto species via an ultrafast barrierless ESIPT and then decays to the ground state in the keto region followed by a reverse ground-state hydrogen transfer (GSHT) involving the same migrating hydrogen atom; (III) the photocycle is the same as in case (II) except that different hydrogen atoms are involved in ESIPT and GSHT. In the following, we present for each photocycle pattern a representative trajectory to illustrate the main photophysical and photochemical events.Fig. 7 shows a typical trajectory for case (I) with direct decay via the S1S0-ENOL conical intersection. Within the first 400 fs, the system starts to rotate around its central C3C5 bond (strong changes in the C2C3C5C6 and C2C3C5C7 dihedral angles; only small fluctuations in the C4C3C2O9 and C3C1C2O9 dihedral angles). During this process, the nonadiabatic coupling remains small and the S1–S0 energy gap remains large, so there is no nonadiabatic transition. After about 400 fs, the C4C3C2O9 dihedral angle starts to decrease from 180° to 40° at ca. 600 fs. The S1 and S0 states now become energetically close to each other (within 4 kcal mol−1) and there is a large nonadiabatic coupling; thus, a nonadiabatic S1–S0 hop takes place, with relaxation of S1 to the S0 state. Thereafter, the C4C3C2O9 and C3C1C2O9 dihedral angles move back towards their original values (from twisted to a more planar arrangement). There is no ESIPT in this trajectory. We emphasize that this photocycle pattern occurs only rarely in our trajectories.
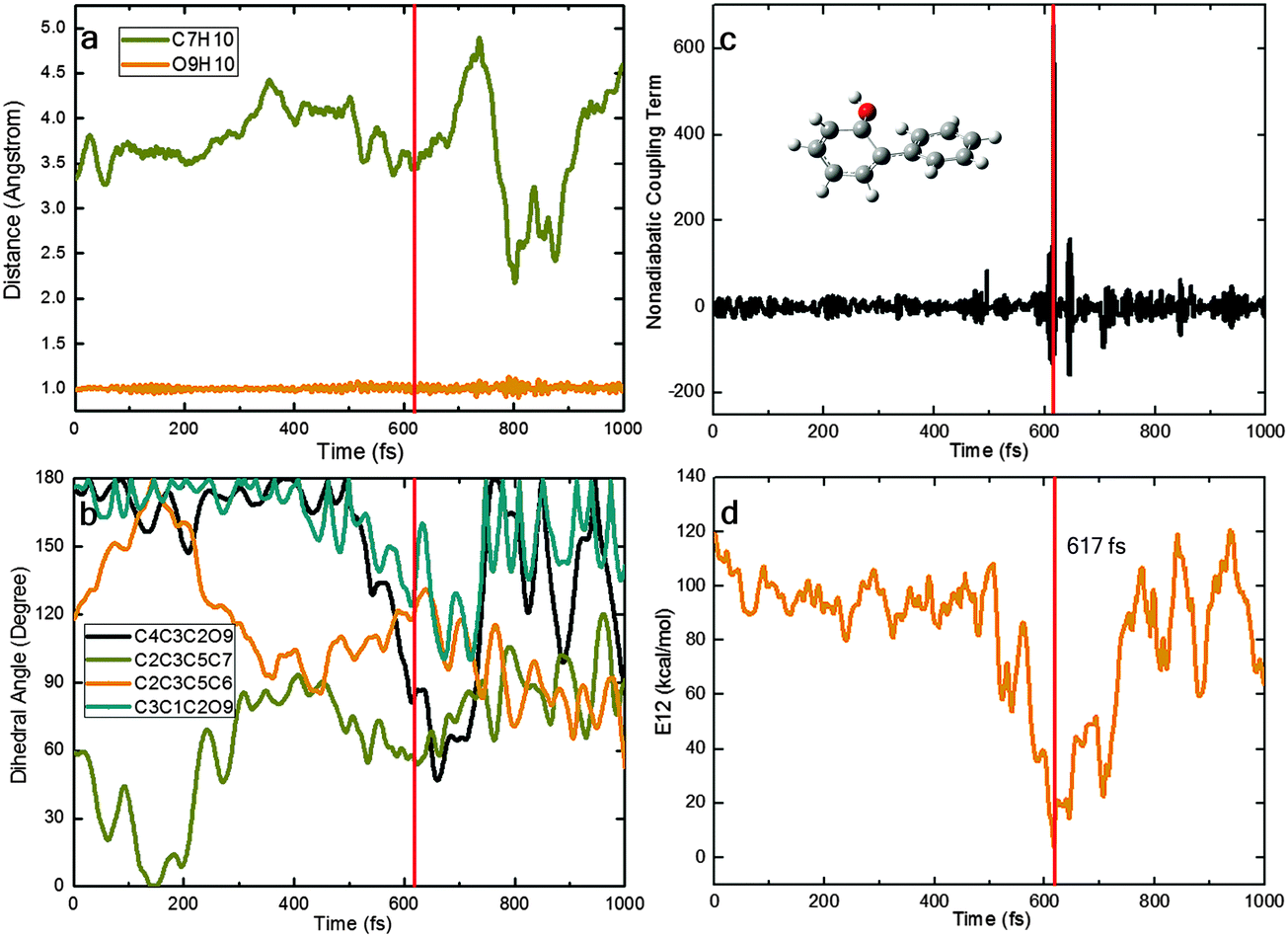 | ||
| Fig. 7 Time-dependent physical variables obtained from a typical OM2/MRCI trajectory of type (I): (a) two key bond lengths; (b) four key dihedral angles; (c) nonadiabatic coupling term; and (d) S1–S0 energy gap. | ||
Fig. 8 depicts a typical trajectory for case (II) with deactivation to the S0 state via the S1S0-KETO conical intersection. In the initial stage of this trajectory, the O9H10 and C7H10 distances quickly increase and decrease, respectively. At ca. 50 fs, the ESIPT is complete and the S1 keto species S1-KETO is formed, which remains in the S1 state for another 150 fs (while retaining a rather short O9H10 distance indicative of excited-state hydrogen bonding interactions). Thereafter, it decays to the S0 state at a point where the S1–S0 nonadiabatic coupling becomes very large (panel c) and the S1–S0 gap is very small (panel d). Interestingly, the generated S0 keto species does not return back to the enol region immediately; instead, it roams the keto region for additional 500 fs. Then, a reverse ground-state hydrogen transfer takes place, regenerating the S0 enol conformer and completing the photocycle. The rotation around the C3–C5 bond starts after ca. 700 fs (see the C2C3C5C6 and C2C3C5C7 dihedral angles in panel b) while the C4C3C2O9 and C3C1C2O9 dihedral angles do not vary much.
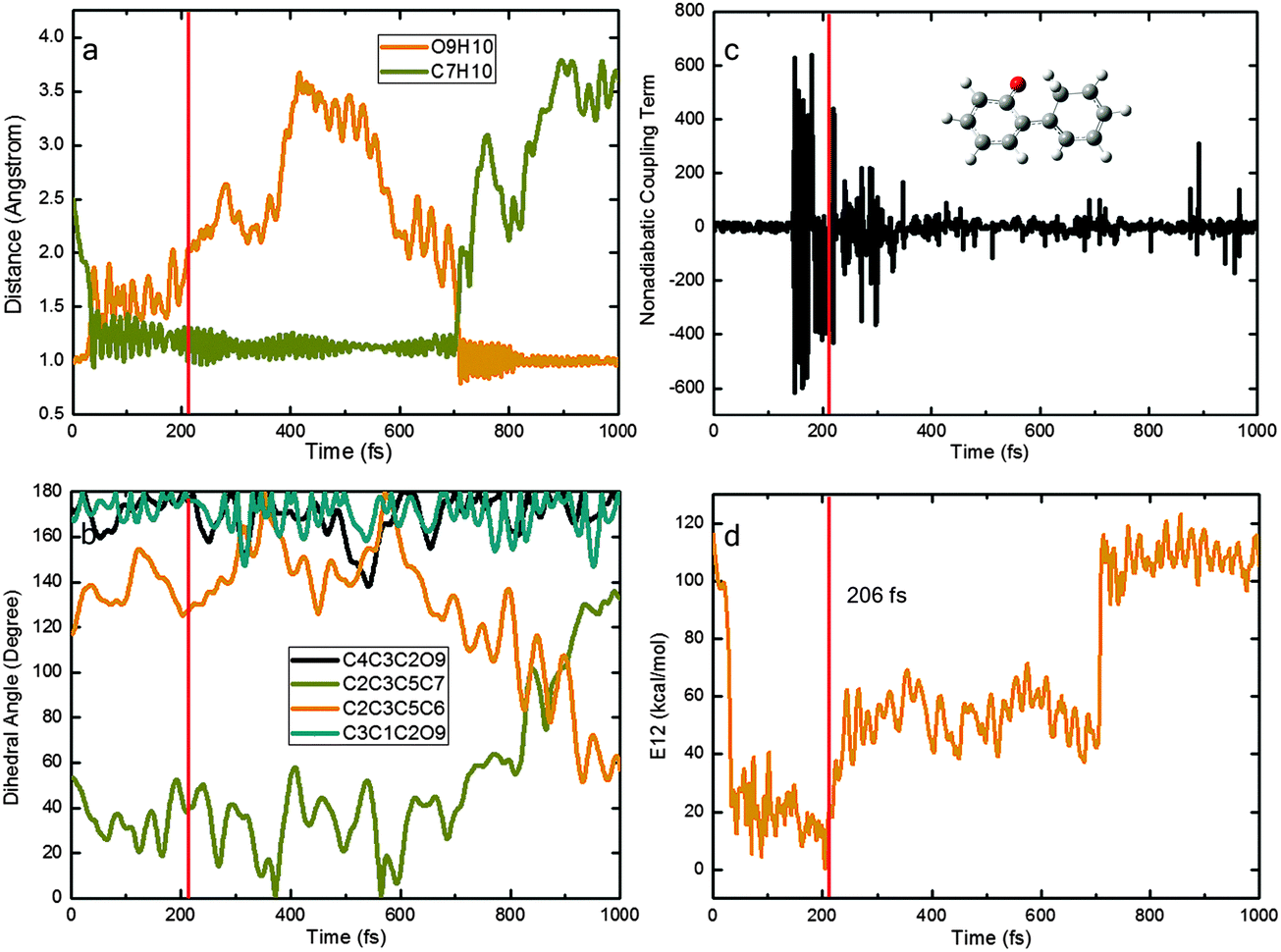 | ||
| Fig. 8 Time-dependent physical variables obtained from a typical OM2/MRCI trajectory of type (II): (a) two key bond lengths; (b) four key dihedral angles; (c) nonadiabatic coupling term; and (d) S1–S0 energy gap. | ||
Fig. 9 presents a typical trajectory for case (III). Here, the O9H10 and C7H10 distances fluctuate around their equilibrium positions in the first 100 fs; then, they start to increase and decrease quickly. At about 110 fs, the S1 keto species S1-KETO is formed, which stays in the S1 state for ca. 50 fs and then decays to the S0 state at 165 fs, when the keto S1/S0 conical intersection is encountered. The generated keto species roams the keto region in the S0 state for a longer time (640 fs). After ca. 800 fs, the most stable S0 phenol conformer is regenerated via a reverse ground-state hydrogen transfer. The rotation around the C3–C5 bond starts after ca. 900 fs (see the C2C3C5C6 and C2C3C5C7 dihedral angles in panel b). Interestingly, the H10 atom bonded to the O9 atom is transferred to the C7 atom in the ESIPT process, while the H8 atom originally bonded to the C7 atom is transferred to the O9 atom in the final GSHT step.
 | ||
| Fig. 9 Time-dependent physical variables obtained from a typical OM2/MRCI trajectory of type (III): (a) three key bond lengths; (b) four key dihedral angles; (c) nonadiabatic coupling term; and (d) S1–S0 energy gap. | ||
Discussion
Our results are consistent with the experiments available for 2-phenylphenol. Lukeman and Wan49 argued that singlet reactivity is major for 2-phenylphenol, which is consistent with our computations. The S1 excited-state proton transfer is nearly barrierless and ultrafast, so it is impossible for the system to efficiently populate triplet states in the Franck–Condon region. In addition, the S1 → T1 intersystem crossing in the keto region is not expected to be competitive with the efficient internal conversion from the S1 keto species to the S0 state. However, this intersystem crossing could become more probable in a rigid environment because the internal conversion involves a large conformational change that could be impeded by steric interactions with the environment. Furthermore, there is experimental evidence that 2-phenylphenol is a strong photoacid in the S1 state. This point is supported by the MS-CASPT2 results (see Fig. 10), which confirm that the S1 excited-state proton transfer is highly exothermic – S1-KETO lies 28.6 kcal mol−1 below S1-ENOL and 40.6 kcal mol−1 below the initially populated S1 Franck–Condon point.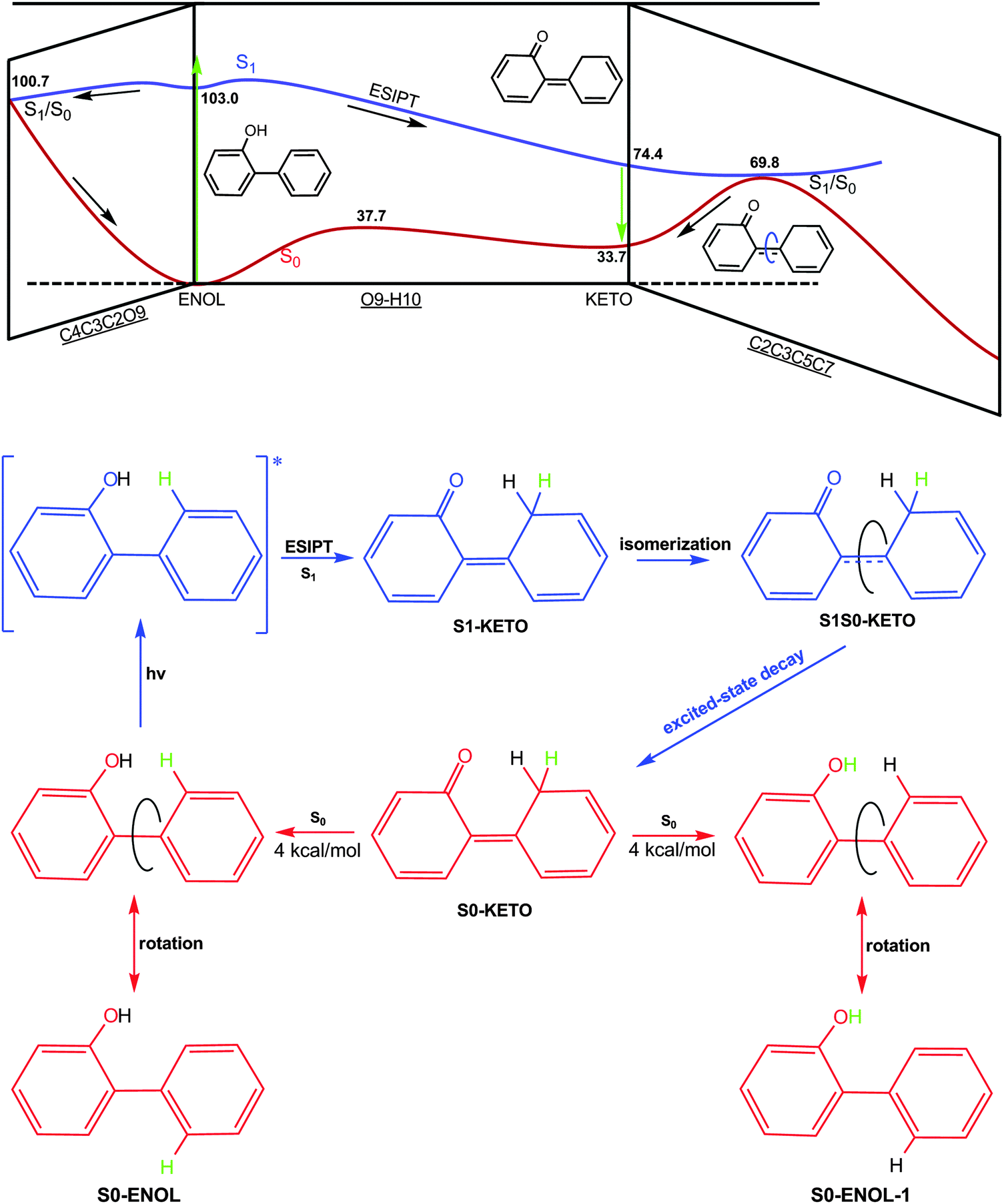 | ||
| Fig. 10 S1 deactivation pathways identified in the present work. Potential energy profiles and structures related to the S0 and S1 states are shown in red and blue, respectively. Also relative energies from single-point MS-CASPT2 computations (in kcal mol−1) are given. See text for discussion. | ||
We emphasize in this context that our present computations are carried out in vacuum and thus only consider the intrinsic photochemistry of 2-phenylphenol, for example, direct ESIPT processes to the ortho-position, without accounting for solvent-assisted intermolecular proton transfer to remote sites such as para-positions.
Previous electronic structure computations on a similar system54 showed that there exists an efficient S1/S0 conical intersection near the keto region, but without optimizing its structure. In this work we precisely located this kind of minimum-energy conical intersection in 2-phenylphenol, both at the OM2/MRCI and CASSCF levels (S1S0-KETO), and we explored its dynamical role in the S1 photodynamics of 2-phenylphenol using full-dimensional surface-hopping dynamics simulations. We find that 67% trajectories decay to the S0 state via this conical intersection in the keto region. In addition, we optimized the S1/S0 conical intersection in the Franck–Condon region (S1S0-ENOL), which also plays an important role in the S1 deactivation (33%). Thus, both conical intersections need to be considered in order to correctly understand the mechanistic photochemistry of 2-phenylphenol and its variants.
How is the S0 isomer S0-ENOL-1 (see the bottom of Fig. 10) generated in the photodynamics of 2-phenylphenol? Experimentally, Lukeman and Wan49 assumed that this species comes from S0-KETO via a concerted reverse hydrogen transfer and 1,5-hydrogen shift. Our present dynamics simulations do not support this scenario – we do not see any 1,5-hydrogen shift in any of the trajectories. Instead, S0-ENOL-1 is generated by a simple single-bond rotation, after the hydrogen atom originally bonded to the phenyl ring has been transferred to the oxygen atom (Fig. 10).
The S0 keto species has not yet been detected spectroscopically when using nanosecond laser flash photolysis.49,54 B3LYP calculations indicate that the tautomerization of S0-KETO to the most stable phenol conformer S0-ENOL has to overcome a small barrier of 4.0 kcal mol−1. It might thus be possible to observe this predicted transient species using ultrafast time-resolved transient spectroscopy.
Summary
With the use of electronic structure computations and trajectory-based surface-hopping dynamics simulations, we have for the first time explored the mechanistic photochemistry of 2-phenylphenol. We have simulated the S1 excited-state proton transfer and deactivation as well as the reverse hydrogen transfer in the S0 state. Mechanistically, some trajectories directly evolve from the Franck–Condon region toward an enol-type S1/S0 conical intersection, followed by an S1 → S0 internal conversion to the ground-state minimum. Most of the trajectories proceed from the Franck–Condon region to the S1 keto species via an essentially barrierless ESIPT; the transient S1 keto species is then de-excited to the ground state via a second S1/S0 conical intersection in the keto region, followed by a quick relaxation back to the most stable phenol minimum via a reverse GSHT process (barrier of ca. 4 kcal mol−1 at the B3LYP level). The nonadiabatic dynamics simulations predict an average lifetime of 118 fs for the ESIPT process.40,49 In these simulations, 67% of the trajectories decay to the S0 state via the keto S1/S0 conical intersection, and 33% decay via the S1/S0 conical intersection in the Franck–Condon region. According to the computed time-dependent state populations, the S1 excited-state lifetime is estimated to be 373 fs in vacuum. We hope that these computational results and mechanistic insights will stimulate further experimental work on 2-phenylphenol, especially by ultrafast time-resolved transient spectroscopy.Acknowledgements
G.C. appreciates the financial support of “The Recruitment Program of Global Youth Experts” and “Youth Scholars Program of Beijing Normal University”; W.T. is grateful for support from an ERC Advanced Grant.References
- E. Kosower and D. Huppert, Annu. Rev. Phys. Chem., 1986, 37, 127–156 CrossRef CAS.
- R. Mathies, S. Lin, J. Ames and W. Pollard, Annu. Rev. Biophys. Biophys. Chem., 1991, 20, 491–518 CrossRef CAS PubMed.
- R. Cukier and D. Nocera, Annu. Rev. Phys. Chem., 1998, 49, 337–369 CrossRef CAS PubMed.
- L. Tolbert and K. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS PubMed.
- D. Stoner-Ma, A. Jaye, K. Ronayne, J. Nappa, S. Meech and P. Tonge, J. Am. Chem. Soc., 2008, 130, 1227–1235 CrossRef CAS PubMed.
- K. Choi and A. Hamilton, Angew. Chem., Int. Ed., 2001, 40, 3912–3915 CrossRef CAS.
- S. Kim, J. Seo, H. Jung, J.-J. Kim and S. Park, Adv. Mater., 2005, 17, 2077–2082 CrossRef CAS.
- S. Park, O.-H. Kwon, S. Kim, S. Park, M.-G. Choi, M. Cha, S. Park and D.-J. Jang, J. Am. Chem. Soc., 2005, 127, 10070–10074 CrossRef CAS PubMed.
- J. Kwon and S. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- A. Bard and M. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- D. Gust, T. Moore and A. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed.
- W. Youngblood, S.-H. Lee, Y. Kobayashi, E. Hernandez-Pagan, P. Hoertz, T. Moore, A. Moore, D. Gust and T. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef CAS PubMed.
- R. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- K.-Y. Chen, Y.-M. Cheng, C.-H. Lai, C.-C. Hsu, M.-L. Ho, G.-H. Lee and P.-T. Chou, J. Am. Chem. Soc., 2007, 129, 4534–4535 CrossRef CAS PubMed.
- S.-J. Lim, J. Seo and S. Park, J. Am. Chem. Soc., 2006, 128, 14542–14547 CrossRef CAS PubMed.
- W.-H. Fang, J. Am. Chem. Soc., 1998, 120, 7568–7576 CrossRef CAS.
- A. Sinicropi, R. Pogni, R. Basosi, M. Robb, G. Gramlich, W. Nau and M. Olivucci, Angew. Chem., Int. Ed., 2001, 40, 4185–4189 CrossRef CAS.
- A. Sobolewski, W. Domcke and C. Hättig, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17903–17906 CrossRef CAS PubMed.
- J. Coe and T. Martínez, J. Am. Chem. Soc., 2005, 127, 4560–4561 CrossRef CAS PubMed.
- A. Sobolewski and W. Domcke, J. Phys. Chem. A, 2007, 111, 11725–11735 CrossRef CAS PubMed.
- A. Migani, L. Blancafort, M. Robb and A. DeBellis, J. Am. Chem. Soc., 2008, 130, 6932–6933 CrossRef CAS PubMed.
- M. Barbatti, A. Aquino, H. Lischka, C. Schriever, S. Lochbrunner and E. Riedle, Phys. Chem. Chem. Phys., 2009, 11, 1406–1415 RSC.
- F. Plasser, M. Barbatti, A. Aquino and H. Lischka, J. Phys. Chem. A, 2009, 113, 8490–8499 CrossRef CAS PubMed.
- D. Shemesh, A. Sobolewski and W. Domcke, J. Am. Chem. Soc., 2009, 131, 1374–1375 CrossRef CAS PubMed.
- S. Olsen, K. Lamothe and T. Martínez, J. Am. Chem. Soc., 2010, 132, 1192–1193 CrossRef CAS PubMed.
- G. Cui, Z. Lan and W. Thiel, J. Am. Chem. Soc., 2012, 134, 1662–1672 CrossRef CAS PubMed.
- G. Cui and W. Thiel, Phys. Chem. Chem. Phys., 2012, 14, 12378–12384 RSC.
- L. Spörkel, G. Cui, A. Koslowski and W. Thiel, J. Phys. Chem. A, 2013, 118, 152–157 CrossRef PubMed.
- L. Spörkel, G. Cui and W. Thiel, J. Phys. Chem. A, 2013, 117, 4574–4583 CrossRef PubMed.
- G. Cui, P.-J. Guan and W.-H. Fang, J. Phys. Chem. A, 2014, 118, 4732–4739 CrossRef CAS PubMed.
- L. Serrano-Andrés and M. Merchán, Chem. Phys. Lett., 2006, 418, 569–575 CrossRef PubMed.
- A. Kyrychenko and J. Waluk, J. Phys. Chem. A, 2006, 110, 11958–11967 CrossRef CAS PubMed.
- H.-H. G. Tsai, H.-L. S. Sun and C.-J. Tan, J. Phys. Chem. A, 2010, 114, 4065–4079 CrossRef CAS PubMed.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- Y. Shigemitsu, T. Mutai, H. Houjou and K. Araki, J. Phys. Chem. A, 2012, 116, 12041–12048 CrossRef CAS PubMed.
- H. Fang and Y. Kim, J. Chem. Theory Comput., 2013, 9, 3557–3566 CrossRef CAS.
- Y. Houari, A. Charaf-Eddin, A. D. Laurent, J. Massue, R. Ziessel, G. Ulrich and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 1319–1321 RSC.
- M. Isaks, K. Yates and P. Kalanderopoulos, J. Am. Chem. Soc., 1984, 106, 2728–2730 CrossRef CAS.
- P. Kalanderopoulos and K. Yates, J. Am. Chem. Soc., 1986, 108, 6290–6295 CrossRef CAS.
- M. Lukeman and P. Wan, Chem. Commun., 2001, 1004–1005 RSC.
- P. Wan and G. Zhang, Res. Chem. Intermed., 1993, 19, 119–129 CrossRef CAS PubMed.
- N. Basarić and P. Wan, Photochem. Photobiol. Sci., 2006, 5, 656–664 Search PubMed.
- N. Aein and P. Wan, J. Photochem. Photobiol., A, 2009, 208, 42–49 CrossRef CAS PubMed.
- N. Basarić, A. Franco-Cea, M. Alesković, K. Mlinarić-Majerski and P. Wan, Photochem. Photobiol. Sci., 2010, 9, 779–790 Search PubMed.
- Y.-H. Wang and P. Wan, Photochem. Photobiol. Sci., 2011, 10, 1934–1944 CAS.
- N. Basarić, N. Doslić, J. Ivković, Y. H. Wang, J. Veljković, K. Mlinarić-Majerski and P. Wan, J. Org. Chem., 2013, 78, 1811–1823 CrossRef PubMed.
- Y.-H. Wang and P. Wan, Photochem. Photobiol. Sci., 2013, 12, 1571–1588 CAS.
- D. Skalamera, K. Mlinarić-Majerski, I. Martin-Kleiner, M. Kralj, P. Wan and N. Basarić, J. Org. Chem., 2014, 79, 4390–4397 CrossRef CAS PubMed.
- M. Lukeman and P. Wan, J. Am. Chem. Soc., 2002, 124, 9458–9464 CrossRef CAS PubMed.
- M. Lukeman and P. Wan, J. Am. Chem. Soc., 2003, 125, 1164–1165 CrossRef CAS PubMed.
- M. Flegel, M. Lukeman, L. Huck and P. Wan, J. Am. Chem. Soc., 2004, 126, 7890–7897 CrossRef CAS PubMed.
- N. Basarić and P. Wan, J. Org. Chem., 2006, 71, 2677–2686 CrossRef PubMed.
- M. Nayak and P. Wan, Photochem. Photobiol. Sci., 2008, 7, 1544–1554 CAS.
- N. Basarić, N. Doslić, J. Ivković, Y. H. Wang, M. Malis and P. Wan, Chem. – Eur. J., 2012, 18, 10617–10623 CrossRef PubMed.
- S. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS PubMed.
- A. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS PubMed.
- J. Schirmer, Phys. Rev. A: At., Mol., Opt. Phys., 1982, 26, 2395–2416 CrossRef CAS.
- A. Trofimov and J. Schirmer, J. Phys. B: At., Mol. Opt. Phys., 1995, 28, 2299–2324 CrossRef CAS.
- A. Köhn and C. Hättig, J. Chem. Phys., 2003, 119, 5021–5036 CrossRef PubMed.
- C. Hättig, Adv. Quantum Chem., 2005, 50, 37–60 CrossRef.
- A. Dreuw and M. Wormit, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 82–95 CrossRef CAS.
- K. Andersson, P. Malmqvist, B. Roos, A. Sadlej and K. Wolinski, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- K. Andersson, P. Malmqvist and B. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS PubMed.
- N. Försberg and P. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef.
- F. Aquilante, R. Lindh and T. Pedersen, J. Chem. Phys., 2007, 127, 114107–114713 CrossRef PubMed.
- M. A. L. Marques, C. A. Ullrich, F. Nogueira, A. Rubio, K. Burke and E. K. U. Gross, Time-dependent Density Functional Theory, Springer, 2006 Search PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- R. Ditchfield, W. Hehre and J. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS PubMed.
- P. Hariharan and J. Pople, Theor. Chem. Acc., 1973, 28, 213–222 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheesem, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision D.02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P. Malmqvist, P. Neogrády, T. Pedersen, M. Pitonák, M. Reiher, B. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- TURBOMOLE V6.5 2013, a development of University of Karlsruhe andForschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- W. Weber, PhD thesis, University of Zürich, 1996.
- W. Weber and W. Thiel, Theor. Chem. Acc., 2000, 103, 495–506 CrossRef CAS.
- A. Koslowski, M. E. Beck and W. Thiel, J. Comput. Chem., 2003, 24, 714–726 CrossRef CAS PubMed.
- W. Thiel, MNDO99 program, version 6.1, Max-Planck-Institut für Kohlenforschung, Mülheim, Germany, 2007 Search PubMed.
- D. Yarkony, J. Chem. Phys., 2001, 114, 2601–2613 CrossRef CAS PubMed.
- T. W. Keal, A. Koslowski and W. Thiel, Theor. Chem. Acc., 2007, 118, 837–844 CrossRef CAS PubMed.
- G. Granucci, M. Persico and A. Zoccante, J. Chem. Phys., 2010, 133, 134111–134119 CrossRef PubMed.
- G. Cui and W. Thiel, Angew. Chem., Int. Ed., 2013, 52, 433–436 CrossRef CAS PubMed.
- O. Weingart, Z. Lan, A. Koslowski and W. Thiel, J. Phys. Chem. Lett., 2011, 2, 1506–1509 CrossRef CAS.
- A. Kazaryan, Z. Lan, L. Schäfer, W. Thiel and M. Filatov, J. Chem. Theory Comput., 2011, 7, 2189–2199 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Active space in OM2/MRCI computations and Cartesian coordinates of all structures. See DOI: 10.1039/c5cp00101c |
| This journal is © the Owner Societies 2015 |