
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excitation and quenching mechanisms in the near-UV photodissociation of CH3Br and CH3Cl adsorbed on D2O or CH3OH on Cu(110)
E. T.
Jensen
Department of Physics, University of Northern BC, 3333 University, Way, Prince George B.C., V2N 4Z9, Canada. E-mail: ejensen@unbc.ca; Tel: +1-250-960-6463
First published on 5th March 2015
Abstract
Photochemical processes for CH3X (X = Cl, Br, I) adsorbed on top of thin films of D2O or CH3OH on a Cu(110) substrate is studied by time-of-flight mass spectrometry for a range of UV wavelengths (351–193 nm). Photodissociation via dissociative electron attachment by photoelectrons and by neutral photodissociation is identified and quantified based on the observed dynamics of the desorbing CH3 fragments. Photoelectron-driven dissociation of CH3X is found to be a maximum for monolayer quantities of the D2O or CH3OH on Cu(110), but with differing kinetic energy release on the two substrates. The dynamics of CH3Br and CH3Cl photodissociation qualitatively differ on CH3OH/Cu(110) as compared to D2O/Cu(110), which is ascribed to differing molecular structures for these systems. Evidence is presented for an efficient inter-molecular quenching mechanism for neutral photoexcitation of CH3Cl and CH3Br on the CH3OH/Cu(110) substrate.
1 Introduction
There has been much recent work reported on UV photon- and electron-driven chemical processes in heterogeneous molecular thin films, particularly for water ices. These studies are driven by interests in astrochemistry and planetary science,1–3 as well as terrestrial processes as seen in radiation chemistry4,5 and photocatalysis.6,7 A wide range of surface science techniques have been applied to studying these systems. To date there have been relatively few studies of photodissociation dynamics in these types of heterogeneous systems. We have studied a range of halomethanes (CH3X, X = Cl, Br, I) adsorbed on thin films of D2O or CH3OH on Cu(110) substrate. The stimulated dissociation properties of these halomethanes have been studied in some detail in both the gas-phase as well as condensed on surfaces. Depending on the context, these molecules can display low-energy photoelectron driven Dissociative Electron Attachment (DEA) or neutral photodissociation processes when in the adsorbed state.The solid ices of D2O and CH3OH have large bandgaps, with the onset of absorption in solid methanol near 6.7 eV (λ < 184 nm),8 while for water the absorption onset is 8.5 eV.9 However these molecules are known to play roles in electron-driven chemistry in a variety of systems, as the dipolar molecules can solvate low-energy electrons such as photoelectrons10 or impinging external electrons.11 The solvation dynamics of electrons at thin water films on metal substrates has been studied extensively by two-photon photoemission.10,12 Several studies have implicated electron transfer at water–halocarbon interfaces as being responsible for halocarbon dissociation.11,13,14 Photoelectron dynamics and solvation on CH3OH15–17 thin films been studied using two-photon photoemission by a variety of research groups, and is known as a ‘hole-getter’ when adsorbed on TiO2.18
The surface chemistry and photochemistry of methyl halides on a variety of metal surfaces has been the subject of many studies,19 as has the adsorption of water20,21 and methanol.22,23 There are several previous studies that have looked at the coadsorption systems of methyl halides with water, most particularly chloromethane and water. Due primarily to the electrostatic dipole moments of these species, the interactions between CH3Cl and D2O on metals are characterized by repulsion, and has been investigated by temperature programmed desorption (TPD) and modelling studies by Maschhoff et al.24 who concluded that the long range electrostatic dipole–dipole interactions are an important factor in the CH3Cl structures. The CH3Cl binding energies were found to decrease with increasing coverage of D2O and CH3Cl, and the repulsive interactions cause CH3Cl islands to form atop the preadsorbed D2O or between D2O islands. Similar conclusions are reached by Lilach and Asscher,25 studying CH3Cl and H2O on Ru(100), and inferred the net orientation of the CH3Cl dipoles from work-function changes during adsorption and TPD. A recent study of CH3Cl and D2O on Pd(111) by Fournier et al. using sum-frequency generation (SFG) spectroscopy26 concluded that the CH3Cl adsorbs onto the D2O through hydrogen bonding between the D and Cl atoms and that the O–D–Cl–CH3 bonds are aligned close to the surface normal, though with wider angular variation than for Cl–CH3 on the bare metal surface. Their SFG results also indicate that the large Cl electronegativity induces a partial charge transfer between the surface and the D2O. There have been several previous studies of photochemistry in these systems – using a UV Hg arc lamp (hν > 5.4 eV) to irradiate CH3Cl/D2O/Pt(111), Jo and White27 highlighted the role of low energy photoelectrons in the observed CH3Cl photodissociation, and the rapid diminution of the transport of the relevant photoelectrons as the D2O layer thickness was increased.
There are relatively few previous studies of the dynamics of photodissociation in these heterogeneous systems, in which the mechanisms of photodissociation can be analyzed from the photofragment translational energies. We recently published a study of CH3I/D2O/Cu(110) photodissociation at λ = 248 nm in which the CH3 photofragment translational energies were analyzed to highlight the varying contributions from neutral photodissociation and photoelectron dissociative electron attachment mechanisms.28 A study of CH3I on thick D2O layers29 analyzed the ground- and excited state I atoms emitted subsequent to λ = 260 nm and 290 nm photodissociation. These studies showed evidence for a proportion of the I-atoms having larger than gas-phase translational energies which was ascribed to “chattering” during dissociation from ‘methyl-down’ oriented molecules on the ice surface as well as fast CH3 photofragments leaving the surface from ‘methyl-up’ oriented molecules.
1.1 Energetics of stimulated dissociation
If one begins by considering the dissociation of a CH3X molecule in free space, then the requirements of momentum and energy conservation put a limit on how the excess kinetic energy is partitioned between the CH3 fragment and the halogen atom. For a DEA process the following can be used to rationalize the CH3 photofragment kinetic energy in terms of the component energy factors: | (1) |
For neutral photodissociation, the analogous equation is:
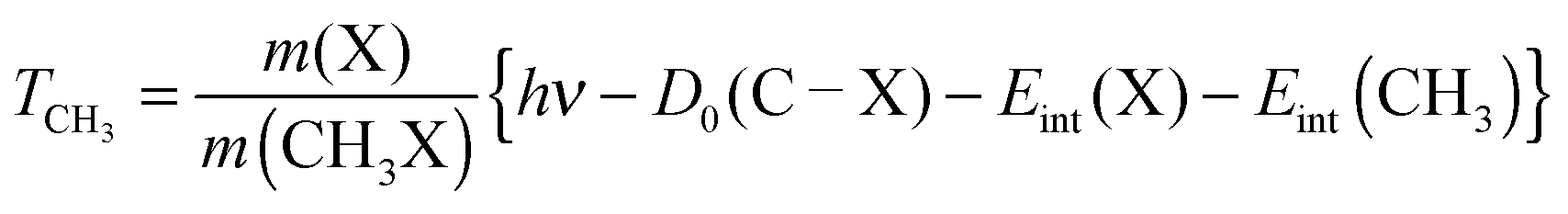 | (2) |
Gas-phase photodissociation of CH3Br33,34 and CH3Cl35 at λ = 193 nm occurs in the ‘A-band’, a set of overlapping excited electronic states. In contrast to the better known case of CH3I A-band photodissociation, the 193 nm photodissociation of CH3Br and CH3Cl is dominated by a perpendicular transition from the ground state, mainly via the 1Q1 state, which correlates to dissociation to CH3 and a ground-state halogen atom. For CH3Br there is evidence34 for non-adiabatic coupling with the 3Q0 state, which leads to CH3 and electronically excited Br*. Given the experimental geometry used in our work, these neutral photodissociation channels are accessible in the experiments we describe below.
For either A-band neutral photodissociation or photoelectron driven DEA of the halomethanes, the dissociation process is direct, with bond-breaking occurring in a few tens of femtoseconds. Photodissociation of halomethanes adsorbed on or close to a metal surface can be inhibited by quenching.36,37 When several layers of halomethanes are adsorbed, or are adsorbed on top of a spacer layer of another species, both DEA and neutral photodissociation have been observed. Quenching of one or both photodissociation pathways by the metal surface at these timescales requires a rapid interaction, such as resonant electron–hole transfer between the excited molecule and the substrate.37
2 Experimental details
The experiments were performed in an ultra-high vacuum system that has been described previously.38 The Cu(110) single crystal sample is cooled by liquid nitrogen (base temperature 90 K) and can be heated by electron bombardment to 920 K for cleaning. Sample cleanliness and order were monitored by Auger electron spectroscopy and low energy electron diffraction measurements respectively.Neutral products from surface photodissociation travel 185 mm to pass through a 4 mm diameter aperture to a differentially pumped Extrel quadrupole mass spectrometer (QMS) with an axial electron bombardment ionizer. The sample to ionizer distance is 203 mm. Ions created in the ionizer then travel through the quadrupole region and are mass selected, in the present experiments using m/z = 15 amu. Ion arrivals are recorded using a multichannel scaler that begins counting 50 μs prior to the initiating laser pulse, and the counts recorded from multiple laser pulses are summed. Unless otherwise indicated, the spectra shown in the present work are the result of summing data from 1000 laser pulses into 1000 1 μs time bins. In order for the ion arrival times to reflect the neutral fragment time-of-flight, they are corrected for the ion flight time (for CH3+, 17 μs at the 50 eV ion energy used in the QMS). This is the leading systematic uncertainty in the recorded flight times (±1.5 μs) which does not affect comparisons between different TOF spectra but does lead to fixed nonlinear systematic uncertainty in the reported fragment kinetic energies (KE ∝ 1/(TOF)2), which is most problematic at short flight times. The TOF spectra N(t) were converted to probability distributions versus CH3 kinetic energy (P(E)) using the Jacobian transformation with a correction factor 1/t to account for the reduced ionization probability of faster neutral CH3 fragments.
The laser pulses (∼5 ns duration) are produced by a small excimer laser (MPB PSX-100) operating at 20 Hz. In this work 351 nm (XeF), 308 nm (XeCl), 248 nm (KrF) and 193 nm (ArF) laser light was used, with laser fluences on the sample of ∼0.8 mJ cm−2 or less per pulse, depending on the wavelength used. Linearly polarized light has been used exclusively in this work. To create polarized light, the beam passes through a birefringent MgF2 crystal to separate p- and s-polarized components, which can then be directed at the sample. All of the TOF spectra shown in the present work were obtained using p-polarized light, though we also acquired data using s-polarized light for comparison.† The laser pulses were collimated using a 6 mm diameter aperture and were unfocussed on the sample. The laser light is incident upon the sample at a fixed angle of 45° from the mass spectrometer axis – for example, when the Cu(110) sample is oriented to collect desorption fragments along the surface normal direction, the light is incident at 45°.
Deposition of molecules on the sample is done using a microcapillary array directed doser,39 with the sample held normal to the doser, 25 mm away. This was found to enhance the deposition by a factor of 10 compared to background dosing. The CH3Br (Aldrich, ≥99.5%) and CH3Cl (Aldrich, ≥99.5%) gas used in this work was transferred via a glass and teflon gas-handling system. The CH3I (Aldrich, 99.5%) dosing used room temperature vapour from the liquid. The D2O (Aldrich, 99.9 atom% D) and CH3OH (Aldrich, ≥99.9%) used in this work was degassed by multiple freeze–pump–thaw cycles and was contained in a pyrex vial a few cm from the precision leak valve used to admit gases to the directed doser. The CH3Br dosing was calibrated by temperature programmed desorption measurements. In this case, 0.95 ± 0.05 L CH3Br was found to correspond to 1.0 ML for that substrate. In the present case of adsorption of CH3Br on D2O thin films in which the surface structure is less well characterized and no distinct TPD or photochemical signatures define what dose corresponds to monolayer CH3Br, we report CH3Br doses in terms of effective monolayers based on this calibration (1.0 ML = 0.95 L). For D2O we determined an effective monolayer calibration based on findings from TPD and titration of CCl4 on top of varying D2O films, which formed atomic chlorine on the metal surface after warming to desorb the molecular layers. From this we found 1.0 ML = 0.30 L for D2O. A similar procedure was used to calibrate the dosing of CH3OH, where 0.35 L was equivalent to 1 ML coverage. In deposition of these molecules, we assumed unit sticking probability on the various substrates for the temperature used for dosing (T < 95 K).
Details of the adsorption structures found for D2O/Cu(110) has been the subject of a number of studies, and is summarized in ref. 21. Less detail is available on the structure of CH3OH/Cu(110), though a study using IR spectroscopy40 indicates that the first layer adsorbs uniformly, and that 3D crystallites grow in the multilayer regime. It is also possible that the D2O or CH3OH structures are perturbed by the coadsorption of the dipolar CH3X molecules.24 In the results reported in the present work, we have looked for changes in photochemical behaviour as the different molecular coverages were varied, in part as a means to understand what structural changes might be occurring.
3 Results and discussion
3.1 Photodissociation at 248 nm on D2O/Cu(110)
Time-of-flight spectra at 15 amu for CH3Br adsorbed on D2O/Cu(110) obtained using 248 nm (hν = 5.0 eV) light show two main features, as illustrated in Fig. 1 – a photodissociation feature that peaks near 60 μs flight time and a broad, slow photodesorption feature centered around 500 μs flight time. Both of the TOF features are the result of photoelectron transfer from the metal substrate41 to the CH3Br layer atop the D2O layer. This photoelectron driven dissociation and desorption is strongly enhanced as the D2O coverage is increased to reach one layer of adsorbed D2O. These features then diminish as the D2O coverage is further increased, as can be seen in the data of Fig. 2. The overall yields observed for both the photodissociation and photodesorption features are plotted in Fig. 3. The yields reach a pronounced maximum for roughly 1–1.5 ML D2O dosed, and then decrease, roughly exponentially with a 1/e attenuation distance of 2.7 ML of D2O. This attenuation of photoelectron driven dissociation and desorption by ultrathin water layers is consistent with earlier photodissociation findings (1/e = 2 ML) of Gilton et al.42 as well as those of Jo and White,43 who measured an attenuation distance of 2.7 ML for D2O/Pt(111) for photoelectrons above the vacuum level. In the present case it is likely that the photoelectrons responsible for the observed TOF features are subvacuum level “hot” photoelectrons, as discussed below. Although the data of Fig. 3 display a smoothly changing photochemical yield as the D2O coverage is increased, this does not necessarily mean that the D2O layer growth is uniform. However the diminution of the photoelectron-driven signal does show that the average distance from the substrate to the CH3Br target that is detected in TOF increases with the D2O coverage. | ||
| Fig. 1 Time of flight spectrum from the photodissociation of 0.7 ML of CH3Br adsorbed on 1.0 ML D2O/Cu(110) detected at 15 amu (CH3+) following 248 nm irradiation. The peak at 58 μs flight time is due to photodissociation of the CH3Br and the slower broad peak (500 μs) is a result of photodesorption of CH3Br molecules that dissociate in the mass spectrometer ionizer. The inset plot shows the same data transformed to a probability distribution as a function of CH3 kinetic energy. | ||
 | ||
| Fig. 2 Sequence of time of flight spectra from 1 ML of CH3Br on D2O/Cu(110) obtained using a varying amount of D2O and 248 nm light. The CH3Br photodissociation feature at 58 μs flight time increases rapidly as the first monolayer of D2O is added, and then diminishes as the D2O layer thickness is increased. | ||
 | ||
| Fig. 3 The yield of CH3 photofragments obtained from 248 nm photodissociation (red circles) and photodesorption (blue triangles) of 1 ML CH3Br on D2O/Cu(110) as a function of D2O dose. The solid line represents a simple exponential decay fit to the data points from 1.0 ML D2O and higher-essentially the same fitted function is obtained from either the photodissociation or the photodesorption data. | ||
The photodissociation of CH3Cl on D2O/Cu(110) at 248 nm has also been studied, with a representative TOF spectrum shown in Fig. 4. The yield of CH3 photofragments from CH3Cl on D2O/Cu(110) is much lower than that seen for CH3Br, and the spectrum shown is an average of 3 spectra, in order to improve the signal-to-noise ratio. In our experiments we could only detect 248 nm photodissociation of CH3Cl on D2O/Cu(110) in TOF spectra for a relatively narrow range of D2O coverages, those close to 1 ML which also gave the largest photofragment yields seen for CH3Br in Fig. 3. For the CH3Cl/D2O/Cu(110), the photodesorption feature near 500 μs is small relative to that that seen for CH3Br. Detailed comparison between the 248 nm photodissociation of CH3Cl and CH3Br on D2O/Cu(110) is shown in Fig. 5 – the CH3 photofragments from CH3Cl are roughly 8 to 9 μs slower than those from CH3Br, corresponding to roughly 0.25 eV lower translational energy. Based on the free molecule dissociation energetics for DEA from eqn (1) both the absolute and the relative CH3 kinetic energies can be considered for the observed photodissociation of CH3Br and CH3Cl. The electron affinities of the halogen atoms are 3.36 eV (Br) and 3.61 eV (Cl), and the dissociation energies D0 are 3.05 eV (CH3–Br) and 3.63 eV (CH3–Cl).‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 We do not know precise values for the energy contributions from the incident electrons or the anion solvation (Ee− and ΔEsolv in eqn (1)), but these can be estimated. For CH3Br condensed in a dielectric medium close to a metal surface, the dissociative anion state is shifted to lower energy45 and results in a DEA process facilitated by very low energy electrons, close to the vacuum level. Using the estimate for electrons causing DEA of Ee− = 0 eV (at the system vacuum level Evac) results in ΔEsolv = 1.06 eV for CH3Br, which is a reasonable value based on estimated values as well as the polarization induced shifts seen experimentally in other similar contexts.46 It may well be the case that the maximum CH3Br DEA cross section occurs for electrons below Evac.47,48 In the case of adsorbed CH3Cl, it is also well-known that the anion state is shifted to much lower energy relative to the ground-state neutral45 so that low energy electrons can cause DEA with a large cross section. Assuming Ee− = 0 eV results in an estimate of ΔEsolv = 1.31 eV. This value for ΔEsolv appears too large in comparison with that for CH3Br, since both anions would be expected to be in similar dielectric environments and have similar dissociation times. The value of ΔEsolv would be reduced if the relevant Ee− for DEA is 0.25 eV larger for the CH3Cl than CH3Br. This magnitude of differing incident electron energy responsible for DEA is compatible with observations in electron beam experiments on CH3Cl and CH3Br condensed on Kr/Pt.45 While the magnitudes of the polarization shifts are likely different between the Kr/Pt and the D2O/Cu(110) substrates, the correspondence between the polarization shifts and the requisite incident electron energies seems to be reasonable.
44 We do not know precise values for the energy contributions from the incident electrons or the anion solvation (Ee− and ΔEsolv in eqn (1)), but these can be estimated. For CH3Br condensed in a dielectric medium close to a metal surface, the dissociative anion state is shifted to lower energy45 and results in a DEA process facilitated by very low energy electrons, close to the vacuum level. Using the estimate for electrons causing DEA of Ee− = 0 eV (at the system vacuum level Evac) results in ΔEsolv = 1.06 eV for CH3Br, which is a reasonable value based on estimated values as well as the polarization induced shifts seen experimentally in other similar contexts.46 It may well be the case that the maximum CH3Br DEA cross section occurs for electrons below Evac.47,48 In the case of adsorbed CH3Cl, it is also well-known that the anion state is shifted to much lower energy relative to the ground-state neutral45 so that low energy electrons can cause DEA with a large cross section. Assuming Ee− = 0 eV results in an estimate of ΔEsolv = 1.31 eV. This value for ΔEsolv appears too large in comparison with that for CH3Br, since both anions would be expected to be in similar dielectric environments and have similar dissociation times. The value of ΔEsolv would be reduced if the relevant Ee− for DEA is 0.25 eV larger for the CH3Cl than CH3Br. This magnitude of differing incident electron energy responsible for DEA is compatible with observations in electron beam experiments on CH3Cl and CH3Br condensed on Kr/Pt.45 While the magnitudes of the polarization shifts are likely different between the Kr/Pt and the D2O/Cu(110) substrates, the correspondence between the polarization shifts and the requisite incident electron energies seems to be reasonable.
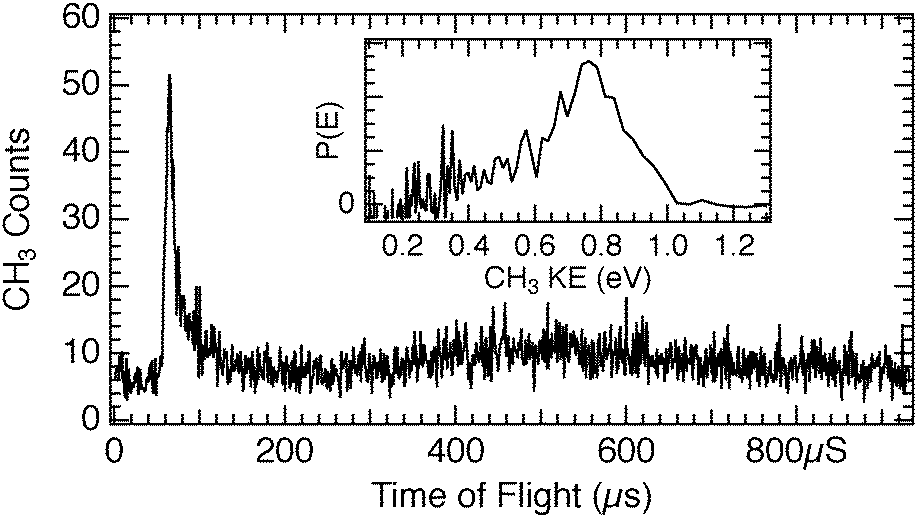 | ||
| Fig. 4 Time of flight spectrum from the photodissociation of 0.5 ML CH3Cl adsorbed on 1.0 ML D2O/Cu(110) obtained using 248 nm light. The spectrum shown is the average of three spectra, in order to improve the signal-to-noise ratio. A photodissociation feature is observed at 65 μs flight time as well as a photodesorption feature centered around 500 μs. The inset shows the same data in a CH3 photofragment kinetic energy distribution, with a peak in P(E) near 0.75 eV. | ||
 | ||
| Fig. 5 Comparison of time-of-flight spectra from 0.5 ML CH3Br (blue data) and CH3Cl (red data) adsorbed on 1.0 ML D2O/Cu(110) obtained using 248 nm light. The inset plot shows this data transformed into the CH3 fragment kinetic energy distributions. | ||
3.2 Photodissociation at 193 nm on D2O/Cu(110)
Irradiation using 193 nm (hν = 6.4 eV) light for CH3Br and CH3Cl adsorbed on D2O/Cu(110) produced TOF spectra that display neutral photodissociation of the methyl halides in addition to the photoelectron driven dissociation seen using 248 nm light. Fig. 6 shows a TOF spectrum for 1 ML CH3Br adsorbed on 1 ML D2O/Cu(110). Although the photon energy is 1.4 eV larger, so that a wider range of photoelectron energies is generated, the CT-DEA photodissociation feature appears at the same flight time (60 μs) and translational energy (1.0 eV) as for the 248 nm data. The 193 nm photons also produce a neutral photodissociation peak seen at 39 μs flight time, which appears at 2.1 eV in the inset photofragment kinetic energy distribution. That the neutral photodissociation is observed for CH3Br using 193 nm light but not for 248 nm is consistent with the gas-phase neutral photodissociation cross section, being 6.0 × 10−19 cm2 at 193 nm, and is at least two order of magnitude smaller at 248 nm.49 Photodissociation TOF features for varying D2O precoverages are shown in Fig. 7 in which similar variation in the CT-DEA dissociation is seen at 193 nm as was the case for 248 nm, while the neutral photodissociation feature is seen to increase with the initial D2O coverage and then remains essentially constant for increasing D2O layer thickness. | ||
| Fig. 6 Time of flight spectrum for 1 ML CH3Br on 1 ML D2O/Cu(110) obtained using 193 nm light. As compared to Fig. 1, there is an additional photodissociation component peaking at 38 μs flight time due to neutral photodissociation of the CH3Br. The inset data shows the distribution of CH3 photofragment kinetic energies, with the neutral photodissociation peak at 2.1 eV and the photoelectron driven DEA feature at 1.0 eV. | ||
 | ||
| Fig. 7 Sequence of TOF spectra from 1 ML CH3Br on D2O/Cu(110) obtained using 193 nm light for varying amounts of D2O coverage on the surface. The photoelectron driven photodissociation feature at 58 μs flight time grows rapidly for low D2O coverages and then diminishes for D2O layer thicknesses beyond 1 ML, while the neutral photodissociation feature at 38 μs grows with D2O coverage and then remains constant for thicker D2O layers. | ||
Irradiation of CH3Cl adsorbed on D2O/Cu(110) using 193 nm light results in TOF spectra such as that shown in Fig. 8, in which both a neutral photodissociation peak (at 46 μs) and a CT-DEA driven dissociation at 62 μs are seen, as well as a small photodesorption feature centered around 500 μs. As for CH3Br, the photoelectron driven photodissociation and photodesorption feature magnitudes are strongly enhanced for roughly 1 ML D2O coverage, and diminish rapidly as the D2O precoverage is increased above 1 ML, as shown in Fig. 9 for the photodissociation peaks. The observation of the neutral photodissociation peak of CH3Cl at 193 nm is reasonable based on the gas-phase cross section of 0.7 × 10−19 cm2, which is at least several orders larger than at 248 nm.49 The observed relative yields from neutral photodissociation for CH3Br and CH3Cl at 193 nm (for example, Fig. 7 and 9) are also consistent with the relative magnitudes (∼9×) of the gas-phase cross sections at this wavelength.
 | ||
| Fig. 8 Time of flight spectrum from 0.7 ML CH3Cl on 1 ML D2O/Cu(110) obtained using 193 nm light. As compared to the same system at 248 nm (Fig. 4), an additional peak is observed at 46 μs flight time, a consequence of neutral photodissociation. The inset plot shows the data plotted to show the CH3 kinetic energy distribution, with peaks at 0.8 eV (photoelectron DEA) and 1.5 eV (neutral photodissociation). | ||
 | ||
| Fig. 9 A sequence of TOF spectra from 0.5 ML CH3Cl adsorbed on varying amounts of D2O on Cu(110), obtained using 193 nm light. The amplitude of the DEA feature is strongly modulated by the D2O coverage, peaking for 1 ML of D2O. The amplitude of the neutral photodissociation peak is relatively unaffected by the D2O coverage, beyond a minimum amount. | ||
3.3 Photodissociation at CH3X on CH3OH/Cu(110)
As a comparison and contrast to the D2O/Cu(110) system, we have also studied the photodissociation of the same small halomethane molecules adsorbed on CH3OH/Cu(110). Methanol has a similar dipole moment44 (1.70 D) to water (1.85 D), and on Cu(110) its adsorption results in a comparable decrease in work function (Δϕ = −1.35 eV for 1 ML at 140 K (ref. 22)) as for water (Δϕ = −1.0 eV for 1 ML (ref. 50)). In some respects our photochemical observations from CH3OH layers are comparable to those from D2O – the photoelectron-driven DEA processes are strongly enhanced for monolayer coverages and diminish for thicker CH3OH layers, however the dynamics of dissociation are significantly altered. Fig. 10 shows comparative TOF spectra for CH3Br adsorbed on CH3OH/Cu(110) and D2O/Cu(110) obtained using 248 nm light. For the CH3OH/Cu(110) substrate, the CH3Br CT-DEA photodissociation signal is slower, reduced in magnitude and broadened, while the photodesorption signal centered around 500 μs is substantially increased. The inset plot of Fig. 10 highlights the observation that the CH3 fragments from photoelectron driven dissociation are slower for CH3Br/CH3OH than those from CH3Br on D2O, with the leading edge of the main TOF peaks separated by ∼8 μs. | ||
| Fig. 10 Comparison of TOF spectra obtained from 0.3 ML CH3Br adsorbed on D2O/Cu(110) (top, blue trace) with CH3OH/Cu(110) (lower, red trace), using 248 nm light. When adsorbed on CH3OH (as compared to D2O), the CH3Br photodissociation feature near 60 μs flight time is reduced in magnitude but broader, whilst the photodesorption feature centered around 500 μs flight time is significantly larger in amplitude. The inset plot shows detail of the CT-DEA photodissociation feature for CH3Br, being slower on CH3OH as compared to on D2O. | ||
Upon changing the photon energy to 193 nm, the TOF spectrum for CH3Br adsorbed on CH3OH/Cu(110) of Fig. 11 displays both photoelectron as well as neutral photodissociation features and allows comparison with that from D2O/Cu(110). The inset plot of the CH3 fragment kinetic energy distributions shows that the slower CH3 photofragments seen from CT-DEA of CH3Br/CH3OH as compared to CH3Br/D2O are unique to the charge-transfer dissociation – the CH3 photofragments from neutral photodissociation at 193 nm have the same translational energy on both molecular films.
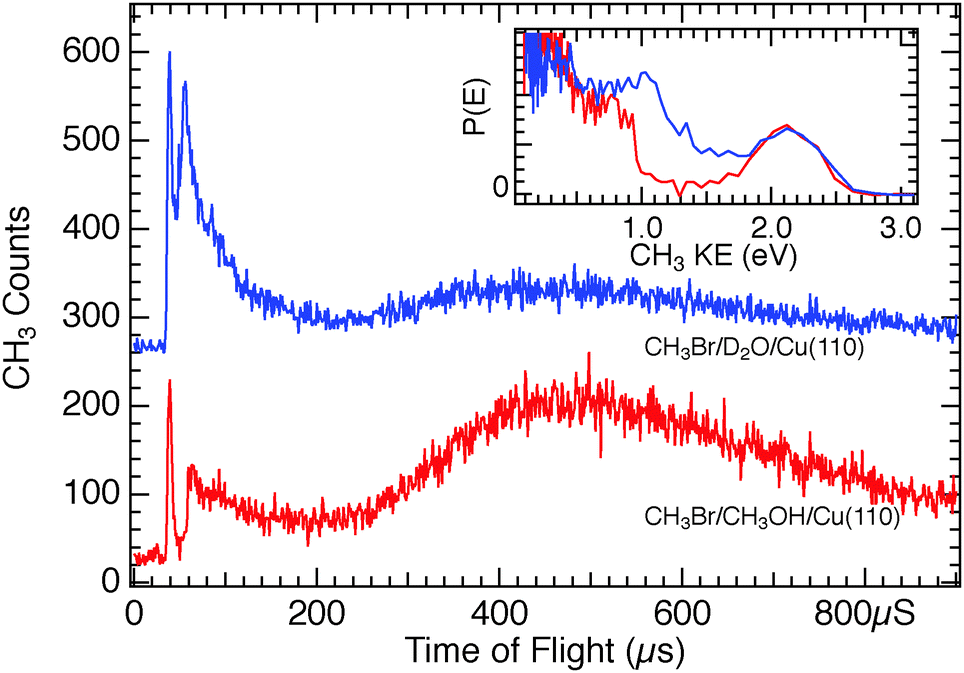 | ||
| Fig. 11 Comparison of TOF spectra obtained for 0.5 ML CH3Br adsorbed on 1 ML D2O/Cu(110) (top, blue trace) and 1.3 ML CH3Br on CH3OH/Cu(110) (lower, red trace), using 193 nm light. The inset plot shows the same data as a function of the CH3 fragment kinetic energy. The neutral photodissociation peak (40 μs; 2.2 eV) is the same on both substrates, while the CT-DEA photodissociation feature is observed to have lower KE on CH3OH as compared to D2O, as also seen at 248 nm in Fig. 10. | ||
Fig. 12 highlights another difference seen for CH3Br adsorbed on CH3OH/Cu(110) – though using 193 nm light has a photon energy sufficient for neutral photodissociation of CH3Br, this is not observed at low coverages of CH3Br on CH3OH and is seen only for CH3Br coverages beyond roughly 1 ML, independent of the precoverage amount of CH3OH. This is in contrast to the analogous situation seen for D2O/Cu(110), in which the 193 nm neutral photodissociation is not observed to have a minimum onset coverage of CH3Br.
 | ||
| Fig. 12 A series of TOF spectra for varying amounts of CH3Br adsorbed on 1 ML CH3OH on Cu(110), obtained using 193 nm light. While the CT-DEA photodissociation and the photodesorption features are observed at all CH3Br coverages, the neutral photodissociation of CH3Br is only observed for coverages of CH3Br above 1 ML. | ||
Based on the findings for CH3Br on CH3OH/Cu(110), the expectations for CH3Cl adsorbed on this substrate would be for a reduced but broadened photoelectron-driven dissociation signal and increased photodesorption. In this light, the observed TOF spectra for this system, such as that of Fig. 13 are surprising. The yield of CH3 photofragments from photoelectron-driven dissociation is very large and also narrow in the TOF spectra compared to the analogous case for CH3Br, and the photodesorption feature is much smaller than that seen in the previously described systems. In common with the findings for CH3Br, the CT-DEA photodissociation peak for CH3Cl on CH3OH/Cu(110) is at a longer flight time than seen for D2O/Cu(110), as shown in Fig. 14. As compared to the D2O case, the CH3 photofragments from CH3Cl photodissociation are ∼6 μs slower, or as shown in the inset plot of Fig. 14, the peak in the P(E) distribution is at 0.15 eV lower kinetic energy.
 | ||
| Fig. 13 Time of flight spectrum from the photodissociation of 0.3 ML CH3Cl adsorbed on 1 ML CH3OH/Cu(110) using 248 nm light. The photodissociation via the DEA mechanism give a large yield of CH3 photofragments (peak at 71 μs), and a very small photodesorption feature (the dashed line indicates the CH3 background count level). | ||
 | ||
| Fig. 14 Comparison of TOF spectra for CH3Cl photodissociation at 248 nm, between that adsorbed on D2O/Cu(110) (blue trace) and on CH3OH/Cu(110) (red trace). The inset plot shows the same data plotting the probability distribution as a function of the CH3 photofragment kinetic energy. While the yield of CH3 photofragments is much larger for CH3Cl adsorbed on CH3OH (note the respective y-axis scales for the TOF spectra), the kinetic energy release is smaller than when adsorbed on D2O/Cu(110). | ||
That the observed photodissociation for CH3Cl on CH3OH/Cu(110) is photoelectron driven is supported by data such as that shown in Fig. 15, in which the amount of CH3OH is varied for a fixed CH3Cl dose. The photodissociation yield is found to increase dramatically from that of the clean surface, to a maximum yield found for roughly 1 ML CH3OH. The yield is then observed to decrease, and a simple exponential fit suggests an attenuation length of 3.0 ML for the relevant photoelectron transport through CH3OH, which is comparable to the 2.7 ML discussed above for D2O. Similar variations with CH3OH dose are also found for CH3Br/CH3OH for the photoelectron driven photodissociation peak as well as for the photodesorption feature. The neutral photodissociation peak for CH3Br seen at 193 nm (the TOF peak at 39 μs in Fig. 12) behaves differently – increasing monotonically with for low CH3OH doses and remaining essentially constant for higher multilayer doses. As the CH3Cl coverage is increased for a fixed amount (1 ML) of CH3OH, the CH3 photoyield due to photoelectron driven dissociation grows monotonically with the CH3Cl dose until 1 ML is dosed, and then remain essentially fixed for higher doses. We interpret this result as being due to having only the first monolayer of CH3Cl able to stick to the CH3OH/Cu(110) substrate at the temperature used for our experiments.
 | ||
| Fig. 15 Yield of CH3 photofragments from photodissociation of CH3Cl/CH3OH/Cu(110) as the CH3OH coverage is varied. The data is obtained using 193 nm light and 1 ML of CH3Cl. The solid line is a simple exponential fit to the data points above 1 ML CH3OH coverage, and represents a 1/e attenuation distance of 3.0 ML for CH3OH. | ||
It is also notable that the CH3 photofragment translational energy distribution from DEA does not change as the CH3OH thickness is varied nor over the range of wavelengths that CH3Cl/CH3OH/Cu(110) photodissociation is observed (308 nm to 193 nm) – TOF spectra from different wavelengths can be overlaid and aside from simple linear scaling, are otherwise identical. This also appears to be the case in our data for the CH3Br on D2O/Cu(110) and CH3OH/Cu(110), though the distributions are more complex. This point is salient as photodissociation via DEA is often interpreted in terms of the ‘3-step model’32,51 in which the energy available for dissociation should reflect the convolution of the photoelectron distribution with the anion resonance attachment energy distribution. This leads to an expectation for lower energy available (eqn (1)) at the red end of the wavelengths used for photodissociation that should be reflected as a suppression of the high-energy side CH3 translational energy distributions, but this is not observed. One possible explanation for this lack of correlation between photon energy and fragment translational energy could be that intermediate electron sates modify the density of states at the vacuum interface, such as image-potential derived states15 that couple with the anion attachment resonance.52
A striking observation for CH3Cl/CH3OH/Cu(110) at 193 nm is that we do not observe CH3 photofragments from neutral photodissociation of the CH3Cl at 193 nm under any of the conditions studied – data obtained at 193 nm is essentially identical to that from 248 nm as shown in Fig. 13. Given the cross sections and intensities measured for CH3Cl/D2O/Cu(110) (e.g.Fig. 9), the neutral photodissociation channel should be easily observable. The absence of neutral photodissociation in the TOF spectra from the first monolayer of CH3Br on CH3OH/Cu(110) that is seen in Fig. 12 indicates that the neutral photodissociation mechanism is suppressed at 193 nm for CH3Cl and CH3Br on the CH3OH/Cu(110) substrate. This observation is discussed further in Section 4.
As described above, we see altered kinetic energy distributions for the CH3 photofragments via CT-DEA for CH3X on CH3OH as compared to D2O. We have also directly compared the observed distributions for CH3Cl to those of CH3Br adsorbed on CH3OH, which are shown in Fig. 16 for 248 nm light. The CH3 photofragments from CT-DEA of CH3Br are slightly faster than those from CH3Cl in the onset of the distribution as well as the most probable time. As shown in the inset plot, the leading edge of the P(E) distribution is at 0.20 eV higher energy for the CH3Br as compared to that of CH3Cl. This is a smaller kinetic energy difference than was seen for these two molecules on D2O in Fig. 5 (∼0.3 eV). This smaller difference in kinetic energies is a result of the larger downward shift in CH3 fragment kinetic energy for CH3Br on CH3OH as compared to D2O (ΔTCH3 = −0.20 eV) as compared to that for CH3Cl (ΔTCH3 = −0.15 eV).
 | ||
| Fig. 16 Comparison of the 248 nm photodissociation of CH3Br (blue trace) and CH3Cl (red trace) adsorbed on 1 ML CH3OH/Cu(110). The inset plot shows the related probability distributions as a function of the CH3 fragment translational energy. | ||
3.4 Cross sections
We have measured depletion cross sections for a selection of the molecular thin films examined in this work. These cross sections are obtained by recording CH3 photofragment yields from photodissociation and/or photodesorption for a sequence of TOF spectra. Time-of-flight spectra are obtained using 200–300 laser pulses per scan, then repeated for 10 or more successive scans. In the systems studied here, the TOF signals are observed to diminish as the net laser photon flux was increased, and the resulting yield vs. flux curves could be fit by a simple exponential decay model. Reasonable fits to the data were obtained, at least in the low flux limit, though this does not exclude the possibility that more complex photochemical processes not seen in the TOF data might be occurring in these heterogeneous thin films. In the cases where multiple features are observed in the TOF spectra (neutral photodissociation, DEA, photodesorption) we measured separate depletion cross sections for each feature. In the cases we examined, we measured the same (within experimental error) cross sections for co-present features. The resulting extracted cross sections are reported in Table 1. The values reported are nearly all obtained using 1 ML of the D2O or CH3OH, with between 0.5 and 1 ML of the methyl halide adsorbed on top. The one exception is for the λ = 193 nm photodissociation of CH3Br/D2O in which we also report the value for 15 ML of D2O (value in brackets) which represents the cross section in which only neutral photodissociation is observed in the TOF spectra. This value of 9.2 × 10−19 cm2 is somewhat larger than that reported for gas-phase49 CH3Br (σ = 6 × 10−19 cm2). This discrepancy may well be due to the rather large error associated with the absolute cross sections we estimate (±50%).§ In several cases we can observe and identify the photochemical processes in the TOF spectra (and some of these are shown and discussed above) but the yields are too low to yield a reliable cross section estimate, so these are denoted by ‘>0’. Situations in which no photochemical signals could be discerned in the TOF spectra are denoted by ‘—’. The tabulated values highlight an interesting contrast between the CH3Br and CH3Cl photodissociation results – the CH3Br photodissociation is observed at longer wavelengths on the D2O/Cu(110) films, while for CH3Cl, the photodissociation is larger and seen at longer wavelengths on the CH3OH/Cu(110) films. We are not aware of any previous observations of CH3Cl photodissociation with substantial cross section for λ = 308 nm (4.02 eV) photons. There are few previously reported photochemical cross-sections for halomethanes co-adsorbed with D2O or CH3OH. A study of CD3Cl caged within H2O layers on Ru(100) measured cross sections of ∼0.5 × 10−18 cm2 at 193 nm and ∼0.02 × 10−18 cm2 at 248 nm by observation of CD3Cl depletion,53 values that are compatible with those reported for CH3Cl/D2O/Cu(110) in Table 1.3.5 Angular distributions
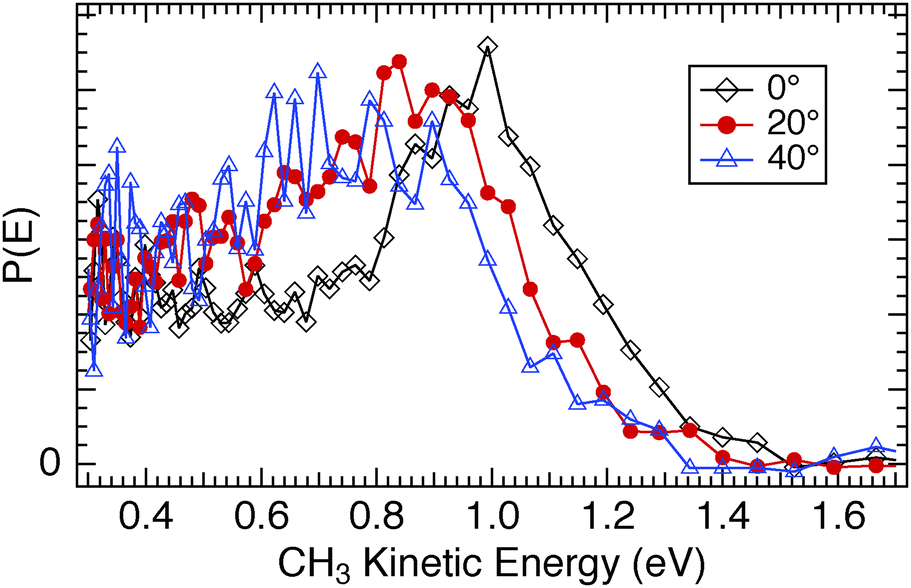
The substantive differences observed in the photodissociation dynamics between CH3Br and CH3Cl when adsorbed on CH3OH/Cu(110) were further investigated by measurements of the angular variation of the signals. For CH3Cl/CH3OH/Cu(110) we found an angular distribution for the CH3 photofragments well fit by a cosNθ functional form with N = 4, which does not change substantially with CH3Cl coverage. As the detection angle from the surface normal increases, the CH3 photofragment kinetic energy distribution is hardly affected, as seen in Fig. 17. There is a small diminution of the fastest CH3 fragments at larger escape angles, but no increase in the proportion of the lower energy inelastic tail even for the 60° detection angle. In contrast, the TOF spectra for CH3Br photodissociation (e.g.Fig. 11) show clear evidence of inelastic interactions on both the D2O/Cu(110) and CH3OH/Cu(110) surfaces. The P(E) distributions for a range of detection angles shown in Fig. 18 for CH3Br/D2O/Cu(110) display a more pronounced inelastic tail for CH3 kinetic energies lower than that of the main CT-DEA peak, and in some spectra a peak can be discerned near TCH3 ≈ 0.4 eV (e.g. insets in Fig. 5 and 16). We ascribed a similar feature seen for CH3I/D2O/Cu(110) to chattering as downward pointing CH3 fragments interact with the substrate before escaping.28 The inelastic signals are seen to grow in relative size as the detection angle from the surface normal is increased. The angular distributions of the CH3 photofragments from CH3Br are peaked in the surface normal direction (∝cosN(θ) with N = 4–5) but also display a shift to lower kinetic energy in both the fastest CH3 photofragments as well as the larger ‘inelastic tail’ as the angle increases.
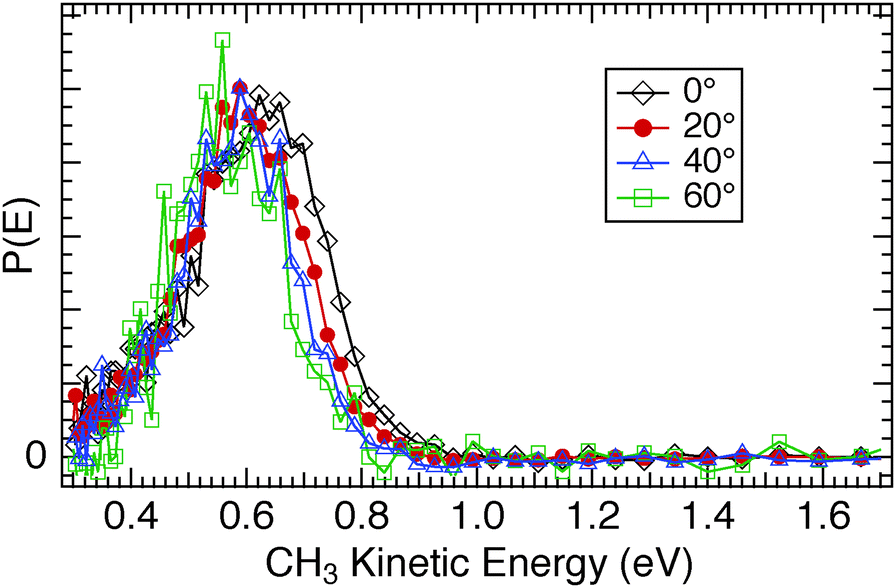 | ||
| Fig. 17 Comparison of the CH3 photofragment translational kinetic energy distributions from 248 nm photodissociation of CH3Cl adsorbed on 1 ML CH3OH/Cu(110) as a function of detection angle from the surface normal. | ||
 | ||
| Fig. 18 Comparison of the CH3 photofragment probability distributions from 248 nm photodissociation of CH3Br adsorbed on 1 ML D2O/Cu(110) as a function of detection angle from the surface normal. | ||
4 Additional discussion
4.1 Structure and dynamics
In many respects the structural properties of the halomethanes are quite similar, with the structure of the solids dominated by the electrostatic dipole interactions (dipole moments: CH3Cl-1.90 D; CH3Br-1.82 D; CH3I-1.64 D).44 In the bulk solid, although the molecular ordering is dominated by dipolar interactions, there are distinct differences between the structure of crystalline CH3Cl54 and CH3Br,55 which is ascribed to differences in the halogen–halogen binding interactions.55 The observed photochemical dynamics for CH3Br and CH3Cl on D2O/Cu(110) do not indicate significant differences in the structural aspects that impact the observed dynamics. For both methyl halides, we observe CT-DEA on thin D2O films over a range of wavelengths in which hot photoelectrons of the appropriate energies would be generated. In addition we observe photodesorption of the intact CH3X molecules that correlate with the CT-DEA dissociation fluxes. The observed dynamics are compatible with an antiferroelectric structure for the methyl halides, and given the net repulsive interactions with the D2O, it is anticipated that CH3X islands are present even at low coverages. These observations are consistent with our previous study of CH3I/D2O/Cu(110).28 At the shorter 193 nm wavelength, we also observe neutral photodissociation of both CH3Br and CH3Cl, with apparent cross sections comparable to those for the gas-phase. That these neutral photodissociation TOF features are very well-defined indicates that the measured CH3 photofragments are from ‘CH3-up’ oriented molecules that do not significantly interact with the surface during or post bond-cleavage.The clean Cu(110) substrate has a work function of 4.48 eV,44 and the D2O and CH3OH adlayers reduce the work function by 1 eV or more for monolayer coverages.22,50 In addition, the CH3X adlayers are known to reduce the work function on D2O25,26 due to the preferred orientation of the dipole. Hence even for the longest wavelength (351 nm, hν = 3.51 eV) used in this work, we expect that free photoelectrons (i.e. above the vacuum level) are present under most conditions studied, in addition to the subvacuum level ‘hot’ photoelectrons. At λ = 351 nm we only observed photoelectron-driven dissociation and photodesorption in the TOF spectra for the CH3Br/D2O/Cu(110) and CH3I/D2O/Cu(110) systems. Photodissociation was observed for a narrow range of D2O coverages around 1 ML, where the work function minimum and maximum CH3 yields (e.g.Fig. 3) are found. Since we observe no photoelectron-driven dissociation of CH3Cl on either D2O/Cu(110) or CH3OH/Cu(110) using 351 nm light, it can be concluded that higher energy photoelectrons are required for DEA of CH3Cl than are substantially available. Upon increasing the photon energy by 0.49 eV using 308 nm light (and consequently producing a similarly increased range of photoelectron energies), we observe not only much stronger dissociation signals from CH3Br/D2O/Cu(110) but also photodissociation of CH3Cl/CH3OH/Cu(110). This indicates a threshold photon energy between 3.53 eV and 4.02 eV for the latter system. The contrasting behaviour of CH3Br and CH3Cl photodissociation yields on D2O/Cu(110) and CH3OH/Cu(110) are highlighted near these thresholds. The CT-DEA photodissociation of CH3Cl is most prominent on the CH3OH/Cu(110) surface, and is only observed at shorter wavelengths on D2O/Cu(110) and with lower CH3 photofragment yields and cross-section. In contrast the CH3Br CT-DEA photodissociation is more prominent on the D2O/Cu(110), and we did not observe CH3 photofragments or photodesorption on CH3OH/Cu(110) using 351 nm light. We believe that these observations are related to the differing molecular structures of CH3Br and CH3Cl on the CH3OH and D2O molecular interfaces.
Further evidence for differing molecular ordering of CH3Br and CH3Cl on the two substrates considered comes from the observed CH3 time-of-flight distributions. The TOF spectra from CH3Cl/CH3OH/Cu(110) display a single narrow features due to CT-DEA of the CH3Cl (Ē = 0.62 eV, ΔE = 0.24 eV FWHM). This system displays almost no ‘inelastic tail’ of lower energy CH3 photofragments (Etrans < 0.4 eV, see Fig. 16 and 17) and there is only a very small photodesorption feature as compared to the other systems studied in this work. These observations for CH3Cl/CH3OH/Cu(110) lead us to propose that the CH3Cl molecules are adsorbed primarily with the Cl–CH3 axis normal to the surface and in the ‘CH3-up’ orientation. Bond-breaking due to DEA leads to the departing CH3 photofragment having little opportunity for inelastic interactions as it departs the surface and hence a relatively narrow translational energy distribution. Due to the solvation shift of the anion, the dissociation probability is high (i.e. low autoionization or quenching probability) so there is a relatively low probability for excited CH3Cl that do not dissociate but might have sufficient energy to break the molecule–surface bond and contribute to photodesorption. On the D2O/Cu(110) surface the photodissociation of CH3Cl yields fewer CH3 photofragments and a significantly lower cross section (Table 1). The data such as in Fig. 4, 8 and 14 show that there is a similarly small inelastic tail (Etrans < 0.5 eV) and a somewhat larger photodesorption signal as compared to the situation on CH3OH/Cu(110). An IR-SFG study of CH3Cl on D2O/Pd(111) indicates that the Cl–CH3 bond is along the surface normal in the ‘CH3-up’ orientation,26 and our own observations of the angular distributions for CH3 photofragments on both CH3OH and D2O are in accord with this. Depending on the dosing order and amount,25 it is possible that some mixed orientations for CH3Cl on D2O can be formed. We do believe that the CH3Cl/D2O/Cu(110) TOF data shows evidence for a minor amount antiferroelectric ordering of the CH3Cl – that downward pointing CH3 photofragments will inelastically scatter from the surface to contribute to the inelastic tail, and that ‘chattering’ type interactions will also lead to more quenching and subsequent molecular photodesorption56 on D2O/Cu(110) than for CH3OH/Cu(110).
On the basis of the observed energy and angular distributions, we conclude that CH3Br on the D2O/Cu(110) and CH3OH/Cu(110) substrates has a more mixed orientational ordering than for CH3Cl, most likely an antiferroelectric structure with both ‘Br-up’ and ‘Br-down’ configurations, though to a lesser extent on D2O/Cu(110) than for CH3OH/Cu(110). The role of local work function modulated by the ordering of surface dipoles at the interface has been noted previously,47,57 and in the case of CH3OH/Cu(110), the preponderance of ‘CH3-up’ ordering of the CH3Cl lowers the barrier for photoelectron interactions to a larger extent than the mixed ordering of the CH3Br. In an analysis of CH3Br electron- and photodissociation on Ru surfaces it was found that the ‘Br-down’ configuration largely results in photodissociation while the ‘Br-up’ configuration leads to photodesorption.56 We also see that there are differences in the solvation of CH3Br and CH3Cl anion states on the two substrates considered, as both are found to have lower CH3 kinetic energies on CH3OH/Cu(110) but with a larger downward energy shift for CH3Br. Based on eqn (1) this indicates that the dissociating CH3Br anion is less well solvated than CH3Cl on the CH3OH, which could be due to differences in the orientational structure as discussed above, or could be due to steric hindrance for the larger neutral CH3Br precursor.
There has been less previous work for the CH3OH thin films, but FT-IR spectroscopy indicates that the equilibrium structure for CH3OH/Cu(110) at low temperatures has the O–H group close to parallel to the surface, with the CH3 group oriented toward the surface normal.40 This is responsible for the sign and magnitude of the surface dipole and the related change in work function that has been observed. This surface dipole structure would most likely cause the preferential orientation of the halomethanes in a similar manner as for the D2O/Cu(110) case.
4.2 Neutral photodissociation and quenching
The neutral photodissociation of CH3Br and CH3Cl in the gas-phase at 193 nm has been well-studied33–35 and occurs with rapid C–X bond scission via a set of excited states referred to as the ‘A-band’. In many respects this photodissociation is similar to that of CH3I at 248 nm, and we recently reported28 on the 248 nm photodissociation of CH3I/D2O/Cu(110) in which neutral photodissociation was prominent, and was identifiable for D2O films from monolayer to many multilayers in thickness. For both CH3Br and CH3Cl on D2O/Cu(110) we do identify the neutral photodissociation pathway using λ = 193 nm light from the characteristic CH3 kinetic energy distributions. When these molecules are adsorbed on CH3OH/Cu(110) however the situation is dramatically different. We do not observe neutral photodissociation of CH3Cl, and neutral photodissociation of CH3Br is not observed for submonolayer CH3Br coverages. We have also examined the 248 nm photodissociation of CH3I/CH3OH/Cu(110) over a range of conditions (e.g.Fig. 19) and do indeed observe characteristic TOF features that can only be a consequence of neutral photodissociation. While in many other respects the behaviour of CH3I on CH3OH/Cu(110) was found to be similar to that of CH3Br, the quenching of neutral photodissociation seen for CH3Br and CH3Cl at 193 nm is absent for the CH3I at 248 nm. In the absence of any detailed calculations for the ground and excited state energy levels of the CH3X/CH3OH/Cu(110) system, it is difficult to pin down the precise mechanism by which the excitations are quenched. We can exclude mechanisms such as orientational or steric blocking on the basis of the observed CT-DEA dissociation dynamics of the CH3X on the same substrate, which is not similarly hindered. It is apparent that an efficient quenching due to a Dexter energy transfer (DET) mechanism is present for CH3Br and CH3Cl using 193 nm photoexcitation. | ||
| Fig. 19 Time of flight spectrum for CH3 photofragments from 1 ML of CH3I adsorbed on a thick (≈10 ML) film of CH3OH on Cu(110) obtained using 248 nm light. The prominent peaks at 44 μs and 54 μs flight times are characteristic of the neutral photodissociation pathways for CH3I in the A-band. | ||
Solid methanol has a bandgap of 6.7 eV, and UPS study of CH3OH/Cu(110)22 places the HOMO 2a′′ level 5.5 eV below EF, while for H2O/Cu(110) the HOMO 1b1 level is located 6.8–7.2 eV below EF.20 As for the methyl halides, a study of CH3X/Ag(111) using UPS placed the halomethane HOMO at 6.0 eV below EF for CH3Cl/Ag(111), and at 5.4 eV and 4.4 eV respectively for CH3Br and CH3I monolayers on the same substrate.58 The other ingredient required for DET is wavefunction overlap, and based on isolated molecule wavefunctions the HOMO of CH3OH has more weight on the CH3 group than the 1b1 level of D2O does on its free D, and so is consistent with this requirement. It is of note that in CH3OH/TiO2 photochemistry, it has been remarked in several studies18,59 that the CH3OH is an effective ‘hole getter’ as compared to H2O/TiO2. This is attractive explanation since the neutral excited state and the DEA anion state both correspond to an excess electron in the CH3X LUMO, so the differences between these two dissociative processes are largely restricted to the differences in time that the excited state lies above the corresponding ground state prior to curve-crossing and the presence of the valence hole for the neutral excitation. However it is unclear if the quenching of the excited state hole for CH3X by CH3OH can be rapid enough to compete with the very rapid bond scission in CH3X A-band photodissociation. We are not aware of similar inter-molecular quenching of rapidly dissociative neutral photoexcitation having been previously identified in the surface photochemistry literature. A somewhat similar quenching mechanism has been identified in X-ray absorption studies of homomolecular clusters of CH3Br,60 although in this case the 3d → 4a1 core–valence excitation has dissociation competing with Auger decay, and the quenching observed in clusters is ascribed to delocalization of the CH3Br LUMO, rather than the HOMO implicated in the present work.
Acknowledgements
The author thanks G. D. Muirhead, C. Miller and E. Schibli, who provided assistance with the experiments as a part of their NSERC Undergraduate Student Research Award tenures in the laboratory. The Natural Sciences and Engineering Research Council (NSERC) of Canada is acknowledged for financial support of this work.References
- G. M. Muñoz Caro and W. A. Schutte, Astron. Astrophys., 2003, 412, 121–132 CrossRef.
- K. I. Öberg, R. T. Garrod, E. F. van Dishoeck and H. Linnartz, Astron. Astrophys., 2009, 504, 891–913 CrossRef PubMed.
- K. I. Öberg, E. F. van Dishoeck, H. Linnartz and S. Andersson, Astrophys. J., 2010, 718, 832–840 CrossRef.
- N. G. Petrik, R. J. Monckton, S. P. K. Koehler and G. A. Kimmel, J. Chem. Phys., 2014, 140, 204710 CrossRef PubMed.
- Q.-B. Lu, Phys. Rep., 2010, 487, 141–167 CrossRef CAS PubMed.
- A. Bumajdad and M. Madkour, Phys. Chem. Chem. Phys., 2014, 16, 7146 RSC.
- B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, H. Bowen, S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. LaVerne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L. S. Wang, C. Wittig and T. S. Zwier, Chem. Rev., 2005, 105, 355–389 CrossRef CAS PubMed.
- Y.-P. Kuo, H.-C. Lu, Y.-J. Wu, B.-M. Cheng and J. F. Ogilvie, Chem. Phys. Lett., 2007, 447, 168–174 CrossRef CAS PubMed.
- M. Denk, M. Hohage, L. D. Sun, P. Zeppenfeld, N. Esser and C. Cobet, Surf. Sci., 2014, 627, 16–22 CrossRef CAS PubMed.
- J. Stähler, U. Bovensiepen, M. Meyer and M. Wolf, Chem. Soc. Rev., 2008, 37, 2180–2190 RSC.
- Q.-B. Lu and L. Sanche, J. Chem. Phys., 2004, 120, 2434–2438 CrossRef CAS PubMed.
- U. Bovensiepen, C. Gahl, J. Stähler, M. Bockstedte, M. Meyer, F. Baletto, S. Scandolo, X.-Y. Zhu, A. Rubio and M. Wolf, J. Phys. Chem. C, 2009, 113, 979–988 CAS.
- S. Ryu, J. Chang, H. Kwon and S. K. Kim, J. Am. Chem. Soc., 2006, 128, 3500–3501 CrossRef CAS PubMed.
- M. Bertin, M. Meyer, J. Stähler, C. Gahl, M. Wolf and U. Bovensiepen, Faraday Discuss., 2009, 141, 293–307 RSC.
- S. H. Liu, A. D. Miller, K. J. Gaffney, S. Garrett-Roe, I. Bezel and C. Harris, J. Phys. Chem. B, 2002, 106, 12908–12915 CrossRef CAS.
- A. D. Miller, I. Bezel, K. J. Gaffney, S. Garrett-Roe, S. H. Liu, P. Szymanski and C. B. Harris, Science, 2002, 297, 1163–1166 CrossRef CAS PubMed.
- J. Zhao, B. Li, K. Onda, M. Feng and H. Petek, Chem. Rev., 2006, 106, 4402–4427 CrossRef CAS PubMed.
- Y. Tamaki, A. Furube, M. Murai, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2006, 128, 416–417 CrossRef CAS PubMed.
- X.-L. Zhou, X.-Y. Zhu and J. M. White, Surf. Sci. Rep., 1991, 13, 73–220 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- A. Hodgson and S. Haq, Surf. Sci. Rep., 2009, 64, 381–451 CrossRef CAS PubMed.
- M. Bowker and R. J. Madix, Surf. Sci., 1980, 95, 190–206 CrossRef CAS.
- C. Ammon, A. Bayer, G. Held, B. Richter, T. Schmidt and H.-P. Steinrück, Surf. Sci., 2002, 507, 845–850 CrossRef.
- B. L. Maschhoff, M. J. Ledema, M. Kwini and J. P. Cowin, Surf. Sci., 1996, 359, 253–268 CrossRef CAS.
- Y. Lilach and M. Asscher, J. Chem. Phys., 2002, 117, 6730–6737 CrossRef CAS PubMed.
- F. Fournier, H. Dubost, S. Carrez, W. Zheng and B. Bourguignon, J. Chem. Phys., 2005, 123, 184705 CrossRef PubMed.
- S. K. Jo and J. M. White, Surf. Sci., 1991, 255, 321–326 CrossRef CAS.
- E. R. Miller, G. D. Muirhead and E. T. Jensen, J. Chem. Phys., 2013, 138, 084702 CrossRef CAS PubMed.
- A. J. DeSimone, B. O. Olanrewaju, G. A. Grieves and T. M. Orlando, J. Chem. Phys., 2013, 138, 084703 CrossRef PubMed.
- Z. J. Sun, A. L. Schwaner and J. M. White, J. Chem. Phys., 1995, 103, 4279–4291 CrossRef CAS PubMed.
- D. Marinica, D. Teillet-Billy, J. Gauyacq, M. Michaud and L. Sanche, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 085408 CrossRef.
- F. Weik, A. de Meijere and E. Hasselbrink, J. Chem. Phys., 1993, 99, 682–694 CrossRef CAS PubMed.
- T. Gougousi, P. C. Samartzis and T. N. Kitsopoulos, J. Chem. Phys., 1998, 108, 5742–5746 CrossRef CAS PubMed.
- F. Wang, M. L. Lipciuc, A. Kartakoullis, P. Glodic, P. C. Samartzis, X. Yangand and T. N. Kitsopoulos, Phys. Chem. Chem. Phys., 2014, 16, 599–606 RSC.
- D. Townsend, S. K. Lee and A. G. Suits, J. Phys. Chem. A, 2004, 108, 8106–8114 CrossRef CAS.
- F. M. Zimmermann and W. Ho, Surf. Sci. Rep., 1995, 22, 127–247 CrossRef CAS.
- X.-L. Zhou and J. M. White, Photodissociation and Photoreaction of Molecules Attached to Metal Surfaces, in Laser Spectroscopy and Photochemistry on Metal Surfaces: Part II, ed. H.-L. Dai and W. Ho, World Scientific, 1995, p. 1173 Search PubMed.
- E. T. Jensen, J. Chem. Phys., 2005, 123, 204709 CrossRef CAS PubMed.
- G. L. Fisher and C. A. Meserole, J. Vac. Sci. Technol., A, 2005, 23, 722–724 CAS.
- A. Peremans, F. Maseri, J. Darville and J.-M. Gilles, J. Vac. Sci. Technol., A, 1990, 8, 3224–3228 CAS.
- P.-T. Howe and H.-L. Dai, J. Chem. Phys., 1998, 108, 7775–7782 CrossRef CAS PubMed.
- T. L. Gilton, C. P. Dehnbostel and J. P. Cowin, J. Chem. Phys., 1989, 91, 1937–1938 CrossRef CAS PubMed.
- S. K. Jo and J. M. White, J. Chem. Phys., 1991, 94, 5761–5764 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, Taylor and Francis, 94th edn, 2013–2014 Search PubMed.
- P. Ayotte, J. Gamache, A. D. Bass, I. Fabrikant and L. Sanche, J. Chem. Phys., 1997, 106, 749–760 CrossRef CAS PubMed.
- M. Michaud and L. Sanche, J. Electron Spectrosc. Relat. Phenom., 1990, 51, 237–248 CrossRef CAS.
- V. A. Ukraintsev, T. J. Long, T. Gowl and I. Harrison, J. Chem. Phys., 1992, 96, 9114–9123 CrossRef CAS PubMed.
- V. A. Ukraintsev, T. J. Long and I. Harrison, J. Chem. Phys., 1992, 96, 3957–3965 CrossRef CAS PubMed.
- H. Keller-Rudek, G. K. Moortgat, R. Sander and R. Sörensen, Earth Syst. Sci. Data, 2013, 5, 365–373 CrossRef.
- D. Lackey, J. Schott, B. Straehler and J. K. Sasss, J. Chem. Phys., 1989, 91, 1365–1376 CrossRef CAS PubMed.
- H. Petek, J. Chem. Phys., 2012, 137, 091704 CrossRef PubMed.
- E. T. Jensen, J. Chem. Phys., 2008, 128, 044301 CrossRef CAS PubMed.
- Y. Lilach and M. Asscher, J. Chem. Phys., 2003, 119, 407–412 CrossRef CAS PubMed.
- R. D. Burbank, J. Am. Chem. Soc., 1953, 75, 1211–1214 CrossRef CAS.
- T. Kawaguchi, M. Hijikigawa, Y. Hayafuji, M. Ikeda, R. Fukushima and Y. Tomiie, Bull. Chem. Soc. Jpn., 1973, 46, 53–56 CrossRef CAS.
- S. Jørgensen, F. Dubnikova, R. Kosloff, Y. Zeiri, Y. Lilach and M. Asscher, J. Phys. Chem. B, 2004, 108, 14056–14061 CrossRef.
- S. J. Dixon-Warren, D. V. Heyd, E. T. Jensen and J. C. Polanyi, J. Chem. Phys., 1993, 98, 5954–5960 CrossRef CAS PubMed.
- X.-L. Zhou, F. Solymosi, P. M. Blass, K. C. Cannon and J. M. White, Surf. Sci., 1989, 219, 294–316 CrossRef CAS.
- A. Yamakata, T. Ishibashi and H. Onishi, J. Phys. Chem. B, 2002, 106, 9122–9125 CrossRef CAS.
- T. Rander, A. Lindblad, I. Bradeanu, G. Öhrwall, S. Svensson and O. Björneholm, J. Chem. Phys., 2014, 141, 224305 CrossRef CAS PubMed.
Footnotes |
| † For work at 248 nm and 308 nm, s-polarized light was derived from the p-polarized beam by inserting an antireflection coated zero order half-waveplate into the beam. For 193 nm and 351 nm, s-polarized light was obtained by rotating the MgF2 crystal to direct the s-polarized beam onto the sample. |
| ‡ We have used D2980 values without correction for T = 90 K of our experiment. |
| § Various geometrical factors as well as the variable laser beam profile uniformity lead to this large absolute error. At a given wavelength, the relative errors of our cross section estimates are quite small – we typically measured cross sections for different molecular combinations on the same day without modifying the laser geometry and using the same laser gas fill, so that the sources of most relative errors are small (<10%) for these values. We do expect that our absolute cross section values and those for comparing values at two different wavelengths have larger errors. |
| This journal is © the Owner Societies 2015 |