
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bracketing subtle conformational energy differences between self-solvated and stretched trifluoropropanol†
Matthias
Heger
 ,
Katharina E.
Otto
,
Ricardo A.
Mata
and
Martin A.
Suhm
*
,
Katharina E.
Otto
,
Ricardo A.
Mata
and
Martin A.
Suhm
*
Institut für Physikalische Chemie, Universität Göttingen, Tammannstr. 6, 37077 Göttingen, Germany. E-mail: msuhm@gwdg.de
First published on 17th March 2015
Abstract
The intramolecular OH⋯F hydrogen bond in 3,3,3-trifluoropropanol (TFP) exerts a subtle stabilizing effect that, when compared to the non-fluorinated analog, reorders the five distinguishable conformers and widens the gap between the two most stable structures. Here, we combine findings from Raman spectroscopy in supersonic expansions and high-level quantum-chemical calculations to bracket the energy difference between the two most stable TFP structures at 1.7(5) kJ mol−1. The torsional potential energy surface suggests consecutive backbone and OH torsional motions for the conformer interconversion, which are discussed in the framework of supersonic jet cooling as a function of nozzle temperature. The picture of a bistable cold molecule with trans or gauche backbone emerges, in which the OH group controls the energy difference and modulates the high barrier separating the heavy atom frames.
1 Introduction
Conformational preferences in flexible chain molecules are influenced by interactions between the chain segments. This is highly relevant for protein folding in aqueous solution,1 but a systematic understanding is more straightforward for elementary prototypes in the gas phase,2 such as the subtle balance between stretched and hairpin conformations in linear alkanes.3 There is a general lack of reliable experimental data on relative energies of chain conformations in the gas phase, because entropy effects come into play at higher temperatures and the vapor pressure becomes too low when the energetics starts to dominate. Adiabatically cooled supersonic jet expansions offer a partial solution, but their non-equilibrium character tends to freeze the conformational equilibrium at higher temperatures, requiring a critical analysis of the findings.3Two particularly elegant approaches exploit nuclear wavefunction mixing between conformations4 and stimulated emission pumping via suitable electronically excited states.5 However, none of them is easily applicable to backbone isomerizations of saturated alkane chains. If one is only interested in the enthalpy difference at non-cryogenic temperatures, He-droplet pickup and freezing is an attractive and fairly universal approach.6
For unsubstituted alkanes, chain segments have to reach a size of about 7 CH2 units before the dispersion forces between them can overcome the intrinsic preference for an all-trans conformation.3 By substituting one CH3 end group for CF3 and the other for CH2OH, one can move the turning point to much smaller chain segments, because a weak hydrogen bond-like CF⋯HO attraction between the OH and CF3 groups can be realized in suitably folded conformations. Indeed, the simplest foldable trifluoroalcohol, 3,3,3-trifluoropropanol (TFP, CF3CH2CH2OH), already prefers a folded conformation, as inferred from microwave data.7 However, the case becomes less clear when taking various related compounds into consideration: mono- and trifluoroethanol still favor a gauche conformation, but the driving force can be attributed more to the “gauche effect” than to traditional hydrogen bonding.8–10 Further, monofluoropropanol shows no distinct preference for a hydrogen-bonded structure,8,11 which can be plausibly explained in terms of dipole–dipole interactions between the C–O and the C–F bonds.11 Increasing the chain length by one CH2 group apparently provides enough conformational flexibility to avoid this unfavorable interaction, and the resulting monofluorobutanol shows a substantial amount of internally hydrogen-bonded conformers.11
Quantitatively, a rough experimental estimate places the folded TFP conformation 3.5(10) kJ mol−1 lower in energy than its stretched conformation.7 This is substantially more than for the non-fluorinated 1-propanol, where an even weaker hydrogen bond-like interaction between a terminal CH group and an electron lone pair of the OH group gives the resulting global minimum conformation an energy advantage of 0.5721 kJ mol−1 over the next OH torsional isomer.4 The energy penalty of the fully stretched conformation is likely even smaller, but also harder to evaluate, because it involves backbone isomerization.12
The goal of the present study is to bracket the energy difference between the two lowest energy conformations of TFP as closely as possible by a combined experimental and theoretical approach, using vibrational spectroscopy in supersonic jets and advanced ab initio methods. For this purpose, we detect and assign both conformers via their spectrally separated low-temperature OH stretching and low frequency modes by spontaneous Raman scattering. In contrast to stimulated Raman techniques,13 this also works for saturated alkyl chains. Assuming that the conformational equilibrium is completely frozen at the pre-expansion temperature, one can derive an upper bound for the energy difference between the two conformations. Comparison to the related case of ethanol, where conformational relaxation only involves a tunneling-assisted rearrangement of the OH group and is thus more feasible in supersonic jets, provides information that is closer to a lower bound. Discrimination between backbone and OH torsion yields a third set of estimates. High-level quantum-chemical predictions with zero-point energy corrections which include adjustments to experimental spectra provide a fourth, independent estimate of the energy difference between the conformations. Taken together, these four estimates indicate that the earlier experimental value of 3.5 ± 1.0 kJ mol−1 for the energy difference derived from microwave spectra7 is too large.
2 Methods
2.1 Raman jet experiment
For measuring Raman spectra in a free jet, we employ the “curry” jet setup3 (“classical unrestricted Raman spectroscopy”). Sample preparation is carried out using a glass saturator at θsat = −30 to +20 °C in which a stream of the carrier gas (typically He) is directed through the liquid alcohol. The prepared gas mixture is stored in a 4.7 L Teflon-coated stainless steel reservoir and conducted through a PTFE tube to the 4 × 0.15 mm2 jet nozzle into a 144 L jet chamber; the tube and the nozzle can be heated up to θn = 150 °C. The jet chamber is evacuated by two Roots blowers and a rotary vane backing pump. The supersonic expansion is crossed by a 532 nm, 18 W cw laser focused to an irradiance on the order of 10 GW m−2. Scattered photons are collimated at a 90° angle by a Nikkor 50 mm f/1.2 photographic lens, focused on the entrance slit of a grating monochromator and detected by a LN2-cooled CCD chip (Princeton Instruments Spec-10). Optionally, a λ/2 plate can be used to control the polarization of the probing laser beam. Typically, six scans of 200 seconds duration are recorded, distinct “spikes” from background events are removed by comparing independent scans, and the obtained data are co-added to yield the final spectrum. Wavenumber calibration of the spectra is carried out using the lines of a Ne lamp. Slight calibration drifts on the order of 1 cm−1 are corrected by linear shifts in the wavenumber domain to fit well-calibrated spectra.3,3,3-Trifluoropropanol (97%) was obtained from ABCR and Manchester Organics and used as received. Ethanol (Merck, 99.8%) was used in the reference measurements for the conformer interconversion.
2.2 Quantum-chemical calculations
The canonical energy and Raman activity calculations presented throughout this work were conducted using the Gaussian0914 software package at the MP2 and B3LYP (without dispersion correction) levels of approximation. All local15 (“L”) and explicitly correlated16,17 (“-F12”) results, including the torsional potential surface, were computed using the Molpro 2010.1 and 2012.1 packages,18 with non-iterated perturbative triples (“(T0)”) used in all local CCSD(T) calculations.19 Local correlation treatments reduce basis set superposition error (BSSE) and are beneficial for vibrational frequencies.20–22 They furthermore speed up the calculations and promise to be applicable to very large molecular systems.23,24 Pipek–Mezey localization25 was employed for all local correlation treatments in this work.All calculations were done using Dunning's correlation-consistent basis sets; for all explicitly correlated calculations, we used the adapted cc-pVTZ-F12 basis set.26 As is common practice, we shorten “aug-cc-pVnZ” to “aVnZ”. The LMP2 structure optimizations for the torsional potential energy surface (TPES) used a Dunning basis set in which diffuse functions were added only on non-hydrogen atoms, abbreviated “a′VnZ”.27
In local and F12 calculations, density fitting approximations were used throughout. The fitting basis sets were the Molpro default aVnZ/JKFIT28 and aVnZ/MP2FIT29 basis sets. For compactness, we omit the “DF-” prefix.
2.3 Nomenclature and conventions
TFP adopts five distinguishable conformations30 (Fig. 1) which may be characterized by two dihedral angles: the O–C1–C2–C3 “backbone torsion” angle, which we denote τ1, and the H–O–C1–C2 “OH torsion” angle, τ2. We use two-letter descriptors for the conformers that qualitatively encode the value of the dihedral angles τ1 with a capital and τ2 with a small letter, using “G/g” for gauche (τ ≈ ±60°) and “T/t” for trans (τ ≈ 180°).30 The most stable conformer shows opposite signs in τ1 and τ2, indicated by a prime. The unprimed counterpart “Gg” turns out to be the least stable conformer.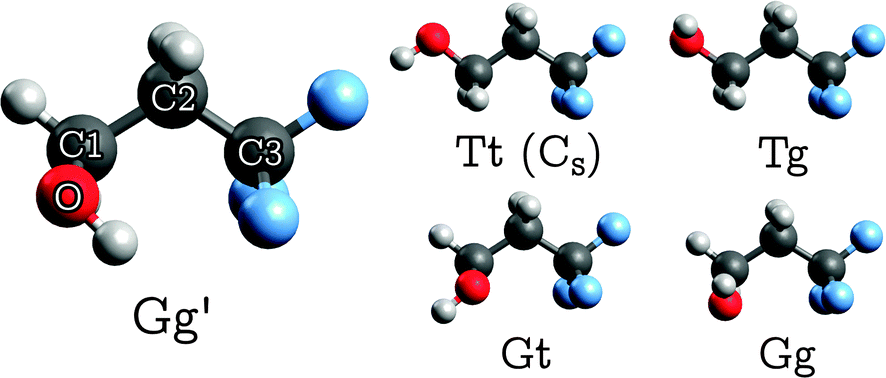 | ||
| Fig. 1 Structures of the five stable TFP conformers (only Tt has a mirror plane, whereas all other conformers are C1-symmetric). See Section 2.3 for details on the labeling scheme. | ||
3 Computational results
3.1 Conformer energies
Quantum-chemical calculations were carried out on the five TFP conformers in order to provide estimates of their relative energies (see Table 1, values in parentheses). Structure optimizations and harmonic frequency calculations were carried out at the B3LYP/aVTZ, MP2/aVTZ, LMP2/aVTZ and LMP2/a′VTZ levels, and the structures were re-used for a number of Coupled-Cluster calculations where optimizations and frequency analyses would be unfeasible. In light of the low temperatures in our supersonic jet experiment, the spectroscopically most interesting feature is the energy difference between the two most stable conformers, Gg′ and Tt. We will first detail on different approaches for benchmarking the electronic energy component, discussing corrections due to zero-point vibrational energy (ZPVE) separately.| B3LYP | MP2 | LMP2 | LCCSD(T0)a,b | CCSD(T)-F12aa,c | |
|---|---|---|---|---|---|
| a ZPVE corrections from B3LYP. b LMP2/a′VTZ-optimized structures. c VTZ-F12 basis set, MP2/aVTZ-optimized structures. d Using “harm + obs” ZPVEs for Gg′ and Tt. | |||||
| Gg′ | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) | 0.00 (0.00) |
| Ttd | 0.80 (1.46) | 1.44 (2.17) | 0.80 (1.49) | 0.59 (1.25) | 1.67 (2.33) |
| Tg | 2.80 (3.28) | 3.73 (4.43) | 3.15 (3.77) | 3.19 (3.67) | 3.88 (4.37) |
| Gt | 5.13 (5.92) | 5.40 (6.49) | 4.94 (5.98) | 4.83 (5.62) | 5.80 (6.59) |
| Gg | 7.01 (7.59) | 7.54 (8.49) | 7.18 (8.09) | 7.05 (7.62) | 7.72 (8.29) |
To estimate the electronic Tt–Gg′ energy difference at the complete basis set (CBS) limit, we employed explicitly correlated CCSD(T)-F12 calculation with local and canonical correlation schemes at the MP2/aVTZ-optimized structures. We find that the results are mostly insensitive to the choice of a specific F12 method31,32 and triples scaling,32 with variations well below 0.1 kJ mol−1; we rely on the F12a energies henceforth. The local and canonical methods yield Tt–Gg′ energy differences of 1.25 and 2.33 kJ mol−1, respectively, and CBS limit extrapolations of LCCSD(T0)/aVTZ and aVQZ calculations agree with the LCCSD(T0)-F12a results within 0.2 kJ mol−1. We tentatively explain the ∼1 kJ mol−1 discrepancy among the results by the intramolecular OH⋯F bond in Gg′ which is treated at the MP2 rather than CCSD(T) level in the local calculations due to default Molpro cut-off thresholds for long-range interactions. Including this interaction at a higher correlation level would thus likely move the locally correlated results closer to the canonical ones. The calculations further provide CABS-corrected SCF energies as CBS limit estimates32 with a Tt–Gg′ difference of −1.39 kJ mol−1, showing that electron correlation is absolutely essential.
We also conducted a comparable series of trial calculations on non-fluorinated propanol, using LMP2/aVTZ-optimized structures and including harmonic ZPVE contributions of +0.03 kJ mol−1 at this level. LMP2/aVTZ and LCCSD(T0)/aVTZ then yield Gg–Gt energy differences of 1.08 and 1.06 kJ mol−1, respectively, while LCCSD(T0)-F12a/VTZ-F12 and CCSD(T)-F12a/VTZ-F12 predict lower energy differences of 0.92 and 0.61 kJ mol−1. These values can be compared to MP2-F12 calculations by Höfener et al.33 and a focal-point analysis by Kahn and Bruice34 which agree on a ZPVE-corrected energy difference of 0.42 kJ mol−1. In this case, the accurate experimental value of 0.5721 kJ mol−1 (ref. 4) also indicates a slightly superior performance of the CCSD(T)-F12 method (0.04 kJ mol−1 difference to the experiment) and suggests variations of at least 0.5 kJ mol−1 for this set of computational analyses. For TFP, this implies a best theoretical estimate of the Tt–Gg′ electronic energy difference of 2.3(5) kJ mol−1.
Zero-point vibrational energies (ZPVEs) for TFP were estimated from harmonic frequency calculations using B3LYP, MP2 and LMP2 methods and the aVTZ basis set and clearly favor the Tt conformation. For some bands, true (anharmonic) wavenumbers ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) i are known from the experiment (see Section 4). Together with their corresponding harmonic predictions ωi, we can estimate better ground-state energy contributions E0,i of these vibrations, assuming a Morse-type energy level spacing and a correct prediction of the harmonic curvature:
i are known from the experiment (see Section 4). Together with their corresponding harmonic predictions ωi, we can estimate better ground-state energy contributions E0,i of these vibrations, assuming a Morse-type energy level spacing and a correct prediction of the harmonic curvature:
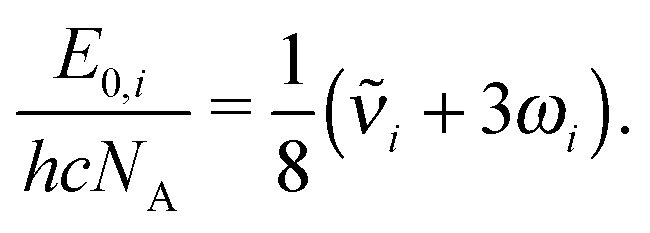 | (1) |
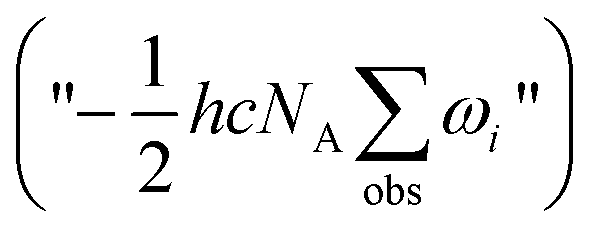 and adding the experimental updates (“
and adding the experimental updates (“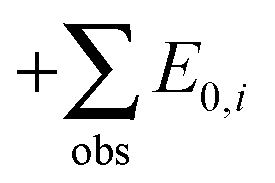 ”, see eqn (1))
”, see eqn (1))
| B3LYP | MP2 | LMP2 | |
|---|---|---|---|
| harm | −0.66 | −0.78 | −0.72 |
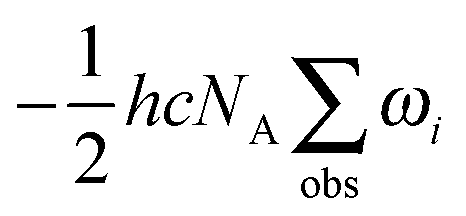
|
+0.02 | +0.20 | +0.15 |
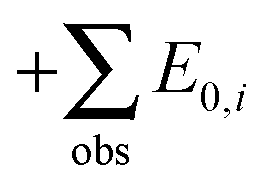
|
−0.02 | −0.15 | −0.12 |
| harm + obs. | −0.66 | −0.73 | −0.69 |
Another simple and popular method to approximate anharmonic effects is to multiply the harmonic frequencies by appropriate scaling factors. However, applying factors as proposed by Merrick et al.35 to the B3LYP and MP2 wavenumbers naturally changes the experimentally updated energy difference very little due to systematic compensation effects. Exploratory VPT2 calculations as implemented in the Gaussian09 package36 at the B3LYP/aVTZ level suffer from irregular behavior for the low-frequency modes, but still serve to confirm a conservative error bar of ±0.5 kJ mol−1 for the zero-point energy difference. We thus obtain a best-estimate Tt–Gg′ ZPVE difference of −0.7(5) kJ mol−1 based on unscaled LMP2/aVTZ harmonic frequencies. Combining this robust ZPVE difference value with the electronic estimate of 2.3(5) kJ mol−1, we obtain a best estimate for the total Tt–Gg′ energy difference of 1.6(7) kJ mol−1.
3.2 Calculated Raman spectra
Harmonic wavenumbers ω, Raman scattering cross-sections37,38σ′ and depolarization ratios δ′ were computed using the Gaussian09 program package, assuming an effective vibrational temperature of 100 K. In our experiment, we control the polarization of the incident radiation, in which case δ′ is equivalent to the “depolar (U)” ratios (≤6/7) reported by the software. Our focus is placed on three band systems which we also characterize spectroscopically in Section 4 below: the O–H, symmetric C1–H and C2–C3 stretching bands. To compensate for the neglect of anharmonicity, we determine scaling factors for these band systems in a customized scheme based on our experimental findings. The harmonic prediction for each observed band is scaled onto its observed anharmonic value; this is done for Gg′ and Tt in all three cases and additionally for Tg in the OH stretching region. The Gg′,Tt,(Tg) average is then applied to all remaining conformers in the corresponding regime.The results from B3LYP, MP2 and LMP2 calculations for the three band systems are given in Table 3, using the scaled wavenumbers (“ωsc”) for the calculation of σ′ predictions. As to the scattering cross-sections, more important than their absolute values are the Tt/Gg′ ratios, since they will be of direct spectroscopic interest in Section 4.3. We find these ratios to be remarkably robust between the B3LYP and MP2 methods, yielding averages of 2.16(3) for the OH, 1.06(2) for the CH and 0.726(4) for the CC bands.
| B3LYP | MP2 | LMP2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ω | ω sc | σ′ | δ′ | ω | ω sc | σ′ | δ′ | ω | ω sc | |
| O–H | ||||||||||
| Gg′ | 3815 | 3668 | 5.36 | 0.23 | 3840 | 3668 | 4.91 | 0.19 | 3841 | 3668 |
| Tt | 3827 | 3682 | 11.75 | 0.36 | 3846 | 3682 | 10.48 | 0.32 | 3844 | 3682 |
| Tg | 3815 | 3673 | 7.66 | 0.31 | 3841 | 3673 | 6.87 | 0.26 | 3838 | 3673 |
| Gt | 3830 | 3685 | 10.94 | 0.36 | 3848 | 3680 | 9.94 | 0.32 | 3847 | 3679 |
| Gg | 3811 | 3666 | 7.34 | 0.30 | 3835 | 3667 | 6.43 | 0.25 | 3834 | 3668 |
| C1–H | ||||||||||
| Gg′ | 3014 | 2904 | 17.47 | 0.22 | 3073 | 2904 | 16.97 | 0.16 | 3073 | 2904 |
| Tt | 3007 | 2919 | 18.75 | 0.04 | 3066 | 2919 | 17.59 | 0.08 | 3065 | 2919 |
| Tg | 3028 | 2928 | 17.59 | 0.18 | 3085 | 2926 | 18.04 | 0.13 | 3083 | 2924 |
| Gt | 2998 | 2899 | 18.94 | 0.19 | 3058 | 2901 | 17.90 | 0.13 | 3056 | 2899 |
| Gg | 3003 | 2905 | 16.96 | 0.33 | 3066 | 2908 | 16.04 | 0.27 | 3065 | 2908 |
| C2–C3 | ||||||||||
| Gg′ | 789 | 797 | 7.37 | 0.03 | 808 | 797 | 7.56 | 0.02 | 805 | 797 |
| Tt | 843 | 848 | 5.38 | 0.03 | 863 | 848 | 5.47 | 0.03 | 859 | 848 |
| Tg | 843 | 850 | 4.62 | 0.05 | 864 | 850 | 4.68 | 0.04 | 860 | 850 |
| Gt | 796 | 802 | 7.61 | 0.01 | 815 | 802 | 7.83 | 0.01 | 811 | 802 |
| Gg | 794 | 801 | 7.91 | 0.02 | 813 | 800 | 7.96 | 0.01 | 809 | 800 |
We note that the unscaled MP2 and LMP2/aVTZ methods underestimate some spacings and in part even fail to reproduce a qualitatively correct ordering of the bands; this is mostly masked by our scaling scheme in which inconsistencies remain only in the position of the Gt band relative to Tt.
The assignment of a small spectral feature near 3673 cm−1 in the OH region to the Tg conformer is still tentative at this point, as we outline in Section 4.1. However, when using only the Gg′ and Tt bands for our scaling scheme, the Tg band still remains slightly blue-shifted relative to the dominant Gg′ band, consistent with the observation. We thus rely on this assignment henceforth.
3.3 Torsional potential energy surface
The two dihedral angles τ1 and τ2 were used to describe the interconversion of the five possible TFP conformers, relaxing all other internal coordinates. The resulting torsional potential energy surface (TPES) can roughly be separated into a “core” region containing the four most stable conformers and a periphery extending to Gg. We sampled the core region (τ1 = 15⋯240°,τ2 = 120⋯360°) with a dense data point grid of 7.5° spacing, while the step size was decreased to 30° in the periphery. The inversion symmetry of the surface allowed to cut down the number of unique surface geometries to 851, including the five well-defined local minima. The TPES was then calculated in a two-step process: first, the molecular structures for all points were optimized at the LMP2/a′VTZ level; their energies were then refined in a second series of single-point energy calculations at the LCCSD(T0)/aVTZ level. LMP2/aVTZ checks revealed that the neglect of hydrogen diffuse functions in the a′VTZ basis set showed no significant effect on the relative energies and structures. Only in the OH stretching wavenumbers, some slight disagreements on the order of a few cm−1 were observed, and all frequency calculations were thus restricted to the full aVTZ basis set. As Table 1 demonstrates, LCCSD(T0)/aVTZ predicts the electronic Tt–Gg′ energy difference in good agreement with the explicitly correlated and extrapolated local calculations, showing the same ∼1 kJ mol−1 error in relation to the more accurate canonical F12 results. This suggests than in order to improve our surface calculations, eliminating this systematic error might be more important than further approaching the CBS limit.The calculated data points were fitted with an inversion-symmetric Fourier series of the form
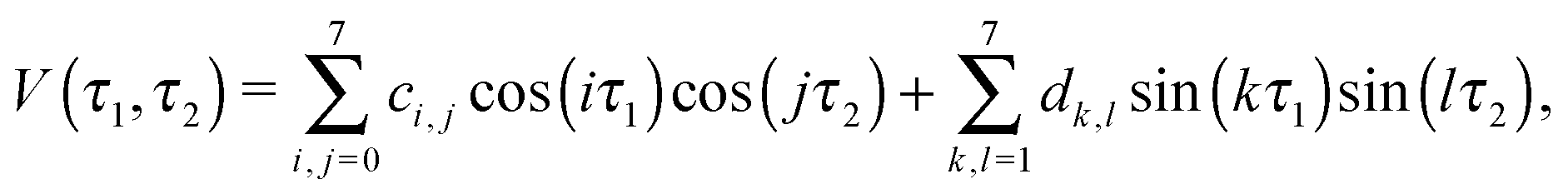 | (2) |
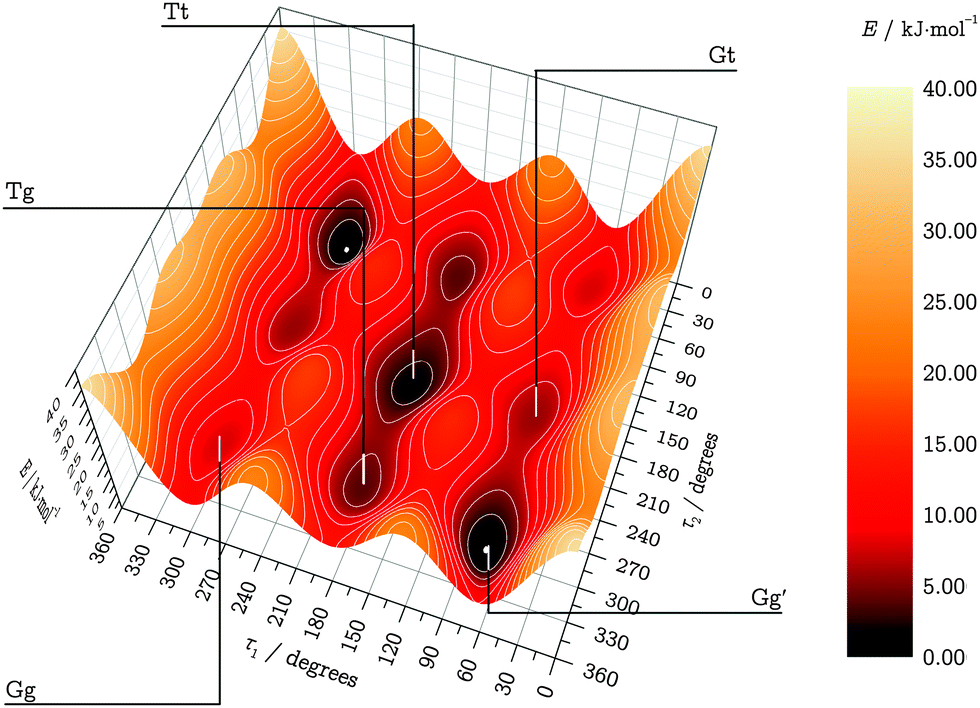 | ||
| Fig. 2 LMP2/a′VTZ-relaxed TPES on LCCSD(T0)/aVTZ level. Energies are given in kJ mol−1 relative to the Gg′ conformer. | ||
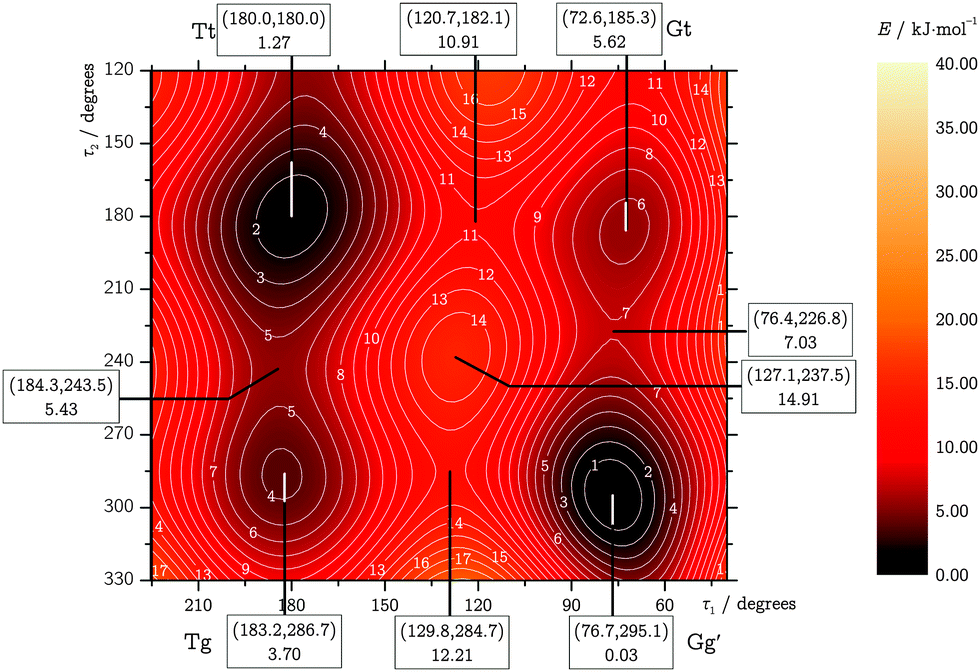 | ||
| Fig. 3 TPES core region. Local minima, saddle points and maxima are labeled with their geometries (τ1, τ2 in degrees) and relative fitted LCCSD(T0)/aVTZ energies in kJ mol−1. | ||
4 Raman jet spectroscopy
4.1 OH stretching region
Jet-spectroscopic signatures of TFP monomers and dimers in the OH stretching region have previously been characterized by Scharge in FTIR studies.30 Two monomer signals were identified at 3667 and 3681 cm−1 and attributed to the Gg′ and Tt conformers, respectively, with an estimated concentration ratio of Tt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Gg′ = 1:4. A small red-shifted 3656 cm−1 band has contributions from the OH stretching modes of acceptor molecules in hydrogen-bonded dimer structures, but may overlap with the symmetric stretching vibration of residual water monomers in Raman experiments.
:Gg′ = 1:4. A small red-shifted 3656 cm−1 band has contributions from the OH stretching modes of acceptor molecules in hydrogen-bonded dimer structures, but may overlap with the symmetric stretching vibration of residual water monomers in Raman experiments.
In order to study the unperturbed conformational preferences of the TFP monomer, we aimed for experimental conditions that essentially suppress the formation of clusters. We thus employed a low saturator temperature θsat of −25 °C, which provides largely dimer-free conditions with the nozzle close to room temperature, given that the expansion is sampled not too far downstream. We were able to reproduce the IR findings of Scharge30 within a calibration accuracy of 1 cm−1, yielding jet Raman Gg′ and Tt band positions of 3668 and 3682 cm−1, respectively. Fig. 4 compares Raman jet spectra in the OH stretching region to B3LYP, MP2 and LMP2 calculations in the aVTZ basis set after application of the observation-based wavenumber scaling procedure (see Section 3.2). The predicted scattering intensities suggest a Tt:Gg′ ratio of about 0.2–0.3, in agreement with the FTIR evidence.30
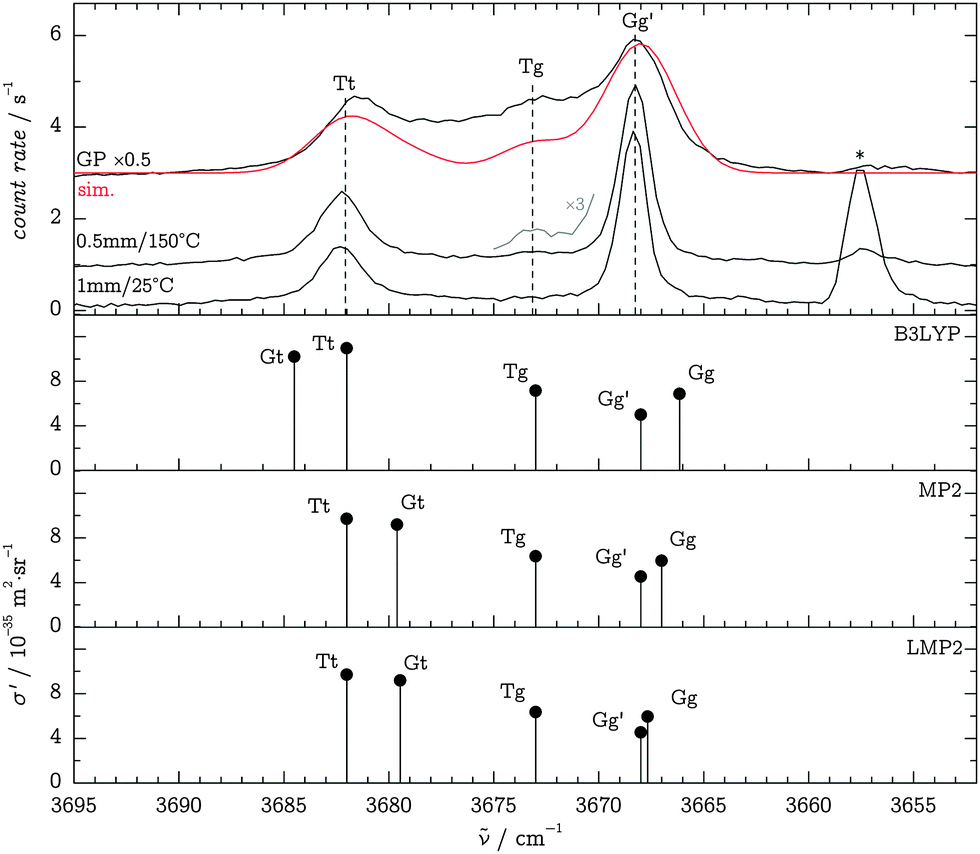 | ||
| Fig. 4 Comparison of experimental and predicted TFP monomer spectra in the OH stretching region (calculations scaled as described in the text). Spectra traces (top panel) from top to bottom: Gas phase at room temperature, scaled by a factor of 0.5 and compared to a simulated spectrum (red trace, see text for details); jet, θn = 150 °C, d = 0.5 mm; jet, θn = 25 °C, d = 1 mm. The band marked with an asterisk stems from variable water impurities. | ||
Heated-nozzle jet spectra at close distances d hint towards the existence of a third feature near 3673 cm−1, slightly blue-shifted to the Gg′ band (Fig. 4). Similarly, spectra recorded at increased background pressures (not shown) indicate a continuous transition from the jet environment to the gas phase while showing an early onset of the 3673 cm−1 feature. On grounds of the calculated energy ordering, we attribute this band to the Tg conformer. In addition, gas-phase spectra were recorded by flooding the test chamber with the sample gas mixture at room temperature. They show a broad scattering contribution between the now less prominent Gg′ and Tt bands, which we attribute to other conformers and anharmonic coupling of the OH oscillators to torsional modes.39Fig. 4 also includes a simulated gas-phase spectrum (red trace) at 298 K, using CCSD(T)-F12a/VTZ-F12 energies for the conformer distribution, B3LYP/aVTZ ZPVE corrections with “harm + obs” values for Tt (see Table 1), B3LYP/aVTZ scattering intensities, scaled LMP2/aVTZ band positions and Gaussian band profiles with a FWHM of 4 cm−1. By construction, the simulation does not capture the broad underlying hot torsional structure of the experimental spectrum, whereas it supports the Tg assignment on grounds of the relative intensity of the 3673 cm−1 band. Further, the simulated Gt band has a considerable intensity contribution, about 1/3 that of Tt. This suggests that the high-wavenumber B3LYP prediction for this band is less likely due to the absence of a separate band in the experiment, while the wave function results are more plausible. Furthermore, the simulation provides initial support for the accuracy of the theoretically predicted conformational energy differences and scattering intensities, which will be elaborated below.
A series of experiments at a nozzle distance of d = 3 mm were also carried out, but analysis of the spectra revealed contamination with dimers, affecting the monomer OH stretching bands in terms of band width and intensity in particular for the Tt feature. We discard these data in favor of the 1 mm spectra which only show traces of dimers.
4.2 CH/CC stretching band systems and fingerprint region
To obtain independent experimental estimates of the Tt/Gg′ conformer ratio, we require pairs of well-isolated bands of both conformers corresponding to comparable vibrational motions together with robust predictions for their scattering strengths. Apart from the OH stretching band system, two other candidates were identified which fulfill these requirements: the symmetric C1–H stretching bands at 2919 (Tt) and 2904 cm−1 (Gg′), and the C2–C3 stretching bands at 848/797 cm−1 (Tt/Gg′). The former bands are in resonance with bending overtones, but for simplicity, we will continue to denote them as “stretching” bands. The spectra are displayed in Fig. 5 for different nozzle temperatures.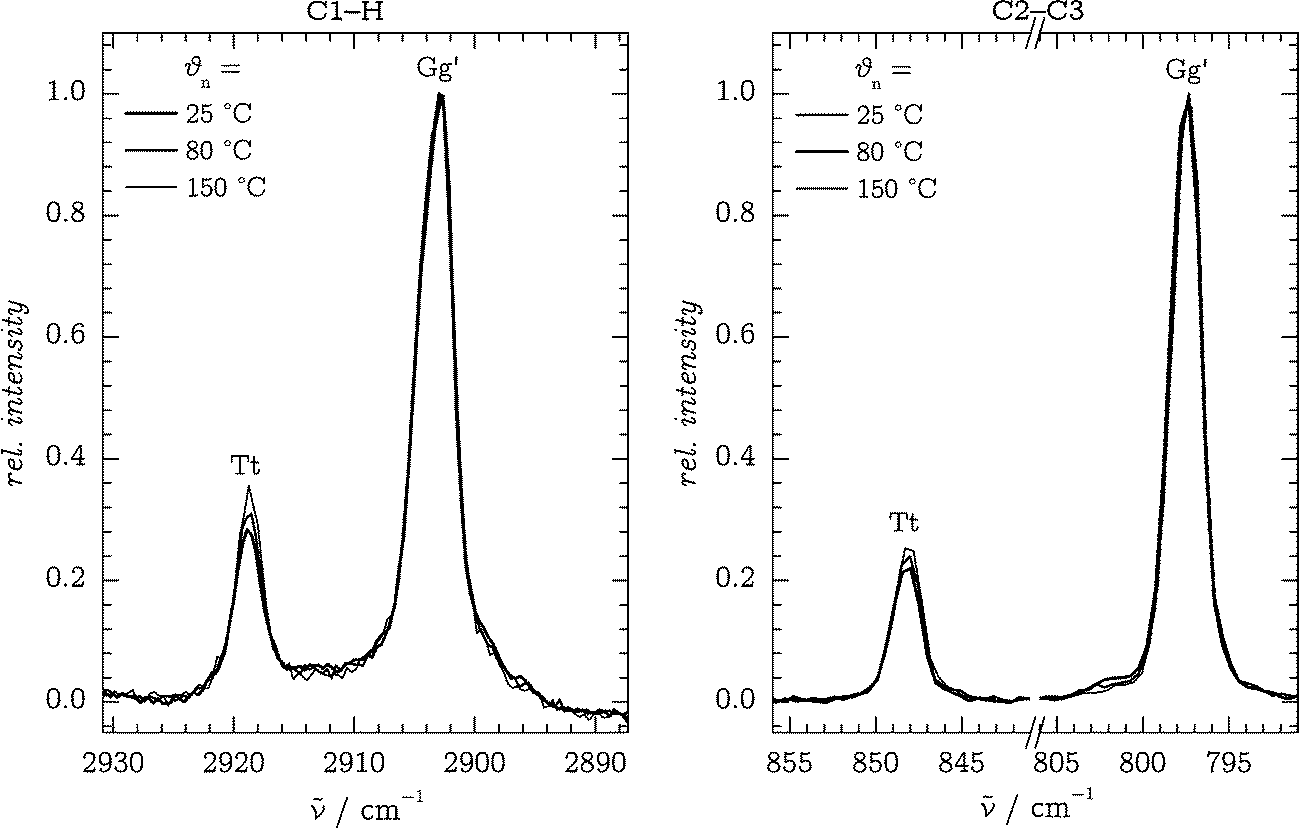 | ||
| Fig. 5 C1–H (left) and C2–C3 (right) stretching band systems at different nozzle temperatures θn, all at d = 1 mm and normalized to the Gg′ bands. | ||
In order to assign the spectral signatures in the fingerprint region to Gg′ and Tt, polarization experiments were carried out as detailed in ref. 38 and 40. The polarization of the incident (“i”) laser radiation was changed from perpendicular (“⊥i”) to parallel (“∥i”) in relation to the scattering plane. Non-totally symmetric modes (A′′ for Tt) are completely depolarized, and their intensity is only reduced by a factor of δ′ = 6/7 upon ⊥i → ∥i polarization change. Totally symmetric modes (A′ for Tt and all for Gg′) are more or less polarized and will be quenched by a factor 0 ≤ δ′ < 6/7. The recorded spectra in the 200–400 cm−1 region are shown in Fig. 6 and are compared to wavenumber-scaled35 B3LYP/aVTZ predictions and unscaled LMP2/aVTZ predictions with an assumed Tt/Gg′ conformer ratio of 0.20. We note that the band positions, σ′ values and depolarization ratios from MP2/aVTZ and scaled B3LYP/aVTZ predictions agree well and can be regarded as interchangeable within our analysis.
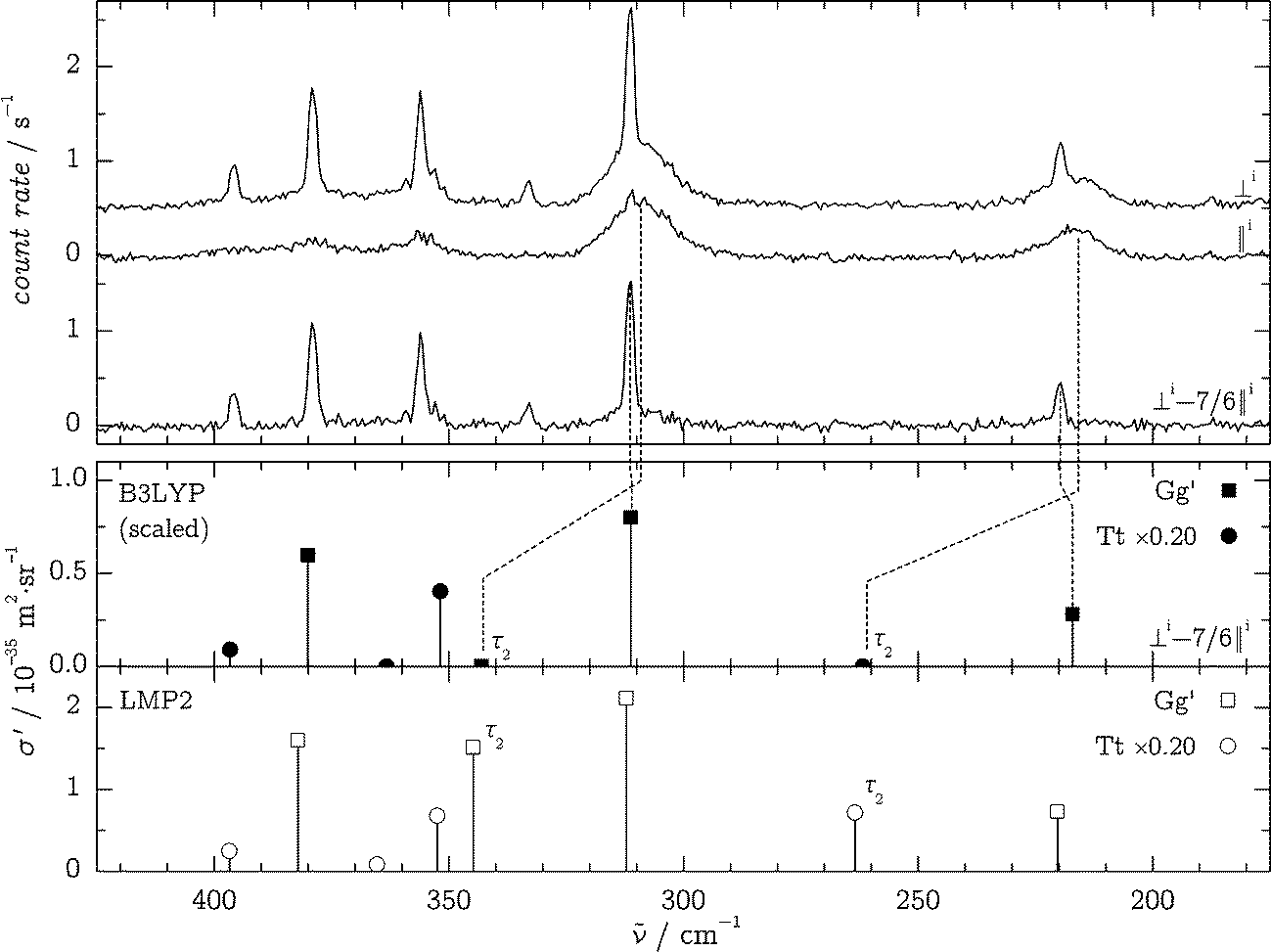 | ||
| Fig. 6 Top: polarization-affected Raman spectra in the OH torsion region: ⊥i, ∥i and ⊥i–7/6∥i spectra at θn = 150 °C, d = 1 mm. Bottom, upper panel: scaled35 B3LYP/aVTZ calculations with an approximate Tt/Gg′ conformer ratio of 0.20 in a simulated depolarization experiment; lower panel: unscaled, polarization-independent LMP2/aVTZ predictions using the isotropic (⊥i) B3LYP intensities (because LMP2 intensities were unavailable and to illustrate the close match between LMP2 and scaled B3LYP band positions). The bands corresponding to approximate τ2 torsional motion are labeled in the theoretical results. | ||
Comparison of predicted and experimental spectra allows for some straightforward assignments, which were used in the “harm + obs” method (see Section 3.1 in this document and the Section S3 of the ESI†). Only two bands show a distinct anharmonic red-shift, consistent with their τ2 (OH) torsional character which is also revealed by the depolarization behavior.
4.3 Experimental conformer interconversion enthalpy
After having established the basic spectral signatures and relative abundances of the two Gg′ and Tt conformers, their ratio can be systematically modified by heating the nozzle and PTFE feeding tube. The idea is to increase the population of the less stable Tt conformer to deduce constraints on its energy difference to Gg′. These constraints depend on the degree of conformational relaxation in the supersonic expansion.In 1982, Felder and Günthard proposed that conformer ratios in supersonic expansions are essentially frozen in at the pre-expansion temperature and formulated that “conformational cooling in supersonic expansions seems to be an exception rather than a rule.”41 This “instant-freezing” assumption seemed to uphold even for small molecular species with only a single internal-rotation degree of freedom governing in the interconversion. Similarly, Ruoff et al. suggested in 1990 that conformer relaxation plays a considerable role only for interconversion barriers below 400 cm−1 (about 4.8 kJ mol−1).42 On the other hand, previous Raman-jet studies have found conformational temperatures as low as 100 K for alkane-folding barriers on the order of 10 kJ mol−1 (ref. 3) and ca. 50 K for the downstream conformer interconversion of ethanol in pure He expansions which profits from hydrogen tunneling through the barriers.37 We can thus expect some conformational cooling for TFP in the supersonic expansion, in particular for the g–t path, which is analogous to ethanol. According to the conformational propensity predictions in Fig. 7, full relaxation down to 50–100 K is inconsistent with the observed quantities of the Tt conformer.
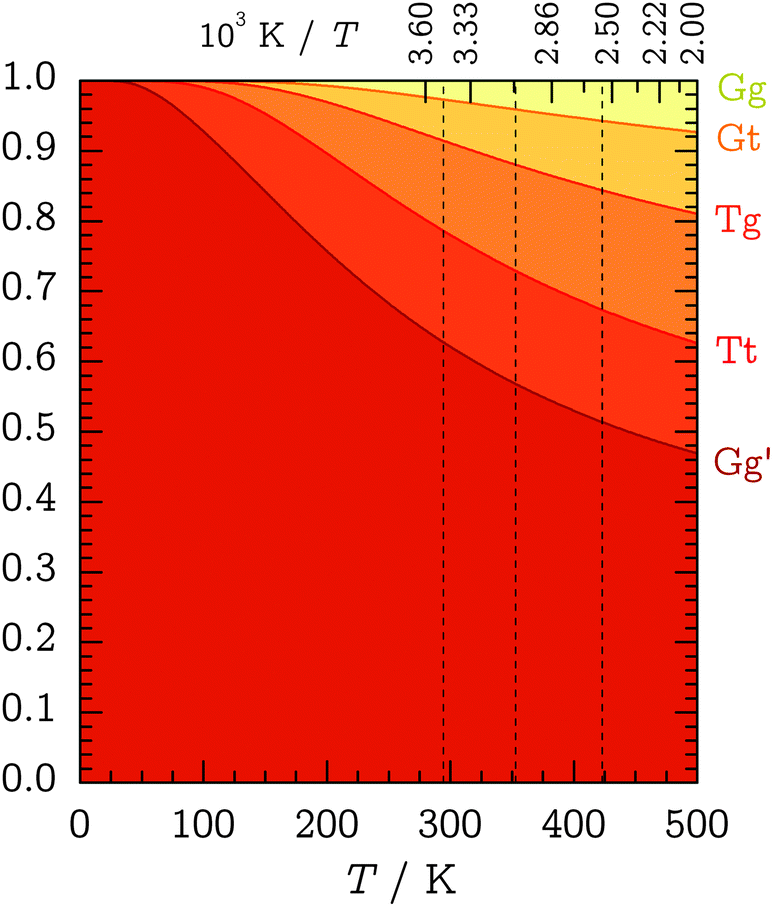 | ||
| Fig. 7 Cumulative Boltzmann conformer populations between 0 and 500 K based on CCSD(T)-F12a/VTZ-F12 relative energies and B3LYP/aVTZ ZPVEs (see Table 1). The dashed vertical lines indicate the experimental nozzle temperatures of θn = 25, 80, 150 °C. | ||
The instant-freezing model effectively keeps the conformational temperature Tconf fixed at the nozzle temperature Tn throughout the entire jet expansion, and the corresponding Tt/Gg′ conformer ratio remains unaltered. For simplicity, we assume that the rotational and vibrational energy levels are spaced equally for all conformers so that all entropic contributions to the interconversion free enthalpy vanish under jet-cooled conditions.41 The spectroscopic Tt/Gg′ band intensity ratio F is then given by
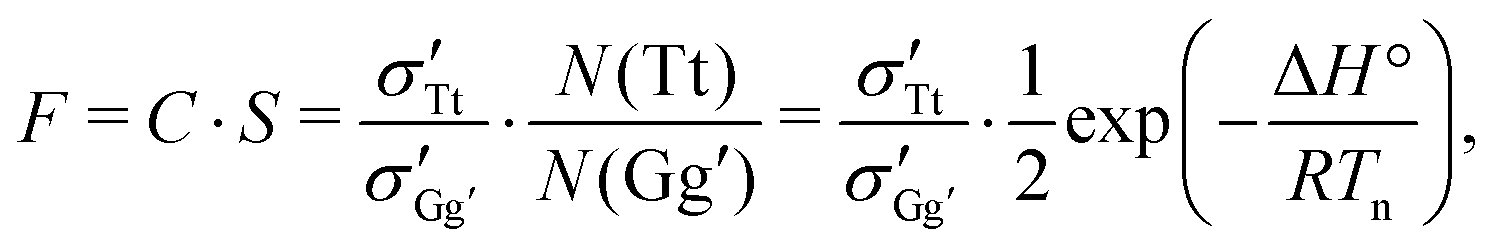 | (3) |
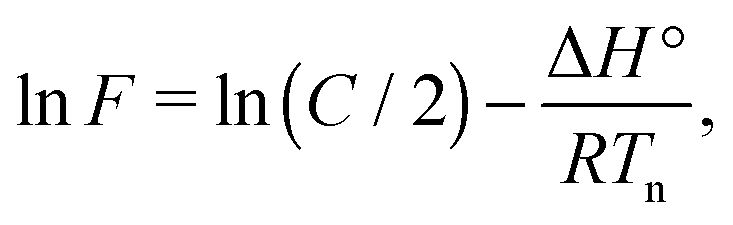 | (4) |
2F/C vs. 1/Tn plots forced to meet the theoretical C value (indicated by a superscript “fix”). The results are displayed in Table 4 and Fig. 8; note that for EtOH, the energy sequence of g and t is reversed. The CH value appears too high and the slope in Fig. 8 visibly deviates from the trend in the data points. We attribute this to stretch-bend Fermi resonance intensity distortions and consider the corresponding fixed value for ΔH° unreliable. Indeed, the transitions are below twice the corresponding CH bending fundamentals observed at 1478 and 1497 cm−1 for Gg′ and Tt, respectively (not shown). They may thus have partial bending character and harmonic stretching intensity estimates are obsolete. If we also relax the ordinate intercept in the regression, the resulting Cfit values are still in moderate agreement with the predictions, and the corresponding ΔH°,fit values – although more affected by statistical errors – do not differ significantly from before. Again, the exception is CH, where the difference amounts to a factor of two. This supports the assumption of unreliable harmonic intensities in this Fermi resonance affected case. The enthalpies from the other fits can be assumed to resemble upper-bound estimates to the true value, also because they assume negligible relaxation.
| ΔH°,fix | C fix | ΔH°,high | ΔH°,fit | C fit | ΔH°,low | ΔH°,τ2 | C τ 2 | |
|---|---|---|---|---|---|---|---|---|
| TFP | ||||||||
| O–H | 1.9(1) | 2.16(3) | 3.4(1) | 2.4(10) | 2.6(9) | 0.57(3) | 2.3 | 2.0 |
| C1–H | [2.7(1)] | [1.06(2)] | [4.1(2)] | 1.37(8) | 0.68(2) | 0.79(3) | 1.3 | 0.6 |
| C2–C3 | 1.44(5) | 0.726(4) | 2.9(1) | 1.5(5) | 0.7(1) | 0.43(1) | 1.3 | 0.6 |
| EtOH O–H | ||||||||
| 1 mm | 1.58(2) | 1/1.5(1)37 | ||||||
| 3 mm | 2.37(10) | |||||||
| 6 mm | 3.16(5) | |||||||
| Lit.43 | 0.47 | |||||||
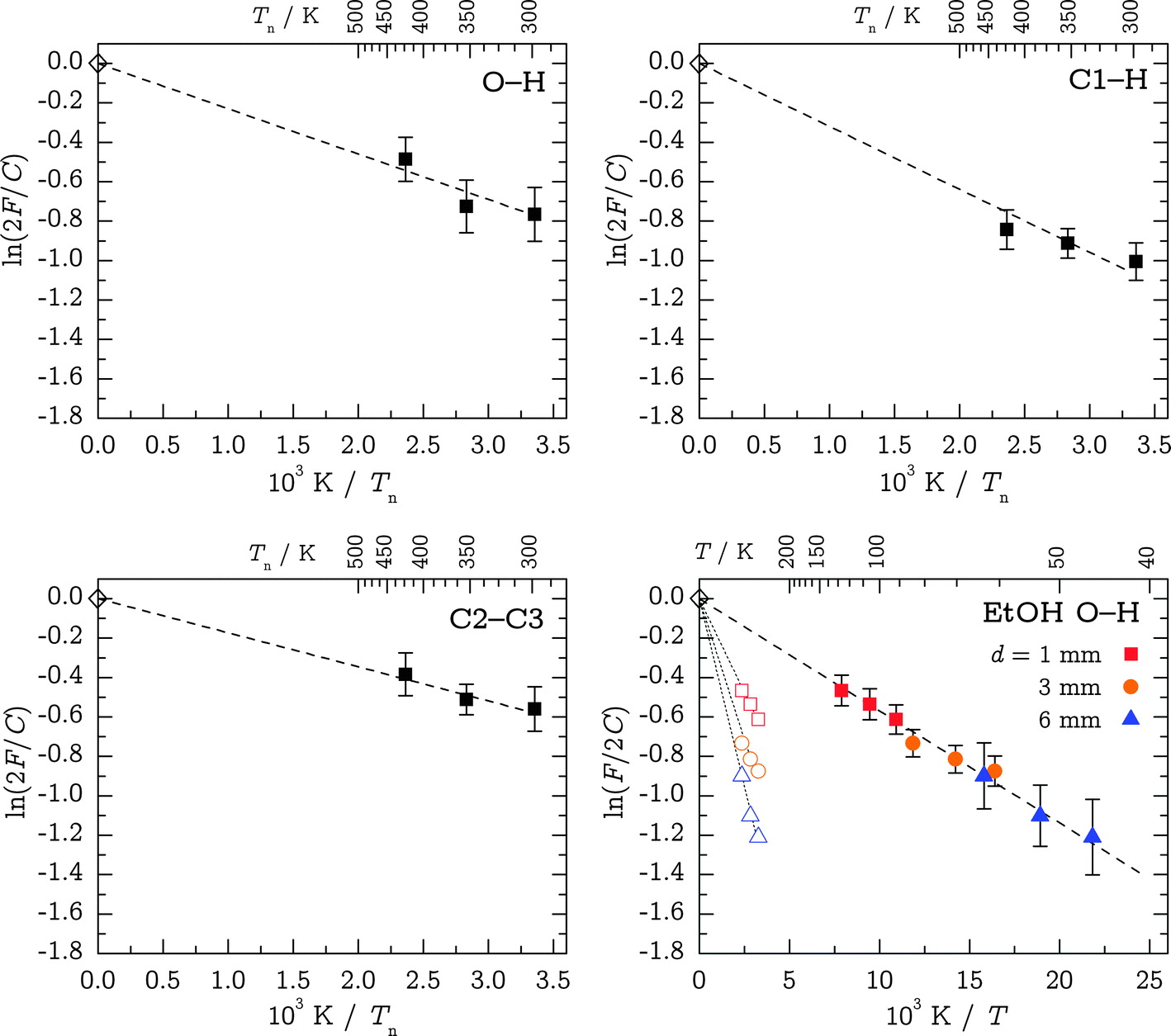 | ||
| Fig. 8 Logarithmic linear regressions of band intensity ratios F for TFP OH, C1–H and C2–C3 stretching band systems. Also given is a benchmark study on the OH stretching bands of ethanol where the conversion is more facile and the value of C is well-established;37 here, hollow symbols are fixed at Tn and solid symbols are scaled in 1/T to reproduce the accurately known energy difference ΔH° = 0.473 kJ mol−1 (ref. 43) at each nozzle distance. All fits were fixed at the origin based on calculated C ratios (indicated by hollow diamonds). EtOH colors only for clarity. | ||
Estimated entropy contributions between 300 and 450 K based on computed partition functions lead to a correction of the linear intercepts in the three TFP panels of Fig. 8 by +0.1 for the rotational contribution and roughly +0.4 for the vibrational contribution in the completely frozen model. In both cases, the Tt conformation is slightly favored by entropy, although the harmonic vibrational partition function estimate is very uncertain at these temperatures due to the large amplitude low frequency vibrations. Any partial relaxation of the Tt conformation will reduce this entropy effect, but as a truly worst case estimate, we add a column ΔH°,high to Table 4 which contains the effect of this intercept increment on the slope of the van't Hoff plot. One can see that the agreement with the fitted values ΔH°,fit stays similar for OH, becomes worse for CC and is very unreasonable for CH, underscoring the Fermi resonance problem for this particular mode when computed harmonic CH scattering cross sections are used. Considering further that the OH stretching region suffers least from band overlap from other conformations, this may provide the most reliable upper bound.
The corresponding ethanol measurements clearly demonstrate a significant cooling of the OH torsion coordinate because they propose far too large values of ΔH°,fix (Table 4, bottom part). As a first step beyond the drastic instant-freezing assumption, we scale the 1/T values of EtOH at each nozzle distance d to reproduce the literature value for ΔH°.43 Conformational cooling in TFP will be less efficient than in EtOH because it involves backbone isomerization. By applying the EtOH scaling factors to our TFP ΔH°,fix values, we obtain safe lower bounds to the interconversion enthalpy, ΔH°,low (Table 4).
The drawback of this indiscriminate scaling approach is that it effectively assumes a uniform relaxation for the τ1 and τ2 degrees of freedom, which appears unrealistic in light of the barriers predicted in Section 3.3. Furthermore, it is a two-state model, whereas at least one or two other conformers come into play at higher nozzle temperatures (cf.Fig. 7). We thus introduce a third model in which the sample mixture is again pre-equilibrated at the nozzle temperature Tn. The less stable and spectrally distinct T-type (Tg) and G-type (Gt, Gg) conformers then undergo exclusive τ2 relaxation by a certain fraction k – assumed to be uniform for all conformers – and pool in the Tt and Gg′ minima, respectively. The relaxed Tt/Gg′ ratio S is thus given by
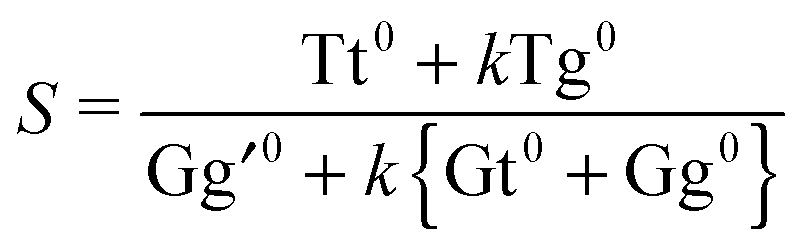 | (5) |
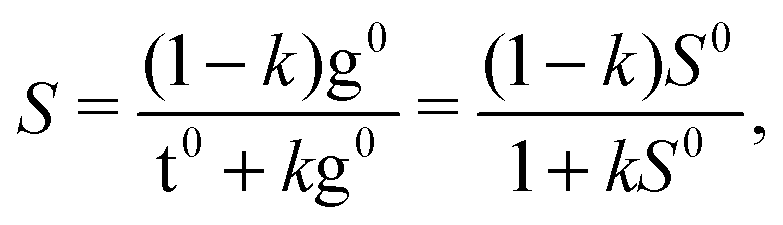 | (6) |
Wrapping up these 13 partially bounded estimates for the Tt–Gg′ energy difference, the energy gap should lie in the 0.5–3.4 kJ mol−1 range, and more likely between 1.3 and 2.3 kJ mol−1. A value of 1.8(5) kJ mol−1 is thus a reasonable best experimental estimate, although one has to keep in mind the imperfection of all employed methods to reproduce the complex partial conformational relaxation in the first 1–2 μs of the expansion. Agreement with the best theoretical estimate of 1.6(7) kJ mol−1 is very satisfactory and a combined best estimate of 1.7(5) kJ mol−1 appears justified in view of the entanglement of different theoretical and experimental inputs for both values.
5 Summary
Working out the quantitative conformational landscape of an aliphatic chain molecule such as trifluoropropanol requires a combined experimental and theoretical effort. As shown in Fig. 3, the key players are an internally hydrogen-bonded Gg′ conformation and an energetically somewhat higher and more polar stretched Tt conformation. Their interconversion involves a facile and tunneling-assisted OH rotation (g/t) followed or preceded by a less facile backbone (G/T) torsion, whereas a concerted rearrangement is energetically unfavorable. Building on the related case of ethanol, the OH reorientation is demonstrated to occur to some extent already 1 mm downstream a supersonic nozzle. However, Tt signals persist in the Raman jet spectra despite a distinct energy penalty of about 1.7(5) kJ mol−1 relative to the global Gg′ minimum, which suggests that backbone torsion is at least partially frozen in the expansion.We have modeled the observed spectral Tt/Gg′ distributions in the jet expansion using three approaches, assuming indiscriminate relaxation of both relevant torsional coordinates, their instantaneous freezing, and exclusive partial relaxation of the more facile OH torsion. Based on these models, one may favor the lower-end, upper-end, or intermediate predictions for the conformational energy gap, respectively. Spectral evidence has been found for at least one further conformer (Tg) being still populated close to the nozzle, corroborated by theoretical energy predictions and underscoring the limits of the instant-freezing model. Further experimental uncertainties arise from the need to include some theoretically calculated cross-sections in the modeling, and from the smallness of the observed relaxation effect. Some degree of backbone interconversion, as demonstrated in alkanes using the same experimental setup,3 cannot be ruled out. We therefore prefer not to narrow down the best estimate for the Tt–Gg′ energy difference beyond stating that it is a factor of two smaller than previously estimated,7 with an error bar that is also reduced by a factor of 2.
On the theoretical side, we provide support for a correct description of the energetics at explicitly correlated CCSD(T) levels, with small but significant deviations for local CCSD(T) approaches due to an approximate treatment of the intramolecular hydrogen bridge. Based on our spectra, there is little evidence for significant anharmonic corrections to the zero-point vibrational energy difference between the dominant conformations. Fig. 7 shows our best estimate for the conformational partitioning of TFP as a function of temperature, underscoring that a two-state model only persists at rather low temperatures, soon extending into the OH torsionally excited states which serve as intermediates in the Tt ↔ Gg′ interconversion.
Overall, it is rewarding to see a balanced error situation between theory and experiment for such a subtle folding equilibrium. More accurate experimental values would probably require either the introduction of an aromatic chromophore or the restriction to OH torsional isomerism. More accurate theoretical values would demand for a smaller system, where anharmonic zero-point energy can be treated more rigorously. What makes the trifluoropropanol system dynamically interesting is the close energy match of two entirely different conformations which are separated from each other by two consecutive barriers and thus fairly stable with respect to collisional interconversion. One could conceive a vibrational above-barrier excitation scheme which switches between these two conformations of different polarity and dipole orientation, making it interesting for electric field deflection experiments. In any case, a microwave reinvestigation of the two or even three lowest-energy conformations of TFP appears rewarding.
Acknowledgements
We thank T. Scharge for providing infrared reference data and the German Research Foundation for financial support (DFG SU 121/2-2).References
- A. D. Mackerell, M. Feig and C. L. Brooks, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- E. Gloaguen, R. Pollet, F. Piuzzi, B. Tardivel and M. Mons, Gas phase folding of an (Ala)4 neutral peptide chain: spectroscopic evidence for the formation of a β-hairpin H-bonding pattern, Phys. Chem. Chem. Phys., 2009, 11, 11385–11388 RSC.
- N. O. B. Lüttschwager and M. A. Suhm, Stretching and folding of 2-nanometer hydrocarbon rods, Soft Matter, 2014, 10, 4885–4901 RSC.
- Z. Kisiel, O. Dorosh, A. Maeda, I. R. Medvedev, F. C. De Lucia, E. Herbst, B. J. Drouin, J. C. Pearson and S. T. Shipman, Determination of precise relative energies of conformers of n-propanol by rotational spectroscopy, Phys. Chem. Chem. Phys., 2010, 12, 8329–8339 RSC.
- T. S. Zwier, Laser Probes of Conformational Isomerization in Flexible Molecules and Complexes, J. Phys. Chem. A, 2006, 110, 4133–4150 CrossRef CAS PubMed.
- D. S. Skvortsov and A. F. Vilesov, Using He droplets for measurements of interconversion enthalpy of conformers in 2-chloroethanol, J. Chem. Phys., 2009, 130, 151101 CrossRef PubMed.
- K. M. Marstokk and H. Møllendal, Microwave Spectrum, Intramolecular Hydrogen Bonding, Conformational Properties and Quantum Chemical Calculations for 3,3,3-Trifluoropropanol, Acta Chem. Scand., 1999, 53, 202–208 CrossRef CAS PubMed.
- R. A. Cormanich, R. Rittner, M. P. Freitas and M. Bühl, The seeming lack of CFHO intramolecular hydrogen bonds in linear aliphatic fluoroalcohols in solution, Phys. Chem. Chem. Phys., 2014, 16, 19212–19217 RSC.
- T. Scharge, C. Emmeluth, T. Häber and M. A. Suhm, Competing hydrogen bond topologies in 2-fluoroethanol dimer, J. Mol. Spectrosc., 2006, 786, 86–95 CAS.
- T. Scharge, C. Cézard, P. Zielke, A. Schutz, C. Emmeluth and M. A. Suhm, A peptide co-solvent under scrutiny: Self-aggregation of 2,2,2-Trifluoroethanol, Phys. Chem. Chem. Phys., 2007, 9, 4472–4490 RSC.
- A. D. Richardson and K. Hedberg, Conformational analysis. 23: A gas-phase electron-diffraction and ab initio molecular orbital investigation of 3-fluoropropan-1-ol. Is there significant internal hydrogen bonding?, J. Mol. Struct., 2001, 567–568, 187–196 CrossRef CAS.
- T. N. Wassermann, M. A. Suhm, P. Roubin and S. Coussan, Isomerization around C–C and C–O bonds in 1-propanol: Collisional relaxation in supersonic jets and selective IR photo-isomerization in cryogenic matrices, J. Mol. Spectrosc., 2012, 1025, 20–32 CAS.
- N. Mayorkas, I. Malka and I. Bar, Ionization-loss stimulated Raman spectroscopy for conformational probing of flexible molecules, Phys. Chem. Chem. Phys., 2011, 13, 6808–6815 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- P. Pulay, Localizability of dynamic electron correlation, Chem. Phys. Lett., 1983, 100, 151–154 CrossRef CAS.
- W. Klopper, F. R. Manby, S. Ten-No and E. F. Valeev, R12 methods in explicitly correlated molecular electronic structure theory, Int. Rev. Phys. Chem., 2006, 25, 427–468 CrossRef CAS.
- T. B. Adler and H.-J. Werner, Local explicitly correlated coupled-cluster methods: Efficient removal of the basis set incompleteness and domain errors, J. Chem. Phys., 2009, 130, 241101 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, 2012. see http://www.molpro.net.
- M. Schütz, Low-order scaling local electron correlation methods. III. Linear scaling local perturbative triples correction (T), J. Chem. Phys., 2000, 113, 9986–10001 CrossRef PubMed.
- R. A. Mata and H.-J. Werner, Local correlation methods with a natural localized molecular orbital basis, Mol. Phys., 2007, 105, 2753–2761 CrossRef CAS.
- T. Hrenar, G. Rauhut and H.-J. Werner, Impact of Local and Density Fitting Approximations on Harmonic Vibrational Frequencies, J. Phys. Chem. A, 2006, 110, 2060–2064 CrossRef CAS PubMed.
- T. Hrenar, H.-J. Werner and G. Rauhut, Accurate calculation of anharmonic vibrational frequencies of medium sized molecules using local coupled cluster methods, J. Chem. Phys., 2007, 126, 134108 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- M. Andrejić, U. Ryde, R. A. Mata and P. Söderhjelm, Coupled-Cluster Interaction Energies for 200-Atom Host-Guest Systems., ChemPhysChem, 2014, 15, 3270–3281 CrossRef PubMed.
- J. Pipek and P. G. Mezey, A fast intrinsic localization procedure applicable for abinitio and semiempirical linear combination of atomic orbital wave functions, J. Chem. Phys., 1989, 90, 4916–4926 CrossRef CAS PubMed.
- K. A. Peterson, T. B. Adler and H.-J. Werner, Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne and Al–Ar, J. Chem. Phys., 2008, 128, 084102 CrossRef PubMed.
- J. E. Del Bene, Proton affinities of ammonia, water, and hydrogen fluoride and their anions: a quest for the basis-set limit using the Dunning augmented correlation-consistent basis sets, J. Phys. Chem., 1993, 97, 107–110 CrossRef CAS.
- F. Weigend, A fully direct RI-HF algorithm: Implementation, optimised auxiliary basis sets, demonstration of accuracy and efficiency, Phys. Chem. Chem. Phys., 2002, 4, 4285–4291 RSC.
- F. Weigend, A. Köhn and C. Hättig, Efficient use of the correlation consistent basis sets in resolution of the identity MP2 calculations, J. Chem. Phys., 2002, 116, 3175–3183 CrossRef CAS PubMed.
- T. Scharge, Schwache Bindungen mit starker Wirkung: Konformation und Aggregation von halogenierten Alkoholen, PhD thesis, Sierke Verlag, Göttingen, 2008 Search PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- S. Höfener, F. A. Bischoff, A. Glöβ and W. Klopper, Slater-type geminals in explicitly-correlated perturbation theory: application to n-alkanols and analysis of errors and basis-set requirements, Phys. Chem. Chem. Phys., 2008, 10, 3390–3399 RSC.
- K. Kahn and T. C. Bruice, Focal-point conformational analysis of ethanol, propanol, and isopropanol, ChemPhysChem, 2005, 6, 487–495 CrossRef CAS PubMed.
- J. P. Merrick, D. Moran and L. Radom, An Evaluation of Harmonic Vibrational Frequency Scale Factors, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- V. Barone, Anharmonic vibrational properties by a fully automated second-order perturbative approach, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- T. N. Wassermann and M. A. Suhm, Ethanol Monomers and Dimers Revisited: A Raman Study of Conformational Preferences and Argon Nanocoating Effects, J. Phys. Chem. A, 2010, 114, 8223–8233 CrossRef CAS PubMed.
- P. Zielke, Ramanstreuung am Überschallstrahl: Wasserstoffbrückendynamik aus neuer Perspektive, PhD thesis, Cuvillier, Göttingen, 2007 Search PubMed.
- D. Schiel and W. Richter, Effect of the internal rotation of the OH group on the OH stretching vibrational Raman profile of ethanol in the gas phase, J. Chem. Phys., 1983, 78, 6559–6562 CrossRef CAS PubMed.
- D. A. Long, The Raman Effect – A Unified Treatment of the Theory of Raman Scattering by Molecules, John Wiley & Sons, West Sussex, England, 2002 Search PubMed.
- P. Felder and H. H. Günthard, Conformational interconversions in supersonic jets: Matrix IR spectroscopy and model calculations, Chem. Phys., 1982, 71, 9–25 CrossRef CAS.
- R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, Relaxation of conformers and isomers in seeded supersonic jets of inert gases, J. Chem. Phys., 1990, 93, 3142–3150 CrossRef CAS PubMed.
- C. Richard Quade, A Note on Internal Rotation-Rotation Interactions in Ethyl Alcohol, J. Mol. Spectrosc., 2000, 203, 200–202 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Parameters of potential energy surface fit; band intensity ratios from Raman-jet experiments; harmonic vibrational frequencies from calculations and assignments of experimental bands for the two main conformers; and Raman jet spectra between 100 and 1600 cm−1. See DOI: 10.1039/c4cp05868b |
| This journal is © the Owner Societies 2015 |