
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of peroxyl radicals in polyester degradation – a mass spectrometric product and kinetic study using the distonic radical ion approach†
B. D. Gervasoni,
G. N. Khairallah,
R. A. J. O'Hair and
U. Wille*
School of Chemistry and Bio21 Institute, The University of Melbourne, Parkville, VIC 3010, Australia. E-mail: uwille@unimelb.edu.au
First published on 5th March 2015
Abstract
Mass spectrometric techniques were used to obtain detailed insight into the reactions of peroxyl radicals with model systems of (damaged) polyesters. Using a distonic radical ion approach, it was shown that N-methylpyridinium peroxyl radical cations, Pyr+OO˙, do not react with non-activated C–H bonds typically present in polyesters that resist degradation. Structural damage in the polymer, for example small amounts of alkene moieties formed during the manufacturing process, is required to enable reaction with Pyr+OO˙, which proceeds with high preference through addition to the π system rather than via allylic hydrogen atom abstraction (kadd/kHAT > 20 for internal alkenes). This is due to the very fast and strongly exothermic subsequent fragmentation of the peroxyl–alkene radical adduct to epoxides and highly reactive Pyr+O˙, which both could promote further degradation of the polymer through non-radical and radical pathways. This work provides essential experimental support that the basic autoxidation mechanism is a too simplistic model to rationalize radical mediated degradation of polymers under ambient conditions.
Introduction
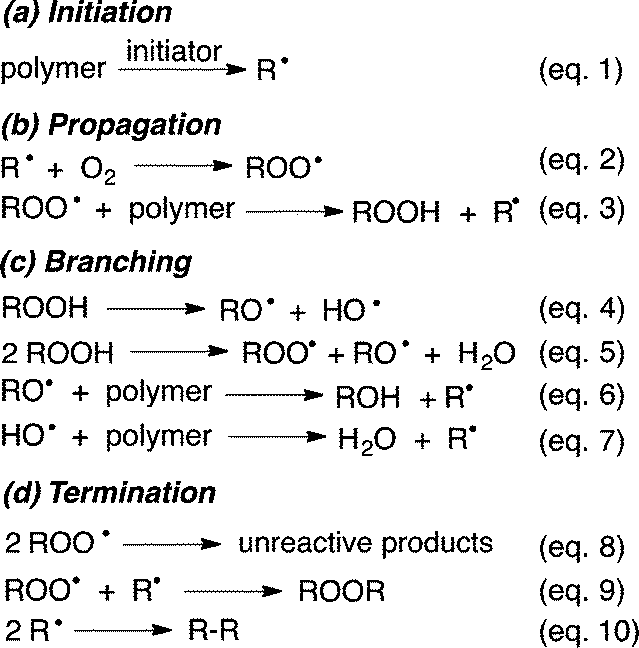
Our society relies heavily on manufactured polymers, which are used in increasingly demanding applications. The lifetime of polymers is strongly influenced by environmental conditions. In particular, weathering and ageing change the polymer's properties dramatically, resulting in decreased service life and limited usage.Upon exposure to light, heat, humidity and air many polymers are believed to undergo degradation through radical-mediated autoxidation (Scheme 1).1
 | ||
| Scheme 1 Polymer autoxidation mechanism proposed by Boland et al.1 | ||
This mechanism, which has not been fundamentally revised since its first proposition in the 1940's, consists of initial formation of C-centred polymer radicals R˙ (eqn (1)) through, for example, mechano-chemical scission of the polymer chain.2 Trapping with oxygen, O2, leads to polymer-derived peroxyl radicals, ROO˙ (eqn (2)), which are key intermediates and believed to propagate the radical chain through hydrogen atom transfer (HAT) from a neighbouring polymer strand (eqn (3)). Although this ‘autoxidation mechanism’ was originally only proposed for polymers that contain activated allylic C–H bonds, for example rubber materials, it has been universally adapted to rationalise radical degradation of any polymer at both ambient and elevated temperatures.
From the thermodynamic point of view this generalisation is problematic, since the bond dissociation energy (BDE) of the ROO–H bond in hydroperoxides is usually about 360–370 kJ mol−1 and therefore less than the BDE for non-activated C–H bonds in saturated alkyl chains (ca. 400–440 kJ mol−1).3 Rate constants of about 0.0015 M−1 s−1 obtained for the abstraction of secondary hydrogens by ROO˙ clearly demonstrate that such reactions are slow.4 Coote et al. recently used high-level kinetic modelling techniques to demonstrate that in polymers without activated C–H bonds the subsequent reactions of ROOH (eqn (4) and (5) in Scheme 1) do not provide a sufficiently strong driving force to compensate for the endothermicity of the radical propagation through eqn (3).5 It was concluded that only hydrogen abstractions by ROO˙, which lead to capto-datively or resonance stabilized radicals should be possible. Thus, at ambient temperature autoxidative degradation of polymers possessing only saturated alkyl chains would require some structural defects, such as terminal or internal double bonds.5
Polyesters used as protective surface coatings are commonly derived from benzene dicarboxylic acids, diols, triols and melamine derivatives for cross-linking. A certain degree of flexibility in most cross-linked and cured polyesters is achieved by incorporating fractions of aliphatic diacids. Although the latter are not durable, such polyesters do not possess sufficiently labile C–H bonds to promote autoxidation by reaction with ROO˙. However, polyesters with β-C–H bonds on the ester moiety can undergo thermal cis elimination (ester pyrolysis) during polymerisation and curing,6 which leads to carboxylic acids and ‘vulnerable’ alkene moieties. This undesired reaction can be reduced through incorporation of neopentyl groups, which lack β-C–H bonds. Even such structurally refined polyesters break down under environmental conditions, which indicates that the degradation mechanism in Scheme 1 is likely too simplistic.‡
Lindsay Smith et al. studied autoxidation of aliphatic polyesters using monomeric neopentyl esters as model system.7 The reaction was initiated by highly reactive O- and C-centred radicals RO˙ and R˙, respectively, and product analysis revealed complex temperature-dependent radical translocations following initial HAT. At elevated temperatures (438 K) and in the presence of O2, formation of ROOH was observed. Although the origin of the hydrogen atoms was not explored, it cannot be excluded that thermal ester degradation could have occurred to some extent, which may have provided a source for labile H.
Assuming that small amounts of ‘damage’, in particular alkene moieties, need to be present to promote radical-mediated degradation in a polyester, it is remarkable that the autoxidation mechanism generally considers hydrogen atom abstraction as the only reaction pathway for ROO˙, despite the fact that ROO˙ is known to react with alkenes also by addition.8 The resulting radical adduct decomposes to epoxides through homolytic O–O bond fragmentation, with concomitant release of highly reactive RO˙.9 A similar radical addition–fragmentation process was proposed to take place also in combustions.10 If such a reaction sequence could occur in polymers as well, the resulting epoxides and RO˙ would both be expected to provide ‘hot spots’ for subsequent degradation.
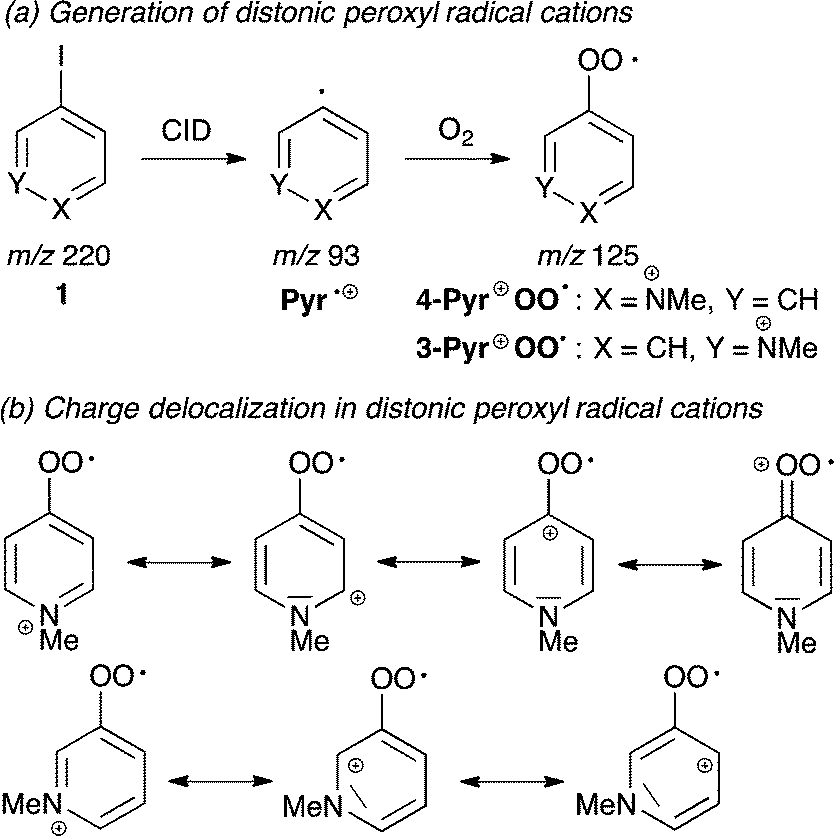
We have recently employed mass spectrometric techniques to study reactions of ROO˙ in the gas phase using the distonic radical ion approach.11 The permanent charge tag in N-methylpyridium peroxyl radical cation Pyr+OO˙ (Scheme 2a) provides the opportunity to monitor its transformations on the millisecond to second time scale and to determine absolute rate data for ROO˙ reactions.12
 | ||
| Scheme 2 | ||
In this work we employed this methodology to provide detailed insight into the role of ROO˙ in radical-mediated polyester degradation under ambient conditions by studying the products and kinetics of the bimolecular reaction of Pyr+OO˙ with structural motifs that could be typically found in (damaged) polyesters.§
Compounds 2–9 (Fig. 1) were selected as simplified models for alkenes, dialkenes, unsaturated esters, alkoxyesters and saturated alkyl systems, which are sufficiently volatile to be studied in the gas phase.¶ High-resolution mass spectrometry (HRMS) and collision-induced dissociation (CID) experiments in combination with density functional theory (DFT) calculations of the potential energy surface were used to identify the chemical nature of the major products and the pathways to their formation.
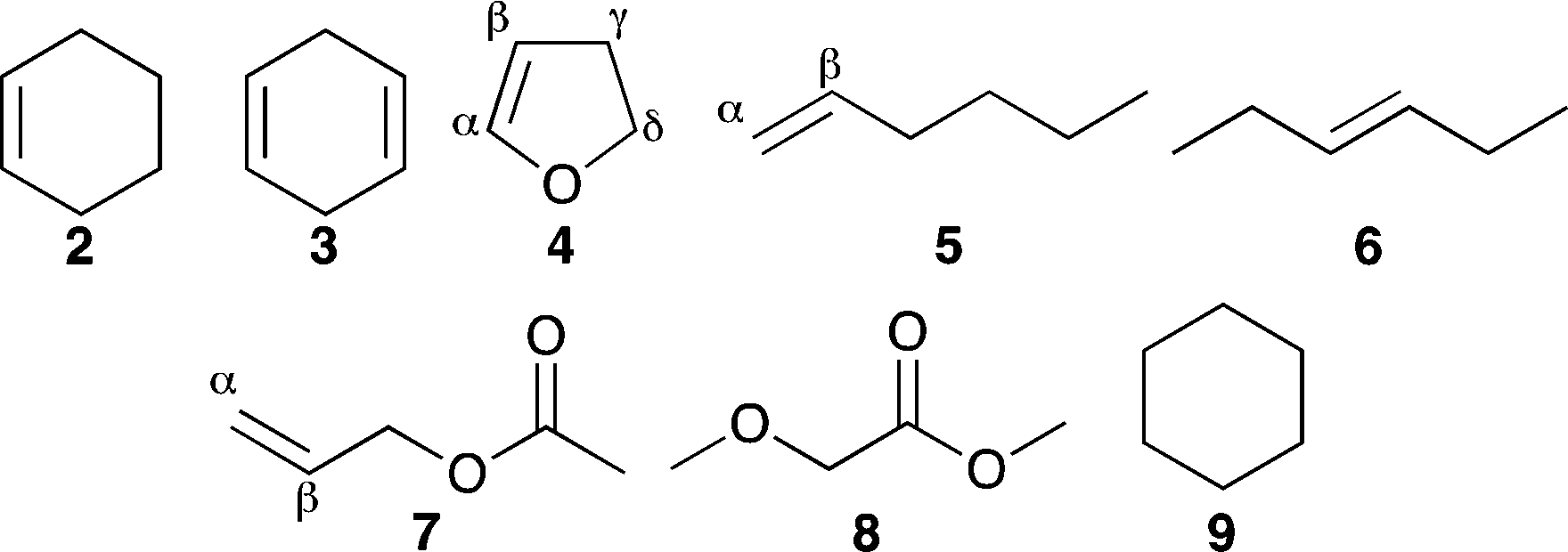 | ||
| Fig. 1 Model systems studied in this work. | ||
Two isomeric Pyr+OO˙ were explored in this study. 4-Pyr+OO˙ is highly electrophilic, because delocalization of the positive charge is assisted by the peroxyl moiety (Scheme 2b). In contrast to this, 3-Pyr+OO˙ is less electrophilic, since the charge is largely contained within the aromatic ring. This approach enables to assess the role of polar effects in these reactions.
Methods
1. Mass spectrometry experiments
The experiments were conducted in a Thermo Scientific (Bremen, Germany) LTQ-FT hybrid mass spectrometer. It consists of a linear ion trap (LTQ) coupled to a Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer, which has been modified to allow introduction of volatile neutral reagents into the ion trap for ion–molecule reaction studies. Details are given in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,13,14 Recent experiments have demonstrated that ions undergoing ion–molecule reactions in the ion trap of the mass spectrometer are quasi thermalized to the temperature of the helium bath gas (298 K).14
11,13,14 Recent experiments have demonstrated that ions undergoing ion–molecule reactions in the ion trap of the mass spectrometer are quasi thermalized to the temperature of the helium bath gas (298 K).14
The rate constants for product formation, e.g. addition of Pyr+OO˙ to π systems (in substrates 2–7) and HAT, were determined from the experimental concentration–time profiles of the various reaction products through kinetic fitting using the software package Dynafit 4, which performs a least-square analysis.15 Mass discrimination is unlikely to be important. Therefore, the peak area for the various ionic species is likely to directly reflect their concentration.
2. Computational studies
The calculations were carried out utilizing the Gaussian 09 program16 at the BHandHLYP/6-311++G** level of theory. Previous single-point benchmarking studies at G3(MP2)//BHandHLYP level confirmed the suitability of this DFT hybrid functional for exploring the potential energy surface of reactions involving Pyr+OO˙.11a All ground and transition states were verified by vibrational frequency analysis at the same level of theory, and all identified transition states showed only one imaginary frequency. The spin expectation value, 〈s2〉, was very close to 0.75 after spin annihilation. It should be noted that the transition states for addition of 4-Pyr+OO˙ to cyclohexene (2) and to dihydrofuran 4 could not be located at the BHandHLYP/6-311++G** level of theory (they were identified at UHF level). All BHandHLYP/6-311++G** electronic energies in this work include zero point vibrational energy correction, which was not scaled. The theoretical ion–molecule collision rate constant kADO was determined from the average dipole orientation (ADO) theory, which was calculated using the Colrate program17 with dipole moments and polarizabilities for compounds 2–8 obtained from the BHandHLYP/6-311++G** computations. The Gaussian archive entries for all optimized ground and transition states, including the imaginary frequencies of the transition states, are given in the ESI.†Results and discussion
1. Generation of Pyr+OO˙
The isomeric distonic peroxyl radical cations 4-Pyr+OO˙ and 3-Pyr+OO˙ were generated in the ion source of the mass spectrometer from the respective fixed-charge aryl iodide cation 1 (with iodide as counter ion),11 which was subjected to low-energy CID (Scheme 2a). The resulting isomeric aryl radical cations, Pyr˙+ (m/z 93) were trapped by ion–molecule reactions with residual O2 present in the ion source to give the corresponding distonic peroxyl radical cations Pyr+OO˙ at m/z 125, according to: Pyr˙+ + O2 → Pyr+OO˙. The identity of Pyr+OO˙ was confirmed by HRMS (calcd for C6H7NO2 125.04713, found 125.04723). Pyr+OO˙ was subsequently purified through mass selection in the ion trap, where the radical ion–molecule reaction with the neutral substrates 2–9 was performed. Both isomeric Pyr+OO˙ were stable in the gas phase, did not further react with O2 and could be stored in the ion trap in the absence of reactants for several seconds without decomposition.11 Most importantly, the electrostatic repulsion caused by the permanent charge tag prevents recombination of Pyr+OO˙, which is a common complication in reactions involving neutral ROO˙.182. Radical ion–molecule reactions
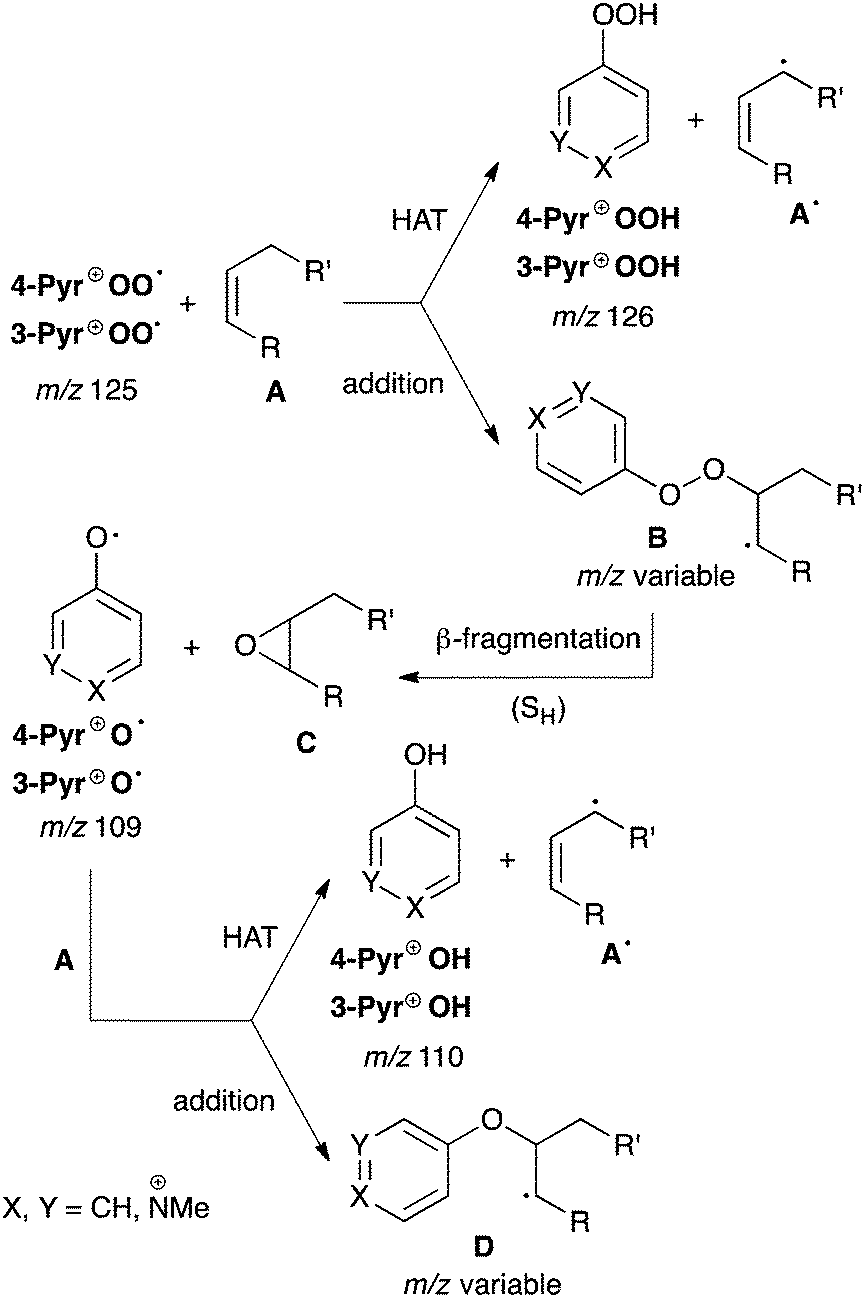 | ||
| Scheme 3 | ||
The mass spectra for the exemplary reactions of 3-Pyr+OO˙ with dihydrofuran 4 and methoxy ester 8 are shown in Fig. 3. The data were acquired with the neutral substrate being present in large excess. The reaction of both isomeric Pyr+OO˙ with cyclohexane (9) was too slow to observe with the experimental set-up. No measurable conversion was observed even after a reaction time of 10 seconds and, therefore, neither product nor kinetic studies could be performed for this substrate. This confirms that HAT from non-activated C–H bonds by Pyr+OO˙ is unfavourable, which is in agreement with the computational predictions by Coote et al.5 The mass spectra of all other reactions are given in Fig. S1–S10 in the ESI.†
In general, the reactions involving 4-Pyr+OO˙ appeared to be faster than those with the isomeric, less electrophilic 3-Pyr+OO˙ (see below). However, because the products of the individual reactions were found to be independent of the nature of the peroxyl radical cation, our discussion will mostly use the general terminology Pyr+OO˙ and Pyr+O˙, respectively, without specifically denoting the position of the charge tag in the pyridine ring.
The mass spectrum of the reaction of 4-Pyr+OO˙ with diene 3 (Fig. 2a) shows formation of Pyr+OOH (calcd for C6H8NO2 126.05495, found 126.05505) and Pyr+O˙ (calcd for C6H7NO 109.05222, found 109.05233). The radical adduct B at m/z 205 was not detected. This is in accordance with a reaction pathway involving both HAT and radical addition, which is rapidly followed by O–O bond fragmentation. When Pyr+O˙ was isolated and kept in the ion trap, a fast reaction with excess diene 3 occurred to give a product at m/z 110. The latter could be identified as Pyr+OH (calcd for C6H8NO 110.06004, found 110.06016), suggesting a reaction via HAT. Thus, in general, the radical addition–fragmentation pathway for Pyr+OO˙ is characterised by formation of the charged product Pyr+O˙ and rapid secondary reactions of the latter with excess neutral A through either HAT (leading to Pyr+OH) and/or addition (leading to the charged adduct D), as outlined in the lower section of Scheme 3. Comparison of the product signals at m/z 126 with the sum of the signals at m/z 109 and 110, which represent the addition–fragmentation reaction channel, highlights that HAT is only a minor pathway in the reaction of Pyr+OO˙ with 3 (see below).
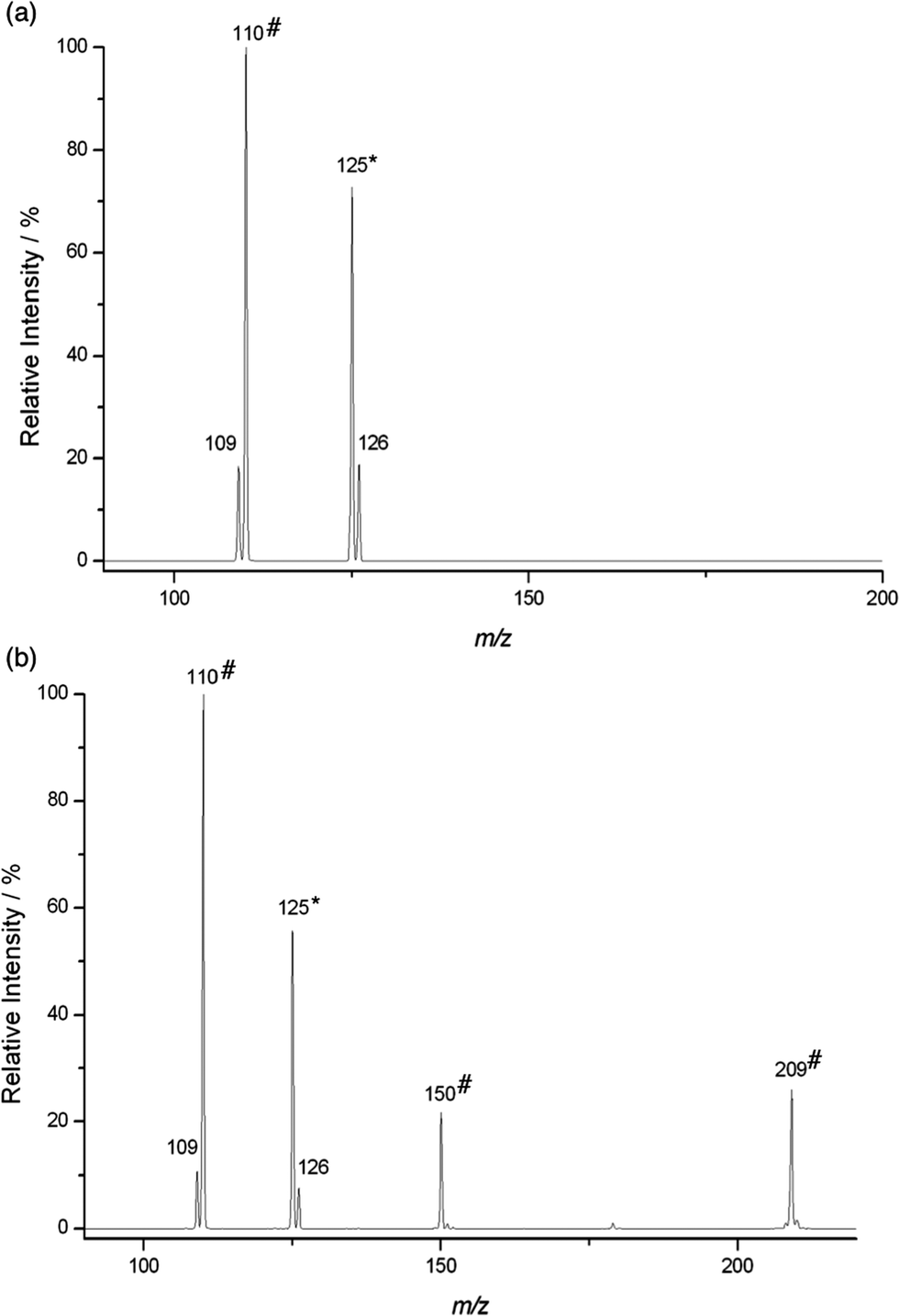 | ||
| Fig. 2 Mass spectrum of the reaction of 4-Pyr+OO˙ with (a) 1,4-cyclohexadiene after 225 ms ([3] ca. 3.4 × 109 molecules cm−3) and with (b) allyl acetate after 500 ms ([7] ca. 1.4 × 1010 molecules cm−3). The mass selected precursor ion is indicated by an asterisk (*). Unlabelled products result from reaction of 4-Pyr+OO˙ with the respective neutral. Products labelled with a hash (#) result from a secondary reaction involving 4-Pyr+O˙ (see text). | ||
In general, due to their lower electron density, terminal π systems are less activated for attack by electrophilic radicals. Despite this, a strong preference for radical addition over HAT was also found in the reaction of Pyr+OO˙ with allyl ester 7 (Fig. 2b). Similar to the reaction with diene 3, the initial radical adduct B at m/z 225 was not observed even at very short reaction times, suggesting a rapid fragmentation into Pyr+O˙ and, likely, the corresponding epoxide C. When Pyr+O˙ was mass selected in the ion trap, a reaction with 7 through both HAT (Pyr+OH at m/z 110) and addition to produce the radical adduct D at m/z 209 was found (calcd for C11H15NO3 209.10464, found 209.10466: CID gave signals at m/z 150, 110, 109). The product at m/z 150 in the mass spectrum likely results through subsequent β-fragmentation of D (presumably Pyr+OCH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, calcd for C9H12NO 150.09134, found 150.09137; CID gave signals at m/z 122, 110, 94, 93).
CH2, calcd for C9H12NO 150.09134, found 150.09137; CID gave signals at m/z 122, 110, 94, 93).
The reaction of Pyr+OO˙ with dihydrofuran 4 also proceeds predominantly via sequential addition/fragmentation, while formation of Pyr+OOH is only a very minor pathway (Fig. 3a). The product at m/z 138 results from a secondary reaction of Pyr+O˙ with 4. HRMS analysis is consistent with the structure Pyr+OCHO (calcd for C7H8NO2 138.05550, found 150.05502; CID gave a signal at m/z 110). It is worth noting that, because of resonance stabilisation of the unpaired electron, neutral aryloxyl radicals, ArO˙, are usually unreactive and considered as radical chain breakers. However, because of their high electrophilicity, Pyr+O˙ are very reactive radical species and, therefore, represent suitable model systems for aliphatic RO˙ formed during polymer degradation (see Scheme 1). We will report on the chemistry of Pyr+O˙ with different π systems in a separate publication.
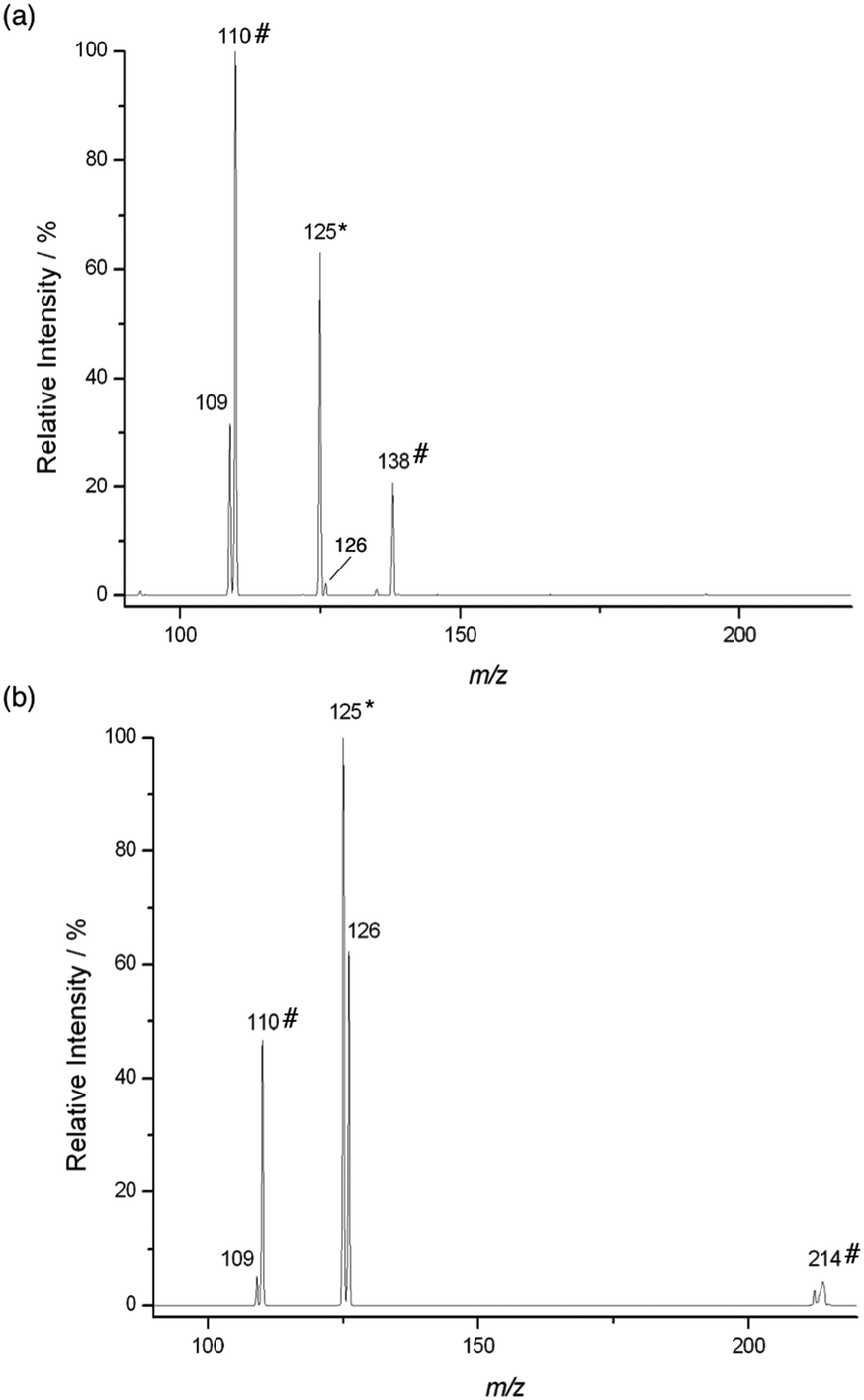 | ||
| Fig. 3 Mass spectrum of the reaction of 3-Pyr+OO˙ with (a) 2,3-dihydrofuran after 100 ms ([4] ca. 6.1 × 109 molecules cm−3) and with (b) methyl methoxyacetate after 1700 ms ([8] ca. 1.8 × 1010 molecules cm−3). The mass selected precursor ion is indicated by an asterisk (*). Unlabelled products result from reaction of 3-Pyr+OO˙ with the respective neutral. Products labelled with a hash (#) result from a secondary reaction involving 3-Pyr+O˙ (see text). | ||
As expected, HAT is an important pathway in the reaction of Pyr+OO˙ with methoxy ester 8, which is apparent from the large signal of Pyr+OOH at m/z 126 (Fig. 3b). However, quite surprisingly, both Pyr+O˙ and Pyr+OH were also formed in this reaction to quite a significant extent. We will discuss this further in Section 2.3. The signal at m/z 214 is not a reaction product, but is believed to be a metastable ion–molecule cluster formed in the ion trap, which requires only little energy to undergo CID to give a fragment at m/z 110.
To conclude, the mass spectrometric data clearly show that the reaction of Pyr+OO˙ with alkenes occurs, rather than by HAT, with high preference by radical addition, which is followed by fragmentation of the radical adduct, (see also Fig. S1–S10 in the ESI†). Even with highly activated bis-allylic C–H bonds, as present in cyclohexadiene 3, HAT is only the minor pathway.
a,b
| Entry | Substrate | [Pyr+OO˙ – A]c | TSHAT | [Pyr+OOH – A˙]c | Pyr+OOH + A˙d |
TSadd | B | TSfrag | [Pyr+O˙ – C]c | Pyr+O˙ + Cd |
|---|---|---|---|---|---|---|---|---|---|---|
| a Electronic energies relative to the free reactants, zero-point vibrational energy correction included. b See Scheme 3 for labelling. c Radical ion–molecule association complex. d Free products. e Transition state could not be located (see text). f See Fig. 1 for labelling. | ||||||||||
| (a) Reaction of 4-Pyr+OO˙ | ||||||||||
| 1 | 2 | −20 | 3 | −88 | −58 | n.d.e | −69 | −57 | −209 | −166 |
| 2 | 3 | −28 | −3 | −123 | −93 | −14 | −72 | −61 | −207 | −165 |
| 3 | 4 | −28 | 3 (γ)f | −45 (γ) | −10 (γ) | n.d. (α) | −98 (α) | −77 (α) | −228 | −177 |
| −3 (δ)f | −117 (δ) | −61 (δ) | n.d. (β) | −89 (β) | −92 (β) | |||||
| 4 | 5 | −20 | 11 | −76 | −56 | −6 | −76 | −58 | −220 | −158 |
| 5 | 6 | −15 | 6 | −78 | −57 | −12 | −79 | −62 | −227 | −173 |
| 6 | 7 | −41 | −3 | −106 | −58 | −2 (α) | −68 (α) | −47 | −234 | −161 |
| 9 (β) | −63 (β) | |||||||||
| 7 | 8 | −80 | 8 | −80 | −41 | — | — | — | — | — |
| (b) Reaction of 3-Pyr+OO˙ | ||||||||||
| 8 | 3 | −23 | 4 | −114 | −82 | 1 | −59 | −44 | −240 | −191 |
| 9 | 5 | −21 | 18 | −68 | −45 | 3 | −65 | −44 | −247 | −183 |
| 10 | 6 | −21 | 12 | −70 | −46 | −5 | −64 | −45 | −245 | −198 |
| (c) Reaction of 4-PyrOO˙ (neutral) | ||||||||||
| 11 | 3 | −5 | 45 | −65 | −51 | 46 | −13 | 25 | −206 | −190 |
According to the computations, all reactions proceed via initial formation of an ion–molecule association complex between the reactants, [Pyr+OO˙ – A], which is 15–80 kJ mol−1 lower in energy, depending on the polarity of the neutral A, than the free reactants. The energy of the free reactants (entrance channel) will be defined as reference point, which is set to 0 kJ mol−1. Because of the low pressure in the ion trap of the mass spectrometer, energy exchange with the surroundings through collision does not occur to a significant extent. [Pyr+OO˙ – A] therefore has excess energy that results from the internal and kinetic energy of the free reactants and the electrostatic energy that is released upon complex formation. Because of this, any subsequent reaction of [PyrOO˙+ – A] is only possible, if the activation barrier for this process is smaller or equal to the energy difference between the entrance channel and the association complex.19 Pathways with energies significantly above the entrance channel, on the other hand, cannot be accessed.
According to the computations, the energy of the transition state for radical addition to the π system in 2–7, TSadd, is generally lower than for allylic hydrogen abstraction, TSHAT. Despite resonance stabilisation in the resulting (bis)allyl radicals, TSHAT for all reactions is located only slightly below or even above the entrance channel, and, therefore, this process should be comparatively slow. This is in excellent agreement with the experimental findings. The predicted more positive energies of both TSHAT and TSadd in the reactions involving the less electrophilic 3-Pyr+OO˙, compared to the reactions of 4-Pyr+OO˙ (entries 8–10 vs. 2, 4 and 5) also qualitatively confirm the experimentally observed lower reactivity of 3-Pyr+OO˙. In terminal alkenes, such as allylester 7, radical addition to the less hindered site (α attack) is associated with a lower barrier than addition to the higher substituted site (β attack), although the energies of the resulting isomeric radical adducts B are similar (entry 6). The impossibility to locate TSadd for both α and β attack in the reaction involving dihydrofuran 4 by DFT methods could be taken as indication for a very fast, practically barrierless process (entry 3). Of all compounds studied in this work (except cycloalkane 9), the lowest reactivity would be expected for the methoxy ester 8. The calculated barrier for HAT from the central secondary methylene group is about 8 kJ mol−1 above the entrance channel, which is therefore a slow, however overall exothermic process, since the most likely product radical MeOC·HC(O)OMe is stabilized by capto-dative effects (entry 7).
The computations predict very rapid and exothermic fragmentation of the O–O bond in radical adduct B for both isomeric Pyr+OO˙. The transition state associated with this process, TSfrag, ranges from virtually non-existent (in the reaction involving β-attack at dihydrofuran 4) to about 20 kJ mol−1. This supports the experimental finding that the radical adduct B was never observed in the reactions involving alkenes 2–7 on the time scale of the mass spectrometric experiments. According to intrinsic reaction coordinate (IRC) calculations, TSfrag is the barrier of a concerted O–O bond scission and epoxide formation process, which should therefore be regarded as an intramolecular homolytic substitution, SH. For simplicity, however, we will use the term ‘fragmentation’ in this work. Under our experimental conditions, the radical leaving group, Pyr+O˙, is rapidly trapped by reaction with excess neutral substrate.
Compared to the HAT pathway, the sequential radical addition–fragmentation process is, overall, not only kinetically, but also thermodynamically considerably more favourable. However, if only the first step is considered, the C-centred radicals A˙ formed by HAT are either stabilized by resonance (substrates 2–7) or by capto-dative effects (substrate 8), and the initially formed product association complex [Pyr+OOH – A˙] is, in fact, more stable than the respective radical adduct B. It is the low barrier for the O–O fragmentation in the latter and the thermodynamic stability of the product association complex [Pyr+O˙ – C], which is located more than 200 kJ mol−1 below the entrance channel, that ultimately provides the driving force for the radical addition pathway. The irreversibility of the sequential radical addition/fragmentation ultimately determines the overall reaction outcome and is particularly important in reactions with similar barriers for both initial HAT and radical addition, for example in the case of allylester 7 (see also Fig. 2b). It should be noted that the data in Table 1 include also the energies of the free products, which are 30–50 kJ mol−1 above the respective product association complexes.
While the charge tag on the peroxyl radicals is a useful tool to explore reaction pathways with polymer model systems in detail on a very short time scale, peroxyl radicals formed during ‘natural’ polymer degradation are most likely neutral species. For comparison, we have therefore also calculated the potential energy surface for the exemplary reaction of cyclohexadiene 3 with the neutral pyridine peroxyl radical 4-PyrOO˙ (entry 11). The most important consequence of the reduced electrophilicity of this peroxyl radical is an increase and equalisation of the activation barriers for HAT and radical addition, thus slowing down both pathways. The barrier for O–O bond fragmentation rises from 11 to 38 kJ mol−1 (entry 2 vs. 11), but the large exothermicity associated with formation of the epoxide and 4-PyrO˙ provides the driving force that renders this pathway irreversible. A similar rate-enhancing effect caused by electron-withdrawing substituents has been observed for HAT and addition reactions of phenyl radicals, which has been rationalized by polarization of the respective transition states.20 Thus, despite the lower reactivity of the less electrophilic, neutral peroxyl radicals, these data clearly show that, even in substrates with highly activated C–H bonds, radical addition to π systems, followed by fragmentation, is a very important pathway that needs to be considered in radical-mediated polymer degradation, in particular since very reactive products are formed. Epoxides are readily attacked by nucleophiles, including oxygen or nitrogen centres that are present in adjacent polymer chains. The highly reactive RO˙ could propagate further radical chain reactions not only through HAT, in accordance with eqn (6) in Scheme 1, but also through addition to π systems, as has been observed in this work. These different processes are expected to lead to significant changes of the polymer structure and morphology.
(a) The rate constant for consumption of Pyr+OO˙ according to eqn (11):
| Pyr+OO˙ + A → products | (11) |
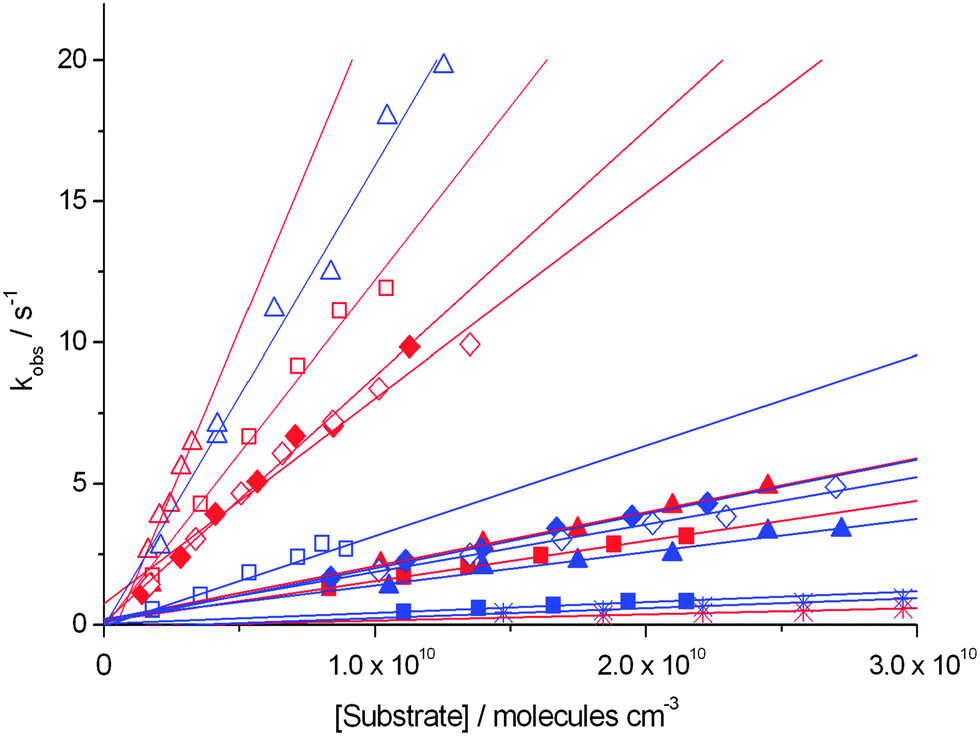 | ||
Fig. 4 Determination of the second order rate constants k for the reaction of Pyr+OO˙ with substrates 2–8, from the time-dependent decay of the Pyr+OO˙ signal at m/z 125. Data in red are reactions involving 4-Pyr+OO˙, data in blue are reactions involving 3-Pyr+OO˙. ◇: cyclohexene (2); □: 1,4-cyclohexadiene (3); △: 2,3-dihydrofuran (4); ■: 1-hexene (5); ◆: E-3-hexene (6); ▲: allyl acetate (7);  : methyl-2-methoxy acetate (8). : methyl-2-methoxy acetate (8). | ||
(b) The rate constants for the two competing pathways of Pyr+OO˙ through HAT and addition (in the case of alkenes 2–7), were obtained from kinetic modelling of the experimental concentration time profiles using the program Dynafit 4 (see Methods). The mass spectrometric data were fitted using eqn (12) for the HAT pathway (formation of Pyr+OOH at m/z 126) and eqn (13) for the radical addition–fragmentation pathway (this was modelled by analysing formation of Pyr+O˙ at m/z 109, since radical adduct B underwent fragmentation on a shorter timescale than our experiments, which is supported by the computational predictions).
| Pyr+OO˙ + A → Pyr+OOH + A˙ | (12) |
| Pyr+OO˙ + A → Pyr+O˙ + C | (13) |
| Pyr+O˙ + A → Pyr+OH + A˙ | (14) |
| Pyr+O˙ + A → D (+fragments) | (15) |
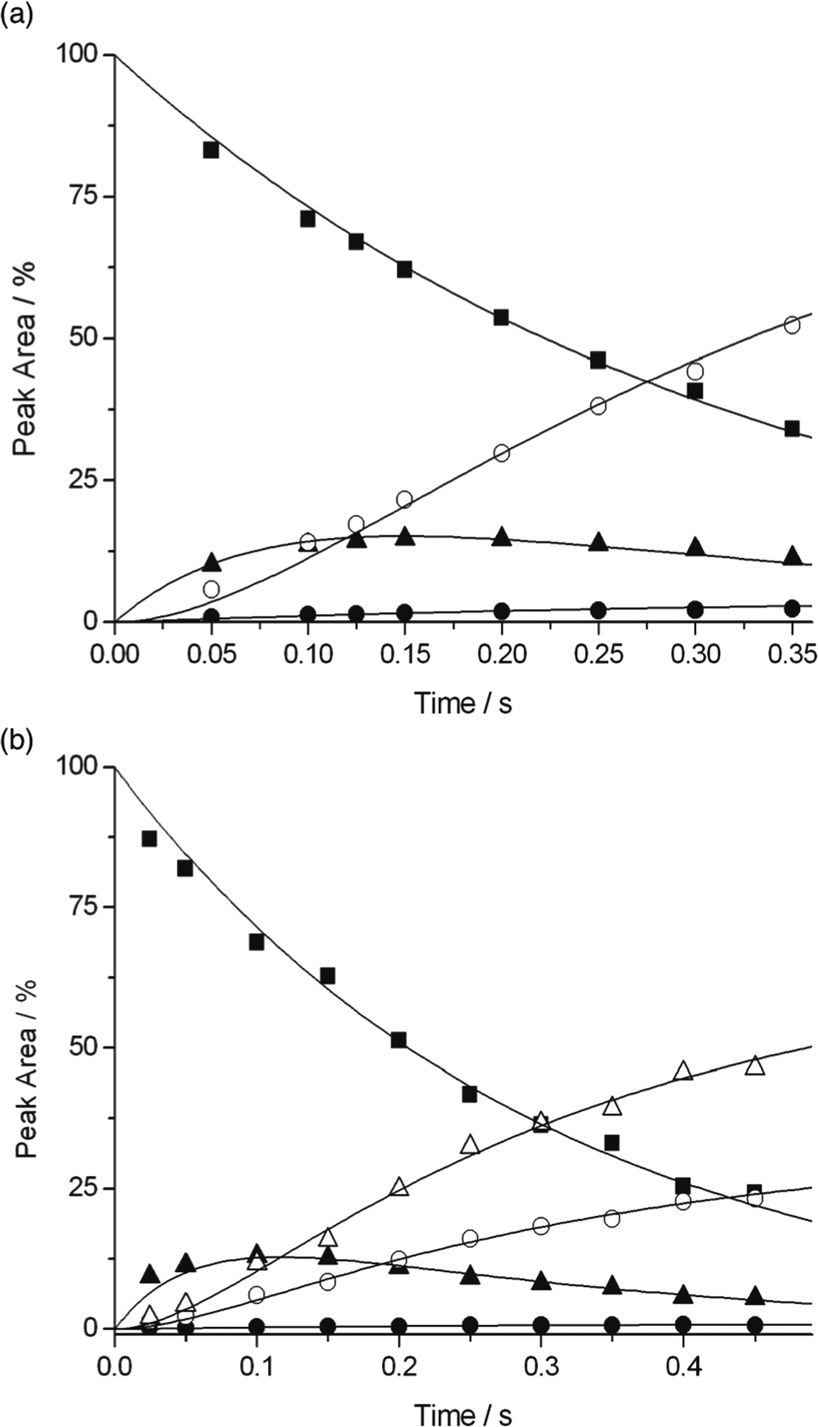 | ||
| Fig. 5 Fitting of the second order rate constants k for the reaction of (a) 4-Pyr+OO˙ with cyclohexene ([2] ca. 3.4 × 109 molecules cm−3) and (b) 3-Pyr+OO˙ with E-3-hexene ([6] ca. 1.7 × 1010 molecules cm−3). ■: Pyr+OO˙ (m/z 125); ▲: Pyr+O˙ (m/z 109); ●: Pyr+OOH (m/z 126); ○: Pyr+OH (m/z 110); △: addition products of Pyr+O˙ (variable m/z). | ||
A compilation of the rate data is given in Table 2.
| Substrate | k (cm3 molecule−1 s−1) | |||||
|---|---|---|---|---|---|---|
| 4-Pyr+OO˙ | 3-Pyr+OO˙ | |||||
| ROO˙ consumptionb | ROO˙ additionc | HATc | ROO˙ consumptionb | ROO˙ additionc | HATc | |
| a Rate data for ROO˙ consumption from pseudo-first order measurements; rate data for ROO˙ addition and HAT were fitted using the Dynafit 4 program (see text). b Experimental error (25%) included. c Standard error included. d Reaction efficiency (in square brackets) = (kexp/kADO) × 100% (see text). e Fitting required inclusion of eqn (16) (see text). | ||||||
| 2 | (7.3 ± 1.8) × 10−10 [70]d | (7.6 ± 0.1) × 10−10 | (3.5 ± 0.6) × 10−11 | (1.7 ± 0.4) × 10−10 [16] | (1.9 ± 0.1) × 10−10 | (3.5 ± 1.4) × 10−12 |
| 3 | (1.2 ± 0.3) × 10−9 [100] | (1.2 ± 0.1) × 10−9 | (2.1 ± 0.1) × 10−10 | (3.4 ± 0.9) × 10−10 [33] | (2.5 ± 0.1) × 10−10 | (8.2 ± 0.2) × 10−11 |
| 4 | (2.3 ± 0.6) × 10−9 [100] | (2.0 ± 0.1) × 10−9 | (1.0 ± 0.2) × 10−10 | (1.7 ± 0.4) × 10−9 [100] | (2.0 ± 0.1) × 10−9 | (4.2 ± 3.3) × 10−11 |
| 5 | (1.4 ± 0.4) × 10−10 [13] | (1.3 ± 0.1) × 10−10 | (1.7 ± 0.2) × 10−11 | (3.6 ± 0.9) × 10−11 [4] | (3.5 ± 0.1) × 10−11 | (3.9 ± 0.1) × 10−12 |
| 6 | (8.8 ± 2.2) × 10−10 [80] | (9.6 ± 0.1) × 10−10 | (5.2 ± 0.6) × 10−11 | (1.9 ± 0.5) × 10−10 [18] | (2.0 ± 0.1) × 10−10 | (2.1 ± 1.4) × 10−12 |
| 7 | (1.9 ± 0.5) × 10−10 [13] | (1.6 ± 0.1) × 10−10 | (7.2 ± 1.0) × 10−12 | (1.2 ± 0.3) × 10−10 [8] | (1.3 ± 0.1) × 10−10 | (4.0 ± 0.8) × 10−12 |
| 8 | (2.1 ± 0.6) × 10−11 [1] | — | (8.7 ± 0.2) × 10−12e |
(3.3 ± 0.9) × 10−11 [2] | — | (1.3 ± 0.1) × 10−11e |
The data clearly show that the more electrophilic 4-Pyr+OO˙ reacts with alkenes 2–7 about 2–4 times faster than the isomeric, less electrophilic 3-Pyr+OO˙. This confirms the role of polar effects in these reactions and provides strong support for the computational predictions in Table 1. In general, the reaction rate for Pyr+OO˙ consumption increases with increasing electron density of the alkene π system, e.g. terminal alkene (5, 7) < internal alkene (2, 3, 6) < enol ether (4). This is in accordance with the computed activation barrier for radical addition, TSadd, shown in Table 1. The efficiency of the reaction of both isomeric Pyr+OO˙ with dihydrofuran 4 is 100%, indicating that every encounter leads to a successful reaction. The efficiency is also quantitative for the reaction of 4-Pyr+OO˙ with cyclohexadiene 3, but drops to around 10% for the reactions of both isomeric Pyr+OO˙ with terminal alkenes. The similarity of the rate data obtained for the reactions with the internal cyclic and linear alkenes 2 and 6 show that conformational restriction in the alkene does not considerably affect the reaction rate (and efficiency). According to Fig. 3b, HAT is a major pathway in the reaction of Pyr+OO˙ with methoxyacetate 8 (see below), and the kinetic data from Pyr+OO˙ consumption clearly confirm that this process is considerably slower than radical addition, which also supports the computational predictions. The rate constants for the reactions of both isomeric Pyr+OO˙ with 8 are similar within error (with reaction efficiencies dropping to 1–2%), suggesting that polar effects are possibly not as important for Pyr+OO˙ induced HAT, compared to Pyr+OO˙ addition to π systems.11,21
The kinetic data for product formation show that radical addition occurs at a significantly faster rate than HAT, with kaddition/kHAT > 20. Only in the reactions involving cyclohexadiene 3, which possesses highly activated, bis-allylic C–H groups, and terminal alkene 5, in which radical addition is comparatively less favourable due to the less electron-rich π system, the relative significance of the HAT pathway increases. However, the ratio kaddition/kHAT of 3–9 shows that radical addition remains the major pathway even in these reactions. Despite the lower rate constant for the reaction of the terminal allylester 7 with Pyr+OO˙, the strong preference for the addition pathway, as indicated by kaddition/kHAT = 22–30, is likely due to the predicted strong exothermicity for formation of the respective product complex [Pyr+O˙ – C] (see Table 1). Overall, the kinetic data are in excellent agreement with the computational studies, which not only predict a generally higher barrier for the HAT process, but also show the strong thermodynamic driving force of the addition–fragmentation pathway.
The unexpected formation of Pyr+O˙ (and its hydrogen abstraction product Pyr+OH) in the reaction involving methoxy ester 8 (see Fig. 3b) clearly indicates that reactions of peroxyl radicals with hydrocarbons are likely more complex. A satisfactory kinetic fit for the decay of Pyr+OO˙, as well as for the rate of formation of Pyr+OOH and Pyr+O˙ could only be derived by including reaction 16 in the kinetic model (see Fig. S16 and S22 in the ESI†).
| Pyr+OO˙ + A → Pyr+O˙ + [A–O˙] | (16) |
O bond in 8, which could be followed by O–O bond fragmentation to yield a dioxirane and Pyr+O˙, similar to the addition to alkenes. However, the O-centred radical adduct resulting from addition to the carbon site of the π system (not shown) is some 46 kJ mol−1 above the entrance channel and cannot be accessed under our experimental conditions.|| Likewise, recombination of Pyr+OO˙ with the capto-datively stabilised radical intermediate MeOC˙HC(O)OMe, followed by fragmentation to produce Pyr+O˙ can also be excluded. Although the computations predict that such a pathway is, in principle, both kinetically and thermodynamically favourable (data not shown), kinetic fitting of the experimental data clearly revealed that formation of MeOC˙HC(O)OMe is too slow to account for the experimentally observed rate of Pyr+O˙ formation. There is no doubt that more work is required in future to fully understand the reaction of aliphatic esters with peroxyl radicals.
Although the chemistry of Pyr+O˙ was not the focus of this work, it is worth noting that its reactions via HAT and addition are very fast, with k14 and k15 being in the range of 10−9–10−10 cm3 molecule−1 s−1.
Conclusions
This work employed a combination of product, computational and kinetic studies to provide detailed insight into bimolecular reactions of peroxyl radicals with model systems of (damaged) polyesters. By using mass spectrometric techniques and the distonic radical ion approach not only could the chemical fate of the peroxyl radicals be directly observed, but also that of any charged reaction product. Because of their intrinsically higher reactivity but similar reaction pattern, the distonic peroxyl radical cations used as model for neutral peroxyl radicals that would be formed in polymers, enabled the study of their reactions at room temperature on the millisecond to second time scale.We have provided clear experimental evidence that non-activated C–H bonds, such as those present in saturated alkyl chains, are practically inert to reaction with peroxyl radicals. This supports the theoretical predictions by Coote et al.5 that the basic autoxidation mechanism (see Scheme 1) is unable to rationalize radical mediated degradation of polymers possessing only saturated alkyl chains under ambient conditions. Such a process requires the presence of small amounts of structural defects as starting point, in particular alkenes, which could result, for example, from the manufacturing process. In the reactions of peroxyl radicals with alkenes, allylic hydrogen abstraction (‘autoxidation’) does indeed occur, but it appears that this is not the exclusive reaction. In fact, the major reaction pathway proceeds through radical addition to the π system. This is immediately followed by fragmentation of the O–O bond through a homolytic substitution mechanism, which leads to formation of an epoxide and highly reactive RO˙. According to our product and kinetic studies, which are supported by the computational data, this sequential addition–fragmentation is both the kinetically and thermodynamically most preferred pathway for the reaction of peroxyl radicals with alkenes. The ease by which O–O bond fragmentation in the peroxyl radical adduct B takes place, suggests that a similar process should readily occur also within the polymer matrix, since bimolecular trapping of B (for example by oxygen) is not likely a competitive pathway. This is supported by the fact that the B–OO˙ adduct resulting from reaction of B with residual oxygen in the ion trap was never observed in this study. The significance of our finding, which has to our knowledge not been considered in the mechanism of radical mediated polymer degradation so far, lays in the fact that epoxides could subsequently be attacked through non-radical pathways by any nucleophile present in the system, including oxygen or nitrogen centres in adjacent polymer chains, which could lead to dramatic alteration of the polymer structure. RO˙, on the other hand, are extremely reactive species, which have been seen in this work to act as radical chain carrier through both HAT and radical addition. In addition to this, depending on their structure, RO˙ could also undergo β-fragmentation, which leads to generation of C-centred radicals that could subsequently be trapped by oxygen.22 In the context of polymer degradation, it can therefore be concluded that, through reaction with alkenes, generally not overly reactive neutral peroxyl radicals are transformed into two highly reactive products, which can promote further degradation through secondary radical and non-radical pathways.
In future work, we will apply this methodology to explore the reaction of different distonic peroxyl and alkoxyl radicals with structural motifs found in other step-growth and chain-growth polymers.
Acknowledgements
This work was supported by the Australian Research Council under the Centre of Excellence Scheme (CE0561607) and the University of Melbourne. Computational resources were provided through the National Computational Merit Allocation Scheme. The authors thank G. Gryn'ova and M. L. Coote for helpful discussions.Notes and references
- Examples: J. L. Bolland, Proc. R. Soc. London, Ser. A, 1946, 186, 218 CrossRef CAS; J. L. Bolland and G. Gee, Trans. Faraday Soc., 1946, 42, 244 RSC; J. L. Bolland and P. ten Have, Discuss. Faraday Soc., 1947, 2, 252 RSC; L. Bateman, Q. Rev., Chem. Soc., 1954, 8, 147 RSC.
- G. Scott, Polym. Degrad. Stab., 1995, 48, 315 CrossRef CAS.
- Y. R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, Florida, 2007 Search PubMed.
- J. A. Howard and J. H. B. Chenier, Can. J. Chem., 1980, 58, 2808 CrossRef CAS.
- G. Gryn'ova, J. L. Hodgson and M. L. Coote, Org. Biomol. Chem., 2011, 9, 480 Search PubMed.
- Examples: H. Zimmermann, Degradation and Stabilisation of Polyesters, in Developments in Polymer Degradation, ed. N. Grassie, 1984, vol. 5, p. 79 CrossRef CAS; F. Samperi, C. Puglisi, R. Alicata and G. Montaudo, Polym. Degrad. Stab., 2004, 83, 3 CrossRef CAS; D. R. Kelsey, K. S. Kiibler and P. N. Tutunjian, Polymer, 2005, 46, 8937 CrossRef PubMed; P. Sivasamy, M. Palaniandavar, C. T. Vijayakumar and K. Lederer, Polym. Degrad. Stab., 1992, 38, 15 CrossRef.
- J. R. Lindsay Smith, E. Nagatomi, A. Stead, D. J. Waddington and S. D. Beviere, J. Chem. Soc., Perkin Trans. 2, 2000, 1193 RSC; J. R. Lindsay Smith, E. Nagatomi and D. J. Waddington, J. Chem. Soc., Perkin Trans. 2, 2000, 2248 RSC; J. R. Lindsay Smith, E. Nagatomi, A. Stead and D. J. Waddington, J. Chem. Soc., Perkin Trans. 2, 2001, 1527 RSC.
- F. R. Mayo, Acc. Chem. Res., 1968, 1, 193 CrossRef CAS.
- Examples: M. S. Stark, J. Am. Chem. Soc., 2000, 122, 4162 CrossRef CAS; U. Neuenschwander and I. Hermans, Phys. Chem. Chem. Phys., 2010, 12, 10542 RSC.
- M. J. Pilling, S. H. Robertson and P. W. Seakins, J. Chem. Soc., Faraday Trans., 1995, 91, 4179 RSC.
- (a) C. H. Li, G. N. Khairallah, A. K. Y. Lam, R. A. J. O'Hair, B. B. Kirk, S. J. Blanksby, G. da Silva and U. Wille, Chem. – Asian J., 2013, 8, 450 CrossRef CAS PubMed; (b) G. N. Khairallah, R. A. J. O'Hair and U. Wille, J. Phys. Chem. A, 2014, 118, 3295 CrossRef CAS PubMed.
- For the use of radical clocks to determine rate constants for ROO˙ reactions, see for example: D. A. Pratt, K. A. Tallman and N. A. Porter, Acc. Chem. Res., 2011, 44, 458 CrossRef CAS PubMed.
- W. A. Donald, C. J. McKenzie and R. A. J. O'Hair, Angew. Chem., Int. Ed., 2011, 50, 8379 CrossRef CAS PubMed.
- W. A. Donald, G. N. Khairallah and R. A. J. O'Hair, J. Am. Soc. Mass Spectrom., 2013, 24, 811 CrossRef CAS PubMed.
- P. Kuzmic, Anal. Biochem., 1996, 237, 260 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, F. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- K. F. Lim, Quantum Chem. Program Exch. Bull., 1994, 14, 3 Search PubMed; T. Su and M. T. Bowers, Int. J. Mass Spectrom. Ion Phys., 1973, 12, 347 CrossRef CAS.
- H. Herrmann, A. Reese, F. Wicktor and R. Zellner, J. Mol. Struct., 1997, 408/409, 539 CrossRef CAS; S. L. Khursan and V. S. Martemyanov, Russ. J. Phys. Chem., 1991, 65, 321 Search PubMed; A. F. Dmitruk, O. M. Zarechnaya and I. A. Opeida, Theor. Exp. Chem., 2003, 39, 283 CrossRef.
- C. H. DePuy, J. Org. Chem., 2002, 67, 2393 CrossRef CAS PubMed; M. Speranza, Mass Spectrom. Rev., 1992, 11, 73 CrossRef.
- P. E. Williams, B. J. Jankiewicz, L. Yang and H. I. Kenttämaa, Chem. Rev., 2013, 113, 6949 CrossRef CAS PubMed.
- W. A. Pryor, D. F. Church, F. Tang and R. Tang, in Frontiers of Free Radical Chemistry, ed. W. A. Pryor, Academic Press Inc. London Ltd, 1980, p. 355 Search PubMed.
- See for example: A. Boto, C. Betancor, T. Prangé and E. Suárez, J. Org. Chem., 1994, 59, 4393 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, mass spectra (Fig. S1–S10), kinetic plots (Fig. S11–S22) and Gaussian archive entries for all ground and transition states. See DOI: 10.1039/c4cp06056c |
| ‡ Photo-oxidation has been deemed as most detrimental for the stability of polyester coatings, but through addition of radical scavengers, such as Hindered Amine Light Stabilizers (HALS) and UV-absorbing agents, their photo-stability can be significantly improved. Another known pathway for polyester degradation involves hydrolysis. However, because this is a non-radical process, it is not a topic of this study. |
| § Studies in the gas phase provide a useful guideline to obtain insight into chemical processes in a polymer, since at the atomistic level bond-breaking and bond-forming processes are remarkably similar in the gas-phase and in solid-state materials, see: R. P. O'Hayre, Materials Kinetics Fundamentals: Principles, processes and applications, John Wiley & Sons, Inc., Hoboken, New Jersey, 2015. |
| ¶ Aromatic systems were not included since we found in a recent study that reaction of Pyr+OO˙ reacts with phenyl acetylene exclusively at the alkyne moiety (see ref. 11b). Therefore, a reaction of ROO˙ with the deactivated aromatic ring of benzene dicarboxylates seems to be not competitive. |
| || A stable ground state for the C-radical resulting from Pyr+OO˙ addition to the carbonyl oxygen could not be located computationally. |
| This journal is © the Owner Societies 2015 |