
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electromers of the benzene dimer radical cation†
Anna
Błoch-Mechkour
and
Thomas
Bally
*
Department of Chemistry, University of Fribourg, Switzerland. E-mail: Thomas.Bally@unifr.ch
First published on 25th March 2015
Abstract
The well-studied benzene dimer radical cation, which is prototypical for this class of species, has been reinvestigated computationally. Thereby it turned out that both the σ-hemibonded and the half-shifted sandwich structures of the benzene dimer cation, which had been independently proposed, represent stationary points on the B2PLYP-D potential energy surfaces. However, these structures belong to distinct electronic states, both of which are associated with potential surfaces that are very flat with regard to rotation of the two benzene rings in an opposite sense relative to each other. The surfaces of these two “electromers” of the benzene dimer cation are separated by only 3–4 kcal mol−1 and do not intersect along the rotation coordinate, which represents a rather unique electronic structure situation. When moving on either of the two surfaces the title complex is an extremely fluxional species, in spite of its being bound by over 20 kcal mol−1.
Introduction
Noncovalent interactions involving aromatic rings are ubiquitous in chemical and biomolecular systems.1 Thus, the benzene dimer, B2, has been the subject of numerous experimental and theoretical investigations, because it serves as a benchmark system to study the π–π interactions that prevail e.g. between the nucleotide bases in RNA and DNA, and are responsible for the strong binding of drug molecules containing planar π-systems by intercalation between base pairs in DNA.2,3 Most computational studies of the benzene dimer arrived at the conclusion that its most stable structures are parallel-displaced sandwich structures such as those labelled XD and YD in Scheme 1 (where the two benzene rings are displaced along the x- or the y-coordinate, respectively), and T-shaped hydrogen bonded ones (TH), whereas the symmetric sandwich structure, SA, does not represent a minimum on the potential surface.4 It has been proposed that the symmetric T-shaped structure is a transition state for the interconversion of two tilted TH structures,5,6 but as all these structures lie within a few tenth of a kcal mol−1 it is clear that the potential surface of the neutral benzene dimer, which is bound by a only 2.5–3 kcal mol−1, is extremely flat, i.e. the system is very fluxional.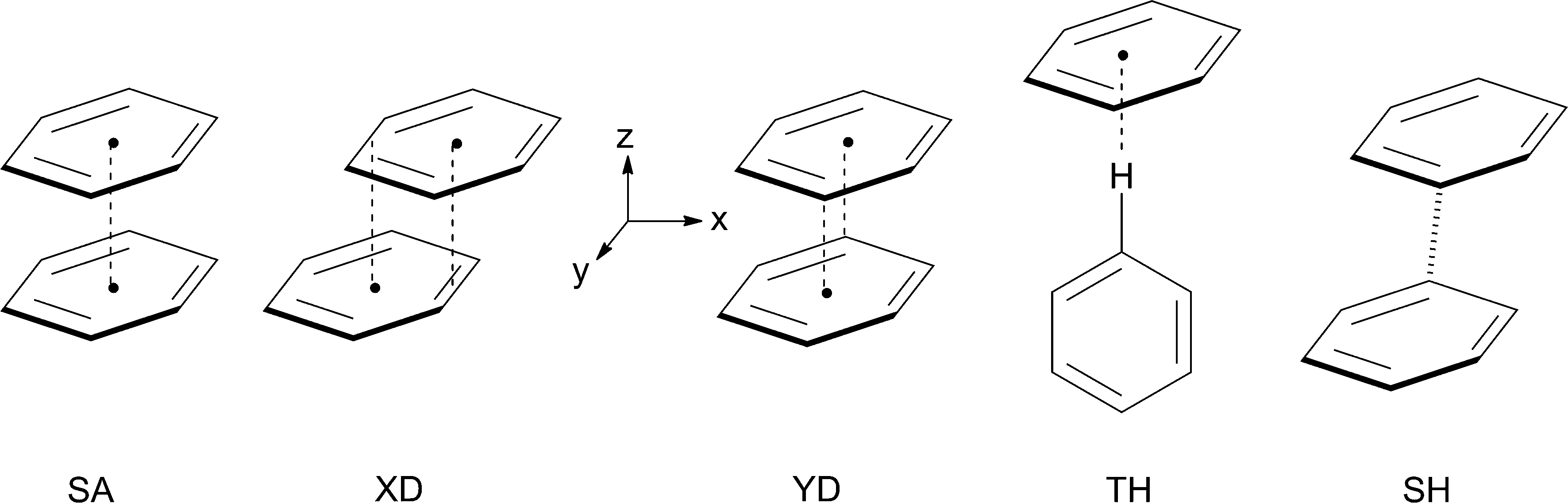 | ||
| Scheme 1 Different structures for the benzene dimer. | ||
Once such π-complexes get ionized, covalent interactions come into play (see below), and these make that the bonding becomes much stronger. The radical cation of the benzene dimer, 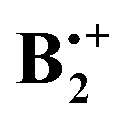 , has also a long history of studies which began with the groundbreaking work of Badger and Brocklehurst who proposed for the first time that the strong, broad NIR bands that had been observed in ionized frozen solutions of benzene (and other planar π-systems) are due to dimer radical cations.7 They also provided a theoretical framework to explain the occurrence of these bands, which they termed “charge resonance (CR) transitions”, and they discussed for the first time possible structures of such dimer cations.8 Later, Nishi et al. observed these CR transitions also upon photoionization in liquid benzene,9 and in gas-phase photodissociation spectra.10 Also in the gas phase, the dissociation enthalpy of
, has also a long history of studies which began with the groundbreaking work of Badger and Brocklehurst who proposed for the first time that the strong, broad NIR bands that had been observed in ionized frozen solutions of benzene (and other planar π-systems) are due to dimer radical cations.7 They also provided a theoretical framework to explain the occurrence of these bands, which they termed “charge resonance (CR) transitions”, and they discussed for the first time possible structures of such dimer cations.8 Later, Nishi et al. observed these CR transitions also upon photoionization in liquid benzene,9 and in gas-phase photodissociation spectra.10 Also in the gas phase, the dissociation enthalpy of 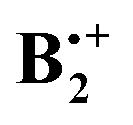 was determined to be 20.6 ± 1 kcal mol−1, i.e. it is much higher than that in the neutral benzene dimer.11
was determined to be 20.6 ± 1 kcal mol−1, i.e. it is much higher than that in the neutral benzene dimer.11
This due to the fact that π-dimer cations are bound by substantial covalent forces that may be regarded as resulting from the interaction of the singly occupied MO of an ionized molecule with the doubly occupied MO of its neutral counterpart, which leads eventually to a symmetric distribution of the spin and charge over both moieties. The extent to which this binding comes to bear in 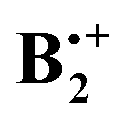 depends on its geometry, and the literature records several computational studies which address this question, studies which have been summarized recently by Krylov et al.12 (ref. 41–47). Similar structures as that of neutral B2 were considered and, depending on the method that was applied, displaced sandwich or T-shaped structures were found to be the most stable ones. However, in addition to the studies cited in ref. 12, a structure bound by a weak one-electron σ-bond between two C-atoms (labelled SH in Scheme 1) had been postulated in a study where the formation of
depends on its geometry, and the literature records several computational studies which address this question, studies which have been summarized recently by Krylov et al.12 (ref. 41–47). Similar structures as that of neutral B2 were considered and, depending on the method that was applied, displaced sandwich or T-shaped structures were found to be the most stable ones. However, in addition to the studies cited in ref. 12, a structure bound by a weak one-electron σ-bond between two C-atoms (labelled SH in Scheme 1) had been postulated in a study where the formation of 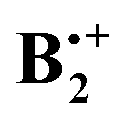 from ionized o,o′-dibenzene was studied.13
from ionized o,o′-dibenzene was studied.13
Pieniazek et al. have recently investigated in great detail the molecular and electronic structure of 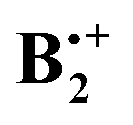 ,12,14 based on a variant of the equation-of-motion coupled cluster method. Within the C2h subspace they located two minima which correspond to structures XD and YD and a C2v structure that corresponds to TH in Scheme 1.14 They noted in particular that in structures XD and YD charge and spin are evenly distributed over the two fragments, whereas in the structure TH (which, according to their calculations, lies ca 6 kcal mol−1 higher in energy than the former ones), ca. 88% of the charge sits on the “stem” and only 12% on the “top” fragment. Consequently, in such TH-type structures, the charge resonance transition turns into a charge transfer transition, which occurs at a much lower energy and with a much lower oscillator strength.
,12,14 based on a variant of the equation-of-motion coupled cluster method. Within the C2h subspace they located two minima which correspond to structures XD and YD and a C2v structure that corresponds to TH in Scheme 1.14 They noted in particular that in structures XD and YD charge and spin are evenly distributed over the two fragments, whereas in the structure TH (which, according to their calculations, lies ca 6 kcal mol−1 higher in energy than the former ones), ca. 88% of the charge sits on the “stem” and only 12% on the “top” fragment. Consequently, in such TH-type structures, the charge resonance transition turns into a charge transfer transition, which occurs at a much lower energy and with a much lower oscillator strength.
However, in the above studies of the USC group, the σ-hemibonded structure which had been found to represent a minimum energy structure on DFT and CASSCF potential surfaces by Schroeter et al.13 was not considered, probably because Pieniazek et al. started from the D6h sandwich structure in which they gradually displaced the two constituent benzene rings in the x- and the y-direction, and stopped when they reached the first potential energy minimum.
This raised the question how this σ-hemibonded structure SH relates to the XD and YD structures found by Pieniazek et al., which are also bound by ca. 20 kcal mol−1. While investigating this problem, we discovered that 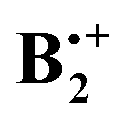 exists actually in the form of different “electromers”,15i.e. structures which differ in the nature (and, in this case, the symmetry) of their wavefunction and cannot be readily interconverted, at least along symmetry-preserving pathways. It is the purpose of the present work to report on these findings which shed new light on the nature of this archetypal π-dimer cation.
exists actually in the form of different “electromers”,15i.e. structures which differ in the nature (and, in this case, the symmetry) of their wavefunction and cannot be readily interconverted, at least along symmetry-preserving pathways. It is the purpose of the present work to report on these findings which shed new light on the nature of this archetypal π-dimer cation.
Methods
To model π-dimer radical cations one should choose a method that allows to account for correlation effects which prevail over the intermolecular distances in such complexes (ca. 2.5 Å). Pure or hybrid DFT methods are very good at modelling short-range (or local) correlation, whereas the MP2 method accounts much better for mid-range correlation effects such as they prevail in van-der-Waals complexes. Finally, it has been found that long-range dispersion effects are best modelled by classical correction terms. The B2PLYP method of Grimme,16 supplemented by corrections for dispersive interactions (-D),17,18 has been designed to judiciously mix the three above techniques,19 and has given very good results in different benchmark studies, some of which also include dimer cations of the type that are investigated in the present paper.20 Also, in a recent study of the indole dimer cation, the B2PLYP method proved to be the only one which gave results in reasonable accord with experiment.21Therefore we chose this method to explore the potential surface of 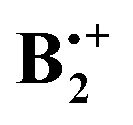 , together with the cc-pVDT basis set (in DFT calculations, going beyond DZ basis sets usually does not improve results significantly).18 As a check, we repeated the above calculations with the range-separated dispersion-corrected wB97X-D functional,22 which is also well suited to study dimer cations. Finally, the stationary points were subjected to single-point calculations by the orbital-optimized spin-component scaled MP2 method (OO-SCS-MP2),23 using the resolution of identity (RI) approximation, and by the G4 method.24 Geometry optimizations and G4 calculations were performed with the Gaussian 09 package of programs,25 while the OO-SCS-MP2 calculations were done with the ORCA program.26
, together with the cc-pVDT basis set (in DFT calculations, going beyond DZ basis sets usually does not improve results significantly).18 As a check, we repeated the above calculations with the range-separated dispersion-corrected wB97X-D functional,22 which is also well suited to study dimer cations. Finally, the stationary points were subjected to single-point calculations by the orbital-optimized spin-component scaled MP2 method (OO-SCS-MP2),23 using the resolution of identity (RI) approximation, and by the G4 method.24 Geometry optimizations and G4 calculations were performed with the Gaussian 09 package of programs,25 while the OO-SCS-MP2 calculations were done with the ORCA program.26
Another methodological question which needs to be addressed in such a study is whether corrections for the basis-set superposition error (BSSE) should be made. Using methods proposed by Dannenberg et al. this can be done, by means of the counterpoise (CP) correction scheme,27 already at the level of the geometry optimization,28 or then in single-point calculations at geometries optimized without CP corrections. After some trials we found that accounting for CP corrections during geometry optimization affected only the interring distance,29 but did not change the nature of stationary points or their relative energies. As CP corrections made it impossible to maintain any molecular symmetry in geometry optimizations we decided to forgo their use in that step. Of course they are needed in predicting correct (absolute) binding energies of dimer cations, which is however not the purpose of this work.
Results and discussion
The benzene radical cation, B•+, is a textbook case of a Jahn–Teller active system.30 By virtue of the degeneracy of the HOMO of D6h benzene, B•+ distorts spontaneously to two structures of D2h symmetry, essentially by elongation or compression, respectively, of two opposite bonds. The two structures have different singly occupied MOs (SOMOs) and hence the two structures represent states of different symmetry (see top of Scheme 2).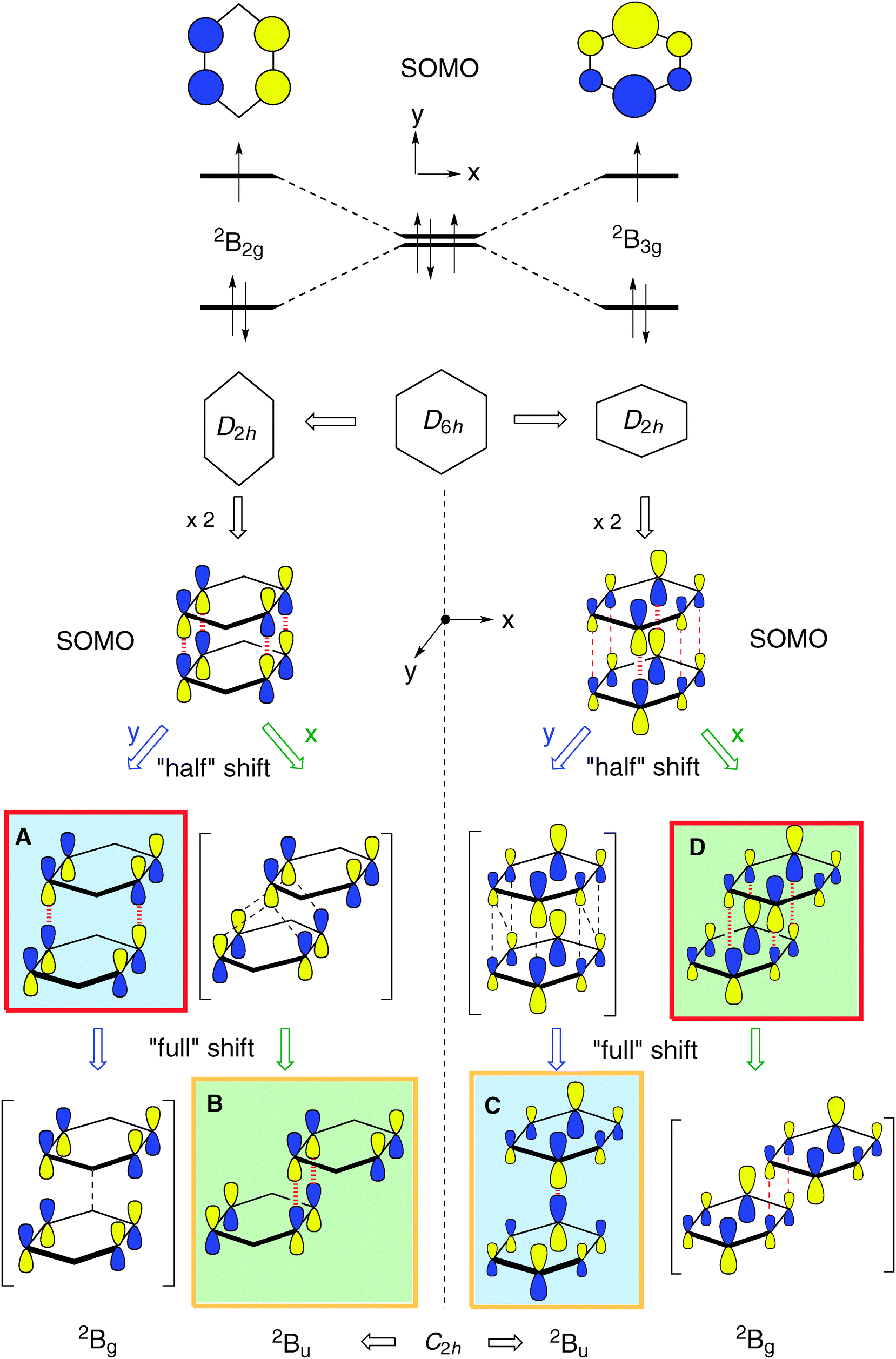 | ||
| Scheme 2 Jahn–Teller distortion of the benzene radical cation (top), formation of dimer radical cations of D2h symmetry (middle), and their distortion to structures of C2h symmetry where the two rings are displaced along the y- (blue arrows and fields) or the x axis (green arrows and fields). The framed structures are those where the highest amount of bonding is expected to prevail (red: 2Bg, orange: 2Bu states). | ||
As shown by Pieniazek et al.14 this pattern of distortions persists in 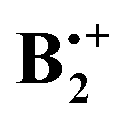 which therefore also comes in pairs of states (2B2g and 2B3g, respectively, in D2h symmetry, if the axes are chosen as in Scheme 2). As the amount of covalent stabilization of dimer radical cations is expected to be highest for structures where the SOMOs show optimal overlap, it would seem that “perfect” D2h sandwich structures should represent the most stable geometries. However, as in neutral B2, all calculations find that these structures undergo spontaneous distortions by displacement of the two rings along the x- or the y-axis (if the z-axis is perpendicular to the rings), which leads, in a first approximation, to geometries of C2h symmetry.
which therefore also comes in pairs of states (2B2g and 2B3g, respectively, in D2h symmetry, if the axes are chosen as in Scheme 2). As the amount of covalent stabilization of dimer radical cations is expected to be highest for structures where the SOMOs show optimal overlap, it would seem that “perfect” D2h sandwich structures should represent the most stable geometries. However, as in neutral B2, all calculations find that these structures undergo spontaneous distortions by displacement of the two rings along the x- or the y-axis (if the z-axis is perpendicular to the rings), which leads, in a first approximation, to geometries of C2h symmetry.
In the lower half of Scheme 2 we show what happens when the two benzene rings in the 2B2g and 2B3g states of D2h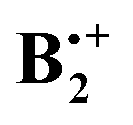 shift by ca. half or by the full length (y-direction) or width (x-direction), respectively, of a benzene ring. Thus one can see that in the 2B2g state a “half shift” along the y-axis leads to a situation of good overlap of the two B2g MOs (cf. red dashed lines), whereas a “full shift” leads to no net bonding (cf. black dashed lines). A similar situation of good overlap prevails when the 2B3g state undergoes a “half shift” along the x-axis (except that in this case a small amount of overlap persists in the fully shifted structure). Conversely half an x-shift in the 2B2g state leads to no net overlap, because the bonding and the antibonding contributions cancel, whereas the full-width shift leads again to similarly good overlap as the half y-shift does. A similar situation prevails for the y-displacement in the 2B3g state, where a half shift leads to no net overlap while a full shift leads to overlap between the centers with the big coefficients (note that in Scheme 2 the SOMO's of the dimer cation are shown, which correspond to the antibonding combination of the benzene HOMOs).
shift by ca. half or by the full length (y-direction) or width (x-direction), respectively, of a benzene ring. Thus one can see that in the 2B2g state a “half shift” along the y-axis leads to a situation of good overlap of the two B2g MOs (cf. red dashed lines), whereas a “full shift” leads to no net bonding (cf. black dashed lines). A similar situation of good overlap prevails when the 2B3g state undergoes a “half shift” along the x-axis (except that in this case a small amount of overlap persists in the fully shifted structure). Conversely half an x-shift in the 2B2g state leads to no net overlap, because the bonding and the antibonding contributions cancel, whereas the full-width shift leads again to similarly good overlap as the half y-shift does. A similar situation prevails for the y-displacement in the 2B3g state, where a half shift leads to no net overlap while a full shift leads to overlap between the centers with the big coefficients (note that in Scheme 2 the SOMO's of the dimer cation are shown, which correspond to the antibonding combination of the benzene HOMOs).
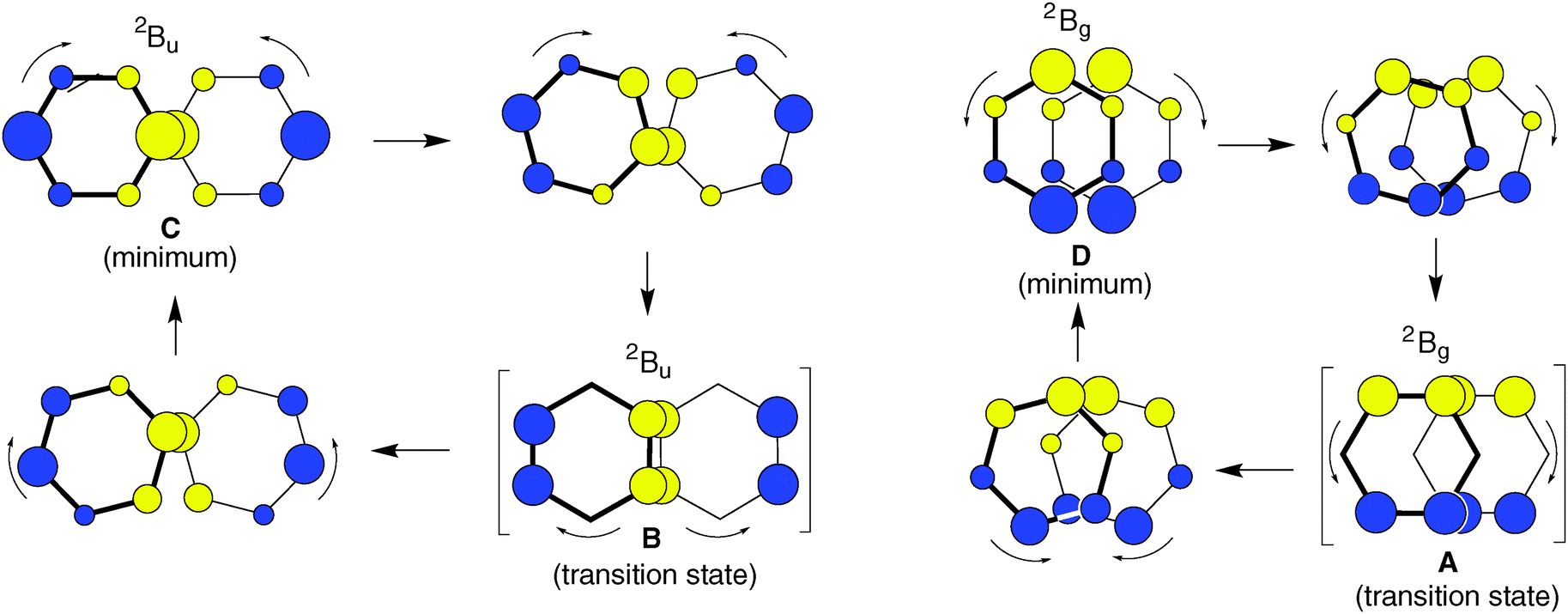
As shown at the bottom of Scheme 2, these distortions are expected to lead to four different structures (y-shifted in blue, x-shifted in green boxes) which have pairwise different state symmetries in C2h (red-framed: 2Bg, orange-framed: 2Bu). To facilitate the discussion, we will henceforth label these structures A–D as indicated in Scheme 2. Note that in the C2h structures that result from x- displacement the y-axis turns into the C2 (i.e. z-) axis, whereas on y-displacement, it is the x-axis which becomes the z-axis (while the x- and y-axes define the horizontal symmetry plane). It becomes evident from Scheme 2 that the half- and fully y-displaced structures A and C, as well as the two x-displaced structures D and B cannot be interconverted along the respective displacement coordinate, because such an interconversion would imply a crossing from a 2Bg to a 2Bu state or vice versa, which cannot be accomplished while maintaining C2h symmetry (the corresponding correlation diagrams and the potential energy curves, which illustrate this, are shown in the ESI†). Thus, 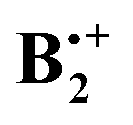 seems to exist in the form of two “electromers”.
seems to exist in the form of two “electromers”.
On the basis of the qualitative considerations illustrated in Scheme 2 we examined whether the above structures A–D represent stationary points on the 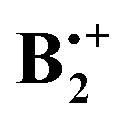 potential energy surface, and, if yes, what type of stationary points those would be. Thereby we found that, on the B2PLYP-D/cc-pVDZ potential surface, only structures C and D, which are derived from the B2g state of the perfect sandwich dimer cation, represent minima, whereas structures A and B, which arise from the corresponding B3g state, are transition states for the automerization of D and C, respectively, by rotation of the two benzene rings in the opposite sense relative to each other, as shown in a simplified fashion in Scheme 3 for the two cases of the “fully shifted” and the “half shifted” structures.31
potential energy surface, and, if yes, what type of stationary points those would be. Thereby we found that, on the B2PLYP-D/cc-pVDZ potential surface, only structures C and D, which are derived from the B2g state of the perfect sandwich dimer cation, represent minima, whereas structures A and B, which arise from the corresponding B3g state, are transition states for the automerization of D and C, respectively, by rotation of the two benzene rings in the opposite sense relative to each other, as shown in a simplified fashion in Scheme 3 for the two cases of the “fully shifted” and the “half shifted” structures.31
 | ||
Scheme 3 Facile interconversion of pairs of 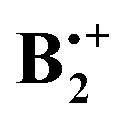 structures by opposite rotation of the two benzene rings relative to each other.31 structures by opposite rotation of the two benzene rings relative to each other.31 | ||
Scheme 3 also illustrates how the SOMOs of the two states change smoothly and continuously, i.e. without any change of the nodal properties of the SOMO, along the reaction coordinate. Thus it is not surprising that the pairs of structures which have the same symmetry in C2h (A and D: Bg, B and C: Bu) lie within 1–2 kcal mol−1, i.e. the potential surface for their interconversion is extremely flat. This is in contrast to the interconversions of the Bg and the Bu states, by small displacements of the two rings along the y- (A → C) or the x-axis (B → D), which involves a complete change of the nodal structure of the SOMOs. Such transformations usually give rise to much higher barriers (cf. Fig. S1–S4 in the ESI†).
Thus it seems that the benzene dimer cation can move on two separate and distinct potential surfaces which do not intersect within the energies that are required for the automerization of the two states. It turns out that the two different 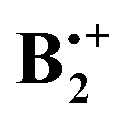 minima which have been located independently by two groups12,13 are in fact both correct structures, but they belong to different electronic states, i.e. they can be called “electromers”.
minima which have been located independently by two groups12,13 are in fact both correct structures, but they belong to different electronic states, i.e. they can be called “electromers”.
At the suggestion of a reviewer, we repeated the above calculations using a different method that is also suitable to treat dimer cations, i.e. the range-separated wB97X-D functional, augmented by a classical dispersion correction.22 The results we obtained by this method do not differ significantly from those we had obtained by B2PLYP-D, except that structure D is no longer a stationary point in C2h symmetry. A very slight distortion leads to a C2 transition state, while the connected minimum lies halfway between structures D and A (the latter also being a transition state). However all these structures lie within 0.12 kcal mol−1, so the surface is as flat as the B2PLYP-D one is. Structure B has three imaginary modes in C2h symmetry, but unfortunately we were unable to find the transition state that connects the minima C on the wB97X-D surface. The energies of structures B and C relative to A and D are, however, very similar to those obtained by B2PLYP-D (see Table 1). The detailed results of the wB97X-D calculations are given in the ESI.†
| Structure | B2PLYP-D/cc-pVDZ18 | wB97X-D/cc-pVDZ | G4a | OO-SCS-MP2/cc-pvTZa |
|---|---|---|---|---|
| a Geometries optimized and stationary points characterized by the B2PLYP-D/cc-pVDZ method. b With this method, the minimum D lies halfway between D and A, while the structure D itself is a transition state. | ||||
| D (min) | (0) | (0)b | 0.89 | 0.21 |
| A (TS) | 0.05 | 0.12 | 0 | 0 |
| C (min) | 2.29 | 2.54 | 4.44 | 3.13 |
| B (TS2)31 | 3.08 | 3.70 | 3.90 | 4.70 |
Finally, we wanted to assess whether high-level wavefunction-based methods would confirm the results that were obtained by the two DFT methods. First we chose the G4 composite method which effectively accounts for most of the dynamic correlation energy,32 but we skipped the B3LYP/6-31G(2df,p) geometry reoptimization step that is part of this protocol, because we thought that the B2PLYP-D method accounts better for the mid- and long range correlation effects on the geometry of 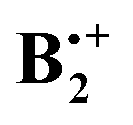 than the B3LYP method, which is part of the G4 protocol, does. At the suggestion of a reviewer we added the results obtained by the orbital-optimized MP2 method (see Methods section). In Table 1 we list the relative energies obtained by the four methods that we applied to above structures A–D. Note that the thermal corrections to the enthalpy that are obtained from the B2PLYP-D frequencies are nearly identical for all four structures. We refrain from quoting free energy differences because, due to the six low-frequency vibrations which involve movements of the two rings in
than the B3LYP method, which is part of the G4 protocol, does. At the suggestion of a reviewer we added the results obtained by the orbital-optimized MP2 method (see Methods section). In Table 1 we list the relative energies obtained by the four methods that we applied to above structures A–D. Note that the thermal corrections to the enthalpy that are obtained from the B2PLYP-D frequencies are nearly identical for all four structures. We refrain from quoting free energy differences because, due to the six low-frequency vibrations which involve movements of the two rings in 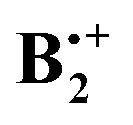 relative to each other, calculated vibrational entropies are associated with potentially large errors.
relative to each other, calculated vibrational entropies are associated with potentially large errors.
The relative ordering of the four states/structures is not the same by all four methods, and the G4 and OO-SCS-MP2 energies of the transition states A lie actually below those of the connected minima D obtained by the DFT methods. However, in view of the extreme flatness of the potential surface that connects A to D, this is not surprising, and also not really relevant, in view of the extreme fluxionality of the benzene dimer cation. G4 also inverts structures C and B but again, this does not affect the conclusion that the surfaces for the interconversion of the two Bg states and that for the two Bu states, respectively, are both very flat, with the latter lying only ca. 3–4 kcal mol−1 above the former.
At the stationary points A–D the Bg and the Bu states are separated by nearly 1 eV (cf. the correlation diagrams and potential surfaces shown in the ESI†), so one is an excited state of the other. At some point on the x- or y-displacement coordinate, respectively, between the “half shifted” and the “full shifted” structures, the two states cross at conical intersections below which the transition states for the interconversion of the two states must lie. However, at these transition states, the unpaired electron must be localized on one of the two benzene rings, so the AO-coefficients of the SOMO on the other ring can go to zero and re-emerge with opposite signs in the other state. Such a localization is energetically quite costly and thus the transition states for the interconversion of the two states lie much higher than the very flat potential surfaces of either of the two states while they undergo the opposite rotation of the two benzene rings (cf.Scheme 3). Thus, the benzene dimer cation can probably even move on the higher-lying of these two surfaces (the one comprising the “fully shifted” structures C and B), without falling onto the lower-lying one (comprising the “half-shifted” structures A and D), which is quite an exceptional situation.
Conclusion
We conclude that the benzene dimer cation, like its neutral homologue, is an extremely fluxional species and that discussing it in terms of stationary points on the potential energy surface, as it has usually been done up to now, makes little practical sense. However, it is conceptually insightful to dissect this potential surface into regions of different state symmetries, as it is done in Scheme 2 because this allows to demonstrate that the benzene dimer cation may move on two distinct potential surfaces which, according to our calculations, are separated by only 3–4 kcal mol−1 but that do not intersect while the complexes are moving on these flat surfaces, because they are associated with states that belong to different irreducible representations in C2h symmetry (2Bg and 2Bu, respectively), and whose singly occupied MOs have therefore different nodal properties. To our best knowledge, this is the first example of such an electronic structure. To interconvert the two states requires full localization of the unpaired electron on one of the two benzene rings, so the AOs of the SOMO on the other ring can change signs. As the charge-resonance transitions of both states are very similar, time-resolved spectroscopic techniques would probably be needed to find experimental proof for the existence of the two close-lying states/surfaces that are predicted by our calculations.Acknowledgements
This work is part of project No. 200020-143410 of the Swiss National Science Foundation.References
- S. E. Wheeler and J. W. G. Bloom, J. Phys. Chem. A, 2014, 118, 6133 CrossRef CAS PubMed.
- A. Mukherjee, R. Lavery, B. Bagchi and J. T. Hynes, J. Am. Chem. Soc., 2008, 130, 9747 CrossRef CAS PubMed.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaroa, Eur. J. Med. Chem., 2014, 74, 95 CrossRef CAS PubMed.
- T. Janowski and P. Pulay, Chem. Phys. Lett., 2007, 447, 27 CrossRef CAS PubMed.
- M. Pitoňák, P. Neogrády, J. Řezáč, P. Jurečka, M. Urban and P. Hobza, J. Chem. Theory Comput., 2008, 4, 1829 CrossRef.
- R. Podeszwa, R. Bukowski and K. Szalewicz, J. Phys. Chem. A, 2006, 110, 10346 CrossRef PubMed.
- B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1969, 65, 2582 RSC.
- B. Badger and B. Brocklehurst, Trans. Faraday Soc., 1969, 66, 2939 RSC.
- Y. Inokuchi, Y. Naitoh, K. Ohashi, K. Saitow, K. Yoshihara and N. Nishi, Chem. Phys. Lett., 1997, 269, 298 CrossRef CAS.
- K. Ohashi, Y. Inokuchi and N. Nishi, Chem. Phys. Lett., 1996, 263, 167 CrossRef CAS , and references cited therein.
- K. Hiraoka, S. Fujimaki, K. Aruga and S. Yamabe, J. Chem. Phys., 1991, 95, 8413 CrossRef CAS PubMed.
- P. A. Pieniazek, A. I. Krylov and S. E. Bradforth, J. Chem. Phys., 2007, 127, 044317 CrossRef PubMed.
- K. Schroeter, D. Schröder, H. Schwarz, G. D. Reddy, O. Wiest, C. Carra and T. Bally, Chem. – Eur. J., 2000, 6, 4422 CrossRef CAS.
- P. A. Pieniazek, S. E. Bradforth and A. I. Krylov, J. Chem. Phys., 2008, 129, 074104 CrossRef PubMed.
- T. Bally, Nat. Chem., 2010, 2, 165 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Phys., 2006, 124, 034108 CrossRef PubMed.
- T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397 RSC.
- At the time when we explored the potential surfaces, the only classical dispersion correction which had been parametrized for B2PLYP and implemented in the Gaussian program was Grimmes D2 method. Meanwhile the D3 correction has also been implemented, so we performed single-point calculation with B2PLYP-D3, employing also the cc-pVTZ basis set (see ESI†). However, the relative energies of the investigated structures changed by less than 0.1 kcal mol−1, so we do not think that the potential surfaces would look different if computed by this more recent method.
- T. Schwabe and S. Grimme, Acc. Chem. Res., 2008, 41, 569 CrossRef CAS PubMed.
- S. N. Steinmann and C. Corminboeuf, J. Chem. Theory Comput., 2012, 8, 4305 CrossRef CAS.
- A. Błoch-Mechkour, T. Bally and A. Marcinek, J. Phys. Chem. A, 2011, 115, 7700 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- S. Kossmann and F. Neese, J. Phys. Chem. A, 2010, 114, 11768 CrossRef CAS PubMed.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed, full citation given in the ESI†.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- S. Simon, M. Duran and J. J. Dannenberg, J. Chem. Phys., 1996, 105, 11024 CrossRef CAS PubMed.
- As the BSSE introduces a nonphysical attraction between the two units, the CP correction generally makes that intermolecular complexes have slightly longer intermolecular distances than predicted by optimization witout CP correction (see, e.g. N. Kobko and J. J. Dannenberg, J. Phys. Chem. A, 2001, 105, 1944 CrossRef CAS ).
- B. E. Applegate and T. A. Miller, J. Chem. Phys., 2002, 117, 10654 CrossRef CAS PubMed.
- Actually, the reaction paths for automerization of structures B and D imply, next to the rotation shown in Scheme 3, motions of the two benzene rings along the viewing axis which cannot be easily visualized in a schematic fashion. In the end the minima and transition states are linked by pathways which imply minimal displacements of the two benzene rings. Furthermore it must be mentioned that, at the B2PLYP-D level, structure B maintains, in addition to the direction of negative curvature (frequency: −37.7 cm−1) that corresponds to the rotation of the two benzene rings a very small additional negative frequency (−9.3 cm−1) that we found impossible to get rid of, even by slight deformations and tight optimization. At the wB97X-D level, structure B has three directions of negative curvature. Unfortunately we were unable to find the transition state that corresponds to B at this level, so the energy of this structure relative to the others must be regarded as an upper limit.
- L. A. Curtiss, P. C. Redfern and K. Rahavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
Footnote |
† Electronic supplementary information (ESI) available: Full citation of the Gaussian program.25 Correlation diagrams and corresponding potential surfaces for the interconversion of the two states along the x- and the y-displacement coordinates, respectively (calculated at the B3LYP/6-31G* level). Cartesians and total energies of all stationary points located on the B2PLYP-D and the wB97X-D potential energy surface of 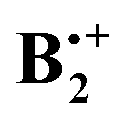 , and total energies obtained by the G4 and the OO-SCS-MP2 method. See DOI: 10.1039/c4cp05784h , and total energies obtained by the G4 and the OO-SCS-MP2 method. See DOI: 10.1039/c4cp05784h |
| This journal is © the Owner Societies 2015 |