
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Buckybowl superatom states: a unique route for electron transport?†
L.
Zoppi
a,
L.
Martin-Samos
bc and
K. K.
Baldridge
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, Zurich, CH-8057, Switzerland. E-mail: kimb@oci.uzh.ch
bUniversity of Nova Gorica, Materials Research Laboratory, vipavska cesta 11C, Ajdovscina 5270, Slovenia
cDEMOCRITOS, Istituto Officina dei Materiali, c/o SISSA Scuola Internazionale Superiore di Studi Avanzati, Via Bonomea 265, 34136 Trieste, Italy
First published on 3rd February 2015
Abstract
A unique paradigm for intermolecular charge transport mediated by diffuse atomic-like orbital (SAMOs), typically present in conjugated hollow shaped molecules, is investigated for C20H10 molecular fragments by means of G0W0 theory. Inclusion of many body screening and polarization effects is seen to be important for accurate prediction of electronic properties involving these diffuse orbitals. Theoretical predictions are made for the series of bowl-shaped fullerene fragments, C20H10, C30H10, C40H10, C50H10. Interesting results are found for the LUMO–SAMO energy gap in C20H10, which is shown to be nearly an order of magnitude lower that that determined for C60. Given the ability to support bowl fragments on metal surfaces, these results suggest the concrete possibility for exploiting SAMO-mediated electron transport in supramolecular conducting layers.
Introduction
Renewed interest in the use of π-conjugated molecules as components in nanoscale electronics and optoelectronic devices1 has motivated recent efforts to construct single-molecule junctions that optimize transport properties.1d,2 Aromatic conjugated fragments are appealing for use in device technology due to their low density, structural stability, and extended-delocalized π networks that support mobile charge carriers.3 As conductor dimensions approach the nanoscale, design principles focus towards producing molecules with tunable functionality4 to enable control of charge transport at the molecular scale.1c,d As such, the need has emerged for improved understanding of the details involved in optimization and control of electronic transport phenomena in these systems.The extended family of curved aromatics based on the smallest bowl-shaped fullerene fragment, corannulene, C20H103,5 (Fig. 1(a)), are key targets of interest. Much effort has been extended towards the design of functionalized building blocks based on this fragment, focusing on tailoring electronic properties such as electrical and optical band gaps.6 Assembled in the solid state, these bowl fragments provide an array of materials supported in varying complex environments, which can be exploited as active molecular layers in optoelectronic applications,7 aggregated as monolayers on metallic surfaces for work-function engineering,8 or, as single molecules in junctions for transport processes (Fig. 1).9
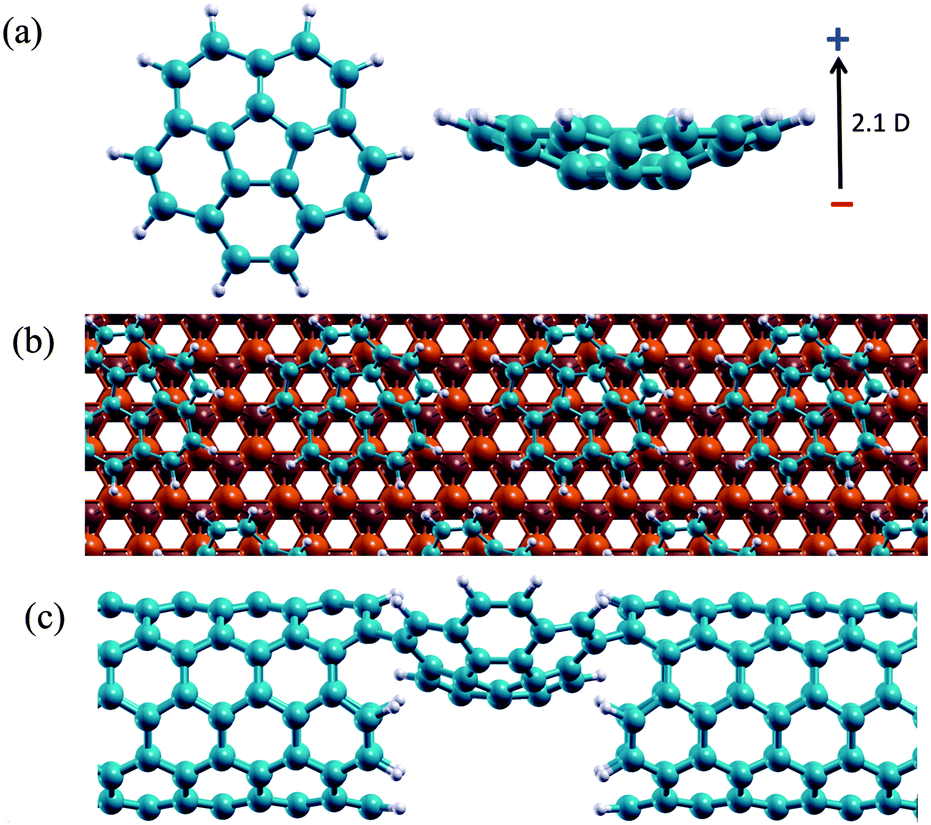 | ||
| Fig. 1 (a) C20H10 molecular structure showing the shallow bowl and intrinsic dipole; (b) C20H10 molecules arranged on a Cu(111) surface; (c) C20H10 molecule assembled into a molecular junction. | ||
Importantly, materials based on curved or hollow aromatic complexes enable exploitation of a unique mechanism of intermolecular charge transport distinct from the conventional mechanism involving tightly bound π molecular orbital overlap. Key to these electron transport routes is evidence of a characteristic set of diffuse molecular orbitals called Super Atomic Molecular Orbitals, SAMOs,10 first investigated in the closed hollow aromatic molecule, C60.10,11 SAMOs are virtual orbitals that arise from the central potential of the hollow molecular core, evoking well-defined hydrogen-like s, p, and d orbital angular momentum shapes. These orbitals extend well beyond the more tightly bound π orbitals, and are also distinct from conventional Rydberg type orbitals that see the core as a point charge.11b
The significance of SAMOs lies in the possibility for exploitation of the nearly free conducting channels, which arise when the molecular units are assembled in series in quantum nanostructures or solids.11c Such channels have been observed in low temperature scanning tunneling microscopy experiments (LT-STM) in the case of C60 molecules assembled on noble metal surfaces.10 Unfortunately, prospects for exploiting the SAMOs in practical applications must be tempered by the fact that typically these orbitals are unoccupied. In the case of C60, the SAMOs lie several eV above the lowest unoccupied molecular orbital, LUMO, and therefore are difficult to exploit for this purpose (Fig. 2).10
 | ||
| Fig. 2 DFT-LDA calculated SAMOs, illustrating the typical hydrogen-like s, p, and d symmetric shapes in C60. | ||
Taking a slightly different view from typically studied spheroid fullerenes, the present work details a theoretical investigation of the electronic properties of SAMOs in a series of curved shaped aromatic constructs, C20H10, C30H10, C40H10, C50H10, with focus on their suitability for applications in molecular circuits (electron transport). The intriguing electronic properties of the parent system, C20H10, can be ascribed to its large intrinsic dipole moment (2.01 D) and shallow bowl depth (0.87 Å),12 which imparts dynamic properties characterized by a bowl-to-bowl inversion barrier of 11.5 kcal mol−1.12 The ability to control curvature (and therefore reactivity and properties) through functionalization of the rim,12 together with the possibility of assembling these building blocks in layers on metallic surfaces,8,13 strongly motivates their creative use as materials for supramolecular conducting layers.
Results and discussion
SAMOs: theoretical description
The physical origin of SAMOs is ascribed to many-body screening and polarization effects, typical of a polarizable assembly (e.g., graphene) that undergoes a topological distortion, such as wrapping or rolling into a nanotube or fullerene.14 At a solid–vacuum interface, these many body interactions give rise to a series of degenerate image potential (IP) states in the near-surface region on both sides of a graphene sheet, which float above and below the molecular plane and undergo free motion parallel to it.14 Topological distortion of the molecular sheet breaks symmetry, lowering/raising the energy of the IP states on the concave/convex side of the resulting material, revealing SAMOs.11bTheoretical investigation of SAMOs requires methods that properly include polarization and correlation effects, which are crucial for describing image-potential states and are typically poorly described with conventional density functional theory (DFT) approaches15 due to the approximate nature of the exchange and correlation (XC) potential.16 In order to capture these effects, the choice of the methodology falls to many body perturbation theory (MBPT)17 electronic structure approaches within the GW scheme.18 In this approximation,17,18 the self-energy, Σ, is the product of a single-particle Green function, G, and a nonlocal and dynamically screened Coulomb potential, W, (Σ = iGW). Due to the high computational demands of fully self-consistent GW (scGW), a range of perturbative GW schemes, from non self-consistent to partially self-consistent have emerged.19 The lowest rung in this hierarchy, standard in practical calculations, is a non-self-consistent scheme, i.e. G0W0,19,20 where the quasi particle (QP) energies are obtained as a perturbative first-order correction to the DFT eigenvalues. It is important to note that GW theory is an approximation for GWΓ theory (former work of Hedin and Lunquist).18 It is not straightforward that full scGW could provide better QP energies than any perturbative GW scheme.
The use and appropriateness of the GW method for prediction of image potential states has raised some skepticism in the QM community. However, as well documented in the literature, we also strongly believe that the GW methodology is suitable to describe many-body screening and long-range polarization effects crucial for treating image-potential states.21 A further criticism can derive from the use of DFT wavefunctions for the purpose of SAMO description. However, differences between the Kohn and Sham and real QP wavefunction come from the incorrect long-range behavior of approximate exchange potentials in conventional DFT schemes, which scarcely effects the GW-corrected QP energies.22 In this respect, it is worth noting that the first theoretical investigation of SAMOs, successfully compared to experiments, was performed within a DFT based scheme.10
Unfortunately, the non-self-consistency in G0W0 can give rise to a dependence of the resulting QP spectra from the starting DFT functional, as recently documented in investigations of QP valence spectra of molecules.19,23 Recent efforts are being directed at the performance of G0W0 schemes across specific benchmark sets of molecules used in organic electronic devices.19,23a Although still controversial, emerging from these investigations is the strategy of including a fraction of exact-exchange (EXX) in hybrid-functionals to mitigate the self-interaction error (SIE), thereby providing an improved starting point for G0W0 calculation.19,23c For example, a recent investigation involving azabenzenes showed good agreement with photoemission experiments with this strategy.19,23c Such studies are still in their infancy, however, with available data primarily involving QP valence spectra, whereas predictions involving the virtual unoccupied space still motivates further investigation.
A second controversial point concerning appropriate methodology for investigation of SAMOs derives from the fact that some QM-based methods (e.g., equation of motion (EOM) methodology24) include higher order correlation than G0W0. However, such approaches are based on series expansions in terms of the bare Coulomb potential and therefore require higher order in the diagrammatic expansion to be able to capture the correlation effects. The GW methodology, on the other hand, is based on a series expansion in terms of a screened Coulomb potential, with the polarizability calculated within RPA and the screening through the inversion of a Dyson equation, which by definition, includes an infinite number of diagrams.18a As such, results coming from QM methods are quite sensitive to the order in the diagrammatic expansion, whereas such sensitivity does not appear in the GW approach. Moreover, the GW method is highly predictive with less computational effort even for systems with high dielectric constants.
In the present work, a customized hybrid methodology is exploited, using standard B97D/Def2-TZVPP25 (GAMESS)26 DFT for full optimization and Hessian characterization of structures, followed by plane-wave DFT formalism (Quantum-ESPRESSO)27 within MBPT17 in the GW approximation18 (SAX).28 This method is applied to a series of C20H10 based systems to investigate and compare SAMOs and SAMO electronic properties. In addition, the performance dependence of the G0W0 scheme on the starting functional for the GW calculation is compared using both a DFT-PZ functional29 (G0W0@LDA) as well as a hybrid PBE0 functional30 (G0W0@PBE0) that includes 25% exact exchange (EXX) (for additional details see ESI†).
SAMOs in C20H10
Calculated SAMOs of corannulene are depicted in Fig. 3. As has been shown for the closed shaped hollow C60 structure, corannulene also manifests the characteristic diffuse molecule-centered hydrogenic-like s, p, and d shapes. To be relevant for charge transport, the SAMO derived states should cross the Fermi level in any resulting material. To facilitate this characteristic, the lowest energy SAMO of the isolated molecule should lie close to the LUMO. Thus, the ability to control the LUMO and SAMOs gap of lowest energy in a molecular system becomes quite important.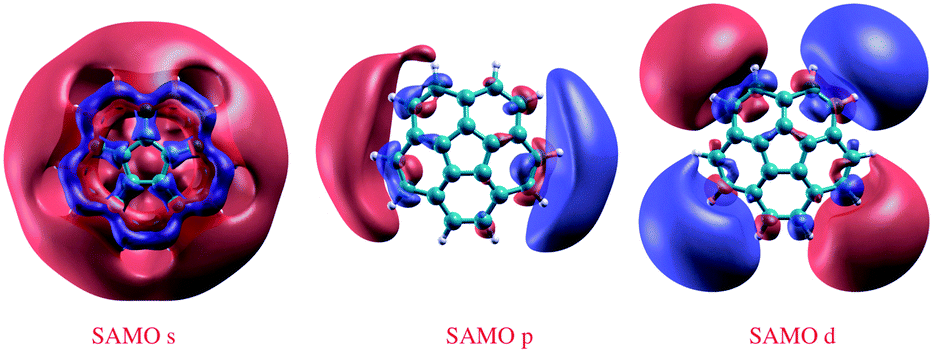 | ||
| Fig. 3 DFT-LDA 3D representation of SAMOs illustrating the typical s, p, and d-like symmetric shapes in corannulene, C20H10. | ||
G 0 W 0 as well as DFT predictions of ΔESAMO–LUMO for C20H10 are summarized in Table 1. As in C60, SAMOs are revealed in the unoccupied part of the spectrum in a simple DFT calculation (LDA or PBE0). Inclusion of many-body effects at the G0W0 level provides a more accurate positioning of SAMO levels with respect to conventional DFT predictions, but still is dependent on the starting functional. Initialization of the GW calculation from a standard DFT-PZ functional (G0W0@LDA) results in SAMO level of s-type symmetry that corresponds to the 1st unoccupied level after the LUMO. On the other hand, initialization of the GW calculation from a hybrid PBE0 functional that includes a percentage of exact exchange, (G0W0@PBE0), predicts all SAMO orbitals to be lower in energy than the LUMO. The origin of the starting point dependence in G0W0 can be traced back to differences in the orbitals and orbital energies used as input for the self-energy calculation. In particular, the Coulomb potential, W, being roughly inversely proportional to the occupied → unoccupied transition energies, is extremely sensitive to any over-(under-)estimation of the HOMO–LUMO gap, which generally results in an under-(over-)estimation of screening.
| SAMO | ΔESAMO–LUMO (eV) | |||
|---|---|---|---|---|
| DFT(LDA) | DFT(PBE0) | G 0 W 0@LDA | G 0 W 0@PBE0 | |
| s | 2.3 | 1.8 | 0.3 | −0.7 |
| p | 2.5 | 2.0 | 0.4 | −0.6 |
| d | 2.8 | 2.3 | 0.5 | −0.3 |
Notably, the ΔESAMO–LUMO energy gap for SAMO orbital of s-type symmetry in C20H10 is predicted to be ∼0.3 eV, nearly an order of magnitude smaller than in C60 (calc. 2.4 eV).11c This proximity of the LUMO with respect to the delocalized SAMOs in C20H10, suggests possibilities for exploiting SAMO-mediated electron transport in a material using these bowl fragments as functional units.
Unfortunately, there are as of yet no reported experimental data concerning the electronic properties of SAMOs for the case of C20H10. However, one can make relevant comparisons of HOMO–LUMO gap given available ionization potential (IP) and electron affinity (EA) measurements that have been made experimentally.31 Depending on the experimental technique used, variations in HOMO–LUMO gap (IP-EA) range from 7.6 to 8.0 eV.31 The corresponding calculated values for the HOMO–(molecular)LUMO gap, considering the different DFT starting points, range from 7.3 eV (G0W0@LDA) to 8.1 eV (G0W0@PBE0). This provides a meaningful comparison with experiment, and establishes the level of accuracy of the described customized hybrid methodology.
Doping effects on SAMOs levels
The optimal size and shape of the small aromatic fragment, C20H10, provides characteristic features complementary, but unique, to that of the fullerenes or graphene structures. In particular, the LUMO–SAMO gap shows an important decrease with respect to C60 (0.3 eV vs. 2.4 eV in C60).11cConsideration of SAMOs for electron transport does not necessarily require one to focus on ‘ad hoc’ charged systems, as are sometimes considered.32 We instead propose a strategy for doping C20H10 in order to occupy the molecular LUMOs, for the purpose of enhancing the SAMO occupation. In fact, a further reduction of the LUMO–SAMO gap can be achieved via a modification of the central hollow potential of the molecular cage, which ultimately defines the SAMO wavefunctions.11a In this respect, internal (endohedral) doping with electron donating metal atoms has been suggested as a means to substantially reduce the LUMO–SAMO gap when the ionization potential of the endohedral atom is sufficiently large, as recently shown for C60.11a Interestingly, external (exohedral) doping is quite ineffective in reducing the SAMO energy, because the molecular cavity actually acts as a Faraday cage, effectively screening the states that are confined by the cage from the presence of any external charge.11a
C20H10 has been previously shown to have strong electron-acceptor character and forms stable complexes with alkaline metals, accommodating up to four electrons in the doubly degenerate LUMOs.13b Photoelectron spectra together with computations have shown C20H10 complexes doped with Cs and deposited on a Cu(111) surface to have full occupation of the original C20H10 degenerate LUMOs.13b Since cage-doping with metals having large ionization potentials (e.g., Li, Na) are suggested as the most effective for tuning the SAMO–LUMO energy gap,11a it is of interest to investigate the corresponding effects in C20H10 complexes with these particular metal atoms.
Three Li-doped C20H10 complexes are considered in this work, differing in the way the Li atoms are complexed to the molecular cage: (1) two Li atoms complexed to the convex side and two on the concave side; (2) all 4 Li atoms complexed to the convex side (exohedral); and (3) all 4 Li atoms complexed to the concave side (endohedral). The B97D//Def2-TZVPP fully optimized gas phase structures are shown in Fig. 4.
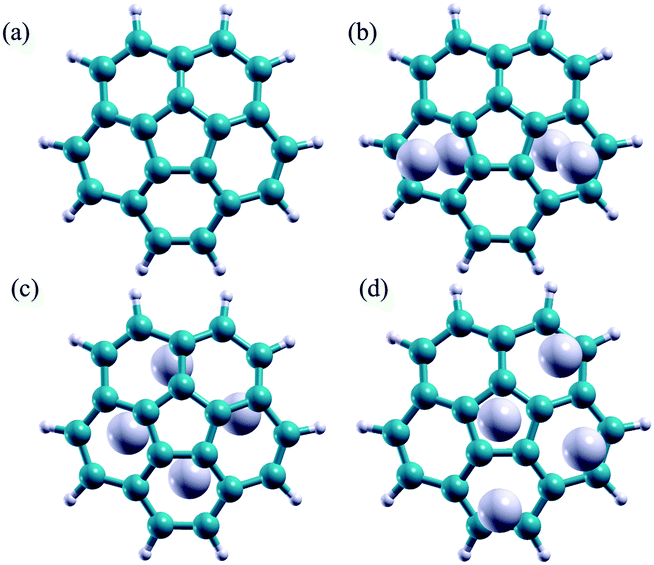 | ||
| Fig. 4 (a) Un-doped C20H10, (b) Li-doped C20H10, 2 Li convex side, 2 Li concave side, (c) Li-doped C20H10 4 Li concave side, (d) Li-doped C20H10 4 Li convex side. | ||
First, it is of interest to compare the basic molecular orbital structure of the un-doped C20H10 with that of the 4 Li-doped complexes. Due to the decrease in symmetry, the two degenerate LUMOs in the un-doped molecule are no longer degenerate in all Li-doped complex. In the doped Li-complexes, these orbitals are now occupied (Li-doped C20H10, 2 Li convex, 2 Li concave and C20H10, 4 Li concave) as HOMO − 1 and HOMO, or frontier molecular orbitals (Li-doped C20H10, 4 Li convex). Fig. 5 illustrates the two degenerate LUMOs in the parent molecule compared to the now-occupied orbitals in the 3 Li-doped complexes.
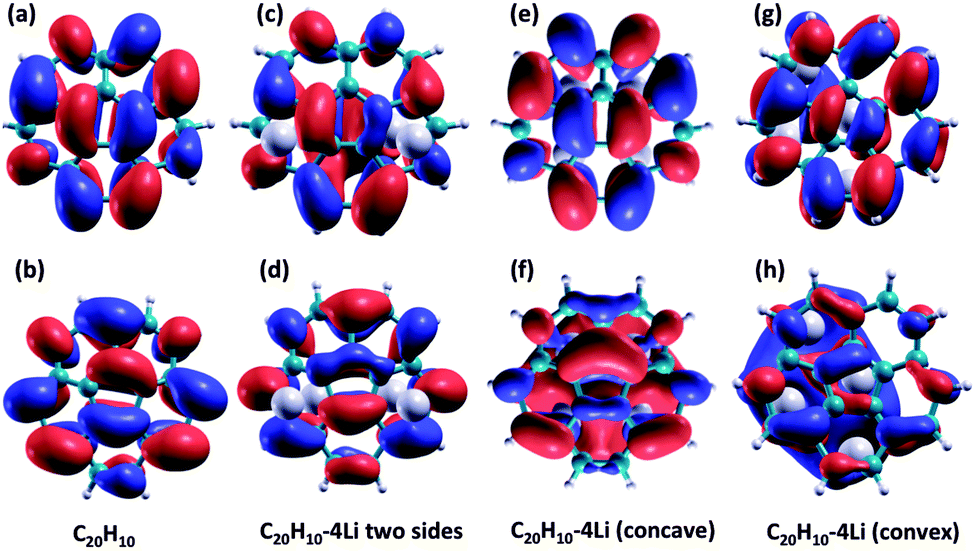 | ||
| Fig. 5 (a) and (b) degenerate LUMOs in C20H10, (c) and (d) HOMO and HOMO − 1 in Li-doped C20H10, 2 Li convex side and 2 Li on concave side, (e) and (f) HOMO and HOMO − 1 in Li-doped C20H10 4 Li on concave side, (e) and (f) HOMO and HOMO − 1 in Li-doped C20H10 4 Li on convex side. | ||
The occupation of the pristine molecular LUMOs in the Li complexes has a dramatic effect in closing the HOMO–LUMO gap with respect to that of the un-doped corannulene. The ‘new HOMOs’ in the hybrid systems are higher in energy, and, considering the GW@LDA HOMO–LUMO gap of C20H10 (7.3 eV) versus the corresponding value of the Li-doped structures (Table 2), one sees a decrease on the order of a factor of 2.
| Structure | HOMO–LUMO GAP (eV) | ||
|---|---|---|---|
| DFT-LDA | PBE0 | G 0 W 0@LDA | |
| C20H10-4Li-convex | 0.2 | 1.2 | 3.7 |
| C20H10-4Li-concave | 0.4 | 1.5 | 3.4 |
| C20H10-4Li-2sides | 0.4 | 1.3 | 3.0 |
A second point of interest is to address the effect of Li doping on the SAMO–LUMO energy gap. In particular, it is of interest to investigate the Li-doped C20H10, 4 Li concave structure, which due to the endohedral doping should be the most effective for tuning purposes in this respect. Moreover, the atomic arrangement of the Li atoms on the concave side of the molecular cage is symmetric and, as such, less perturbing to the SAMOs of p and d symmetry, which extend outside the central molecular core and are easily recognizable.
The calculated ΔESAMO–LUMO data is reported in Table 3. According to these results, the Li doping is seen not to decrease the SAMO–LUMO gap compared to the parent molecule at the G0W0 level, independent of starting functional. In fact, considering the G0W0@LDA (G0W0@PBE0) SAMOs–LUMO gap of C20H10 (Table 1) versus the corresponding values of the Li-doped structures (Table 3), one sees that C20H10 ΔESAMO–LUMO values are lower than those of the Li-doped complex by 0.5 eV (0.8 eV) for p symmetry SAMOs and by 0.7 eV (0.9 eV) for d symmetry SAMOs.
| SAMO | ΔESAMO–LUMO (eV) | |||
|---|---|---|---|---|
| DFT(LDA) | DFT(PBE0) | G 0 W 0@LDA | G 0 W 0@PBE0 | |
| p | 2.5 | 2.1 | 0.9 | 0.2 |
| d | 2.7 | 2.0 | 1.2 | 0.6 |
The above analysis shows again the strong electron-acceptor character of C20H10, which accommodates 4 electrons in the double-degenerate LUMOs when complexed with Li. Other than the dramatic decrease in HOMO–LUMO gap and loss of the p, d quasi degeneracy, doping with Li does not significantly affect the LUMO–SAMOs gap.
SAMOs in fullerene fragments
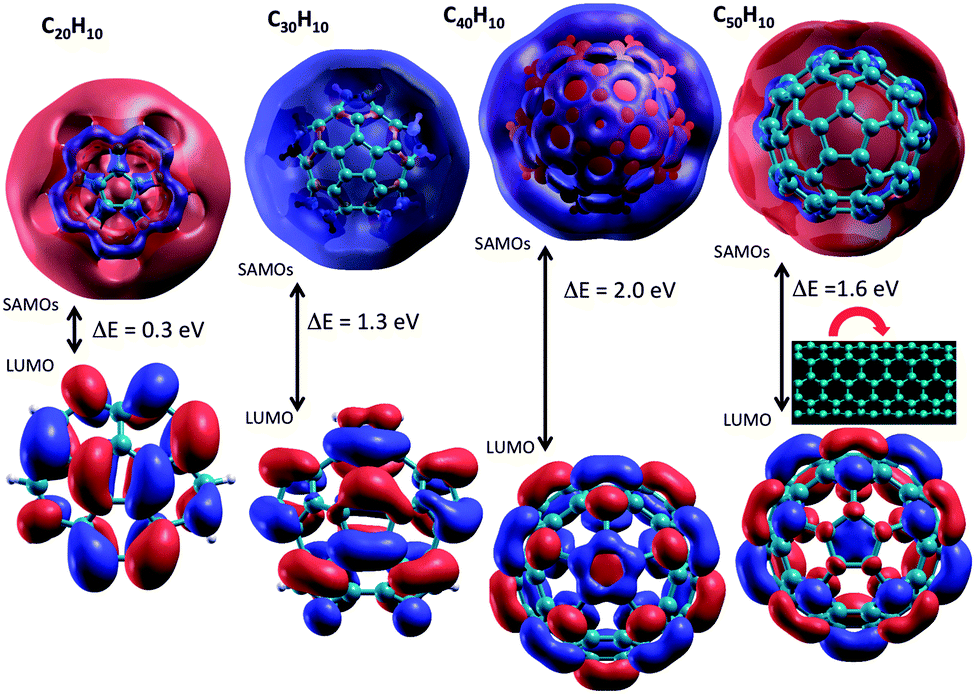
Consideration of electronic and transport properties across the full series of molecules of increased curvature, C20H10–C50H10, becomes of interest based on the results for the smallest of the series, C20H10, shown above. The presence of strong intrinsic molecular dipoles manifested by the curvature in these molecular fragments, together with the increasingly large polarizable surface of π electron density, are fundamental to unlocking and exploiting such systems for material devices.9 The increasing bowl depth and change in curvature across the series shows systematic trends in structure and property towards a tube structure (Fig. 6).9 It is of interest, therefore, to see if the same trends can be observed in SAMOs related electronic properties. | ||
| Fig. 6 Curved aromatic bowl constructs of increasing size and depth, based on the smallest corannulene unit (left most). | ||
As shown in previous work for shallow molecular bowls,9 dipoles induced across the relatively large surface area of the cap (pentagon) region may become comparable or larger than the intrinsic molecular dipole (perpendicular to the cap). On the other hand, for deeper bowls such an effect is not so apparent, reflecting the increased conjugated area of the belt region while approaching what one might find in a tube-like structure. To fully understand how the structural and electronic transition between the bowl-like structure (C20H10) to the tube-like (C50H10) structure can possibly affect the SAMO electronic properties, G0W0 calculations have been undertaken for investigating SAMOs across the series C30H10–C50H10. The G0W0@LDA predicted SAMOs–LUMO gap energy values for the series are summarized in Table 4 and Fig. 7. The corresponding orbital depictions are illustrated in Fig. 8.
| System | SAMO type | ΔESAMO–LUMO |
|---|---|---|
| C20H10 | s | 0.3 |
| p | 0.4 | |
| d | 0.5 | |
| C30H10 | s | 1.3 |
| p | 1.9 | |
| d | 1.7 | |
| C40H10 | s | 2.0 |
| p | 2.2 | |
| d | 2.6 | |
| C50H10 | s | 1.6 |
| p | 2.1 | |
| d | 2.5 | |
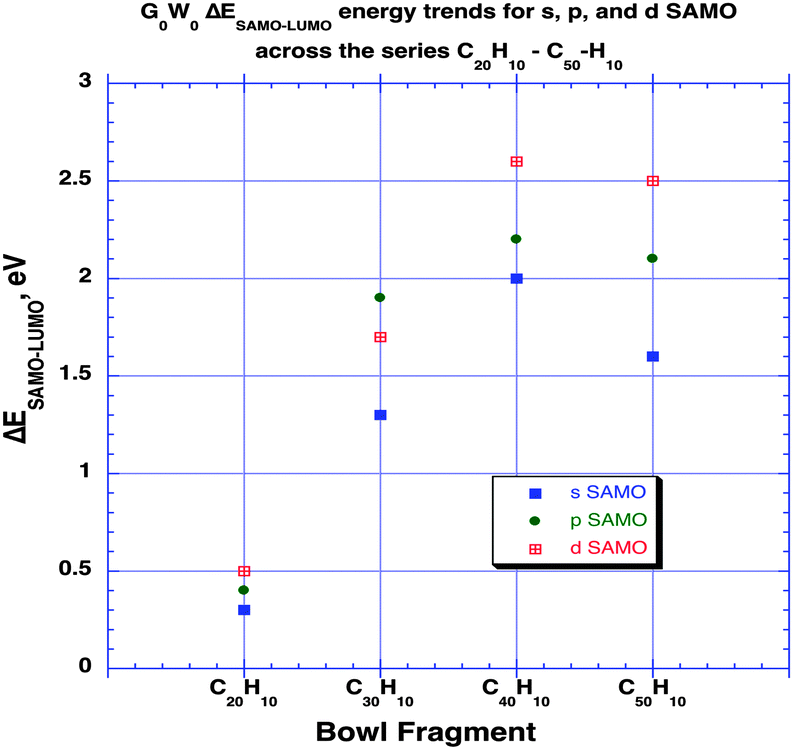 | ||
| Fig. 7 Trends in G0W0@LDA predicted ΔESAMO–LUMO across the series of aromatic bowl constructs of increasing size and depth, C20H10, C30H10, C40H10, C50H10, in eV. | ||
 | ||
| Fig. 8 DFT-LDA representation of SAMOs illustrating the typical s, p, d-like symmetric shapes in (a) C30H10, (b) C40H10, (c) C50H10. | ||
Comparison of the SAMO–LUMO energy gaps shows a sharp increase from C20H10 to C30H10, followed by a more modest increase to C40H10, and then a significant drop for the more tube-like structure, C50H10 (e.g., Fig. 7). For the lowest energy s-type SAMO, the gap relative to C20H10 (0.3 eV) increases by 1.0 eV for C30H10, by 1.7 eV for C40H10, and then levels off to an increase of 1.3 eV for C50H10. Importantly, in the case of the smallest bowl, C20H10, this energy gap is still one order of magnitude lower than the next higher analogue, C30H10. SAMOs of p and d-type symmetry are significantly higher in energy than the LUMO (from 1.7 eV up to 2.6 eV), and do not show promise for enhancing the occupation of the delocalized orbitals.
Finally, one can observe where C60 fits in the series in terms of ‘curvature’ and resulting ΔESAMO–LUMO with respect to the series, as shown in Fig. 9. One finds that the value of ΔESAMO–LUMO at 2.4 eV is still on the dramatic increase in trend of the bowl structures shown in Fig. 7, before the falloff towards the tube construct of C50H10. The local curvature of C60 also places it between C40H10 and C50H10, but closer to the latter.
 | ||
| Fig. 9 G 0 W 0@LDA predicted trends across the series of buckybowls, C20H10, C30H10, C40H10, C50H10. | ||
Conclusions
SAMO-type orbitals, shown to exist in conjugated hollow-shaped molecule, offer an important opportunity to exploit new mechanisms for intermolecular electron transport. Taking a slightly different view from the typically studied C60, the present investigation reports theoretical evidence of the electronic properties of SAMOs in a series of bowl shape aromatic constructs, C20H10–C50H10, with focus on the suitability of such orbitals in electron transport applications. SAMO electronic properties are revealed using accurate many body screening and polarization effects at the G0W0 level, with consideration of the dependence of calculated results on the DFT starting functional.G 0 W 0 predictions of SAMO levels in C20H10 reveal a LUMO–SAMO energy gap nearly an order of magnitude lower that that found in C60.11c This finding is extremely important in supporting design of C20H10-based materials to enhance occupation of these diffuse orbitals in experimental investigations. Contrary to literature proposals for C60,33 Li doping in C20H10 does not reduce the LUMO–SAMO gap over that of the un-doped corannulene. However, the promising results for un-doped corannulene, as well as other prospects for doping in these constructs, warrants further investigation towards this direction.
Further analysis of SAMOs in higher order analogues of C20H10, revealed a sharply increasing trend in ΔESAMO–LUMO energy gap up to C40H10, followed by a levelling off when the bowl structure reaches the morphology of tube structure (e.g., C50H10). Results of the present work have motivated experimental efforts to look into SAMO structure of C20H10. However, investigation of C20H10 assembled on metallic surface with established LT-STM methods is nontrivial, and considerable efforts are still necessary to determine the appropriate surface type and associated experimental protocols to achieve the necessary resolution.
Studies such as the present investigation provide an important opportunity to establish strengths and limitations of customized and enhanced hybrid methodologies for accurate predictions of materials phenomenon. Given the scant literature on performance of theoretical strategies for prediction of transport phenomenon, it is desirable to continue efforts in this direction, as established theoretical approaches hold an important role in the improved understanding of fundamental mechanisms.
Acknowledgements
L.Z. and K.K.B. acknowledge the University of Zürich, the Swiss National Science Foundation, and the UZH-UFSP for support of this research. L. M.-S. acknowledges Prof. Erio Tosatti for useful discussions. CSCS supercomputing center is gratefully acknowledged for a grant of computer time.References
- (a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234–240 CrossRef CAS PubMed; (b) D. l. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. V. Voorhis and M. A. Baldo, External Quantum Efficiency Above 100% in a Singlet-Exciton-Fission-Based Organic Photovoltaic Cell, Science, 2013, 340, 334–337 CrossRef CAS PubMed; (c) H. Song, M. A. Reed and T. Lee, Single Molecule Electronic Devices, Adv. Mater., 2011, 23, 1583–1608 CrossRef CAS PubMed; (d) S. V. Aradhya and L. Venkataraman, Single-molecule junctions beyond electronic transport, Nat. Nanotechnol., 2013, 8, 399–410 CrossRef CAS PubMed.
- (a) H. Vazquez, R. Skouta, S. Schneebeli, M. Kamenetska, R. Breslow, L. Venkataraman and M. S. Hybertsen, Probing the conductance superposition law in single-molecule circuits with parallel paths, Nat. Nanotechnol., 2012, 7, 663–667 CrossRef CAS PubMed; (b) C. M. Guédon, H. Valkenier, T. Markussen, K. S. Thygesen, J. C. Hummelen and S. J. v. d. Molen, Observation of quantum interference in molecular charge transport, Nat. Nanotechnol., 2012, 7, 305–309 CrossRef PubMed.
- L. Zoppi, J. S. Siegel and K. K. Baldridge, Electron transport and optical properties of curved aromatics, WIREs Comput. Mol. Sci., 2013, 3, 1–12 CrossRef CAS.
- (a) D. Chaudhuri, H. Wettach, K. J. V. Schooten, S. Liu, E. Sigmund, S. Höger and J. M. Lupton, Tuning the Singlet–Triplet Gap in Metal-Free Phosphorescent p-Conjugated Polymers, Angew. Chem., Int. Ed., 2010, 49, 7714–7717 CrossRef CAS PubMed; (b) M. D. Valle, R. Gutiérez, C. Tejedor and G. Cuniberti, Tuning the conductance of a molecular switch, Nat. Nanotechnol., 2007, 2, 176–179 CrossRef PubMed; (c) G. Valenti, C. Bruno, S. Rapino, A. Fiorani, E. A. Jackson, L. T. Scott, F. Paolucci and M. Marcaccio, Intense and Tunable Electrochemiluminescence of Corannulene, J. Phys. Chem. C, 2010, 114, 19467–19472 CrossRef CAS.
- L. T. Scott, H. E. Bronstein, D. V. Preda, R. B. M. Ansems, M. S. Bratcher and S. Hagen, Geodesic polyarenes with exposed concave surfaces, Pure Appl. Chem., 1999, 71, 209–219 CrossRef CAS.
- Y.-T. Wu, D. Bandera, R. Maag, A. Linden, K. K. Baldridge and J. S. Siegel, Multi-ethynyl Corannulenes: Synthesis, Structure and Properties, J. Am. Chem. Soc., 2008, 130, 10729–10739 CrossRef CAS PubMed.
- (a) J. Mack, P. Vogel, D. Jones, N. Kaval and A. Sutton, The development of corannulene-based blue emitters, Org. Biomol. Chem., 2007, 5, 2448–2452 RSC; (b) L. Zoppi, L. Martin-Samos and K. K. Baldridge, Effect of Molecular Packing on Corannulene-Based Materials Electroluminescence, J. Am. Chem. Soc., 2011, 133, 14002–14009 CrossRef CAS PubMed.
- T. Bauert, L. Zoppi, G. Koller, A. Garcia, K. K. Baldridge and K.-H. Ernst, Large interface dipole moments without charge transfer: buckybowls on metal surfaces, J. Phys. Chem. Lett., 2011, 2, 2805–2809 CrossRef CAS.
- L. Zoppi, A. Ferretti and K. K. Baldridge, Static and Field-Oriented Properties of Bowl-Shaped Polynuclear Aromatic Hydrocarbon Fragments, J. Chem. Theory Comput., 2013, 9, 4797–4804 CrossRef CAS.
- M. Feng, J. Zhao and H. Petek, Atomlike, hollow-core-bound molecular orbitals of C60, Science, 2008, 320, 359–362 CrossRef CAS PubMed.
- (a) J. Zhao, M. Feng, J. L. Yang and H. Petek, The Superatom States of Fullerenes and Their Hybridization into the Nearly Free Electron Bands of Fullerites, ACS Nano, 2009, 3, 853–864 CrossRef CAS PubMed; (b) M. Feng, J. Zhao, T. Huang, X. Zhu and H. Petek, The Electronic Properties of Superatom States of Hollow Molecules, Acc. Chem. Res., 2011, 44, 360–368 CrossRef CAS PubMed; (c) L. Zoppi, L. Martin-Samos and K. K. Baldridge, Structure–Property Relationships of Curved Aromatic Materials from First Principles, Acc. Chem. Res., 2014, 47, 3310–3320 CrossRef CAS PubMed.
- T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, Structure/Energy Correlation of Bowl Depth and Inversion Barrier in Corannulene Derivatives: Combined Experimental, and Quantum Mechanical Analysis, J. Am. Chem. Soc., 2001, 123, 517–525 CrossRef CAS PubMed.
- (a) L. Merz, M. Parschau, L. Zoppi, K. K. Baldridge, J. S. Siegel and K. H. Ernst, Reversible phase transitions in a buckybowl monolayer, Angew. Chem., Int. Ed., 2009, 48, 1966–1969 CrossRef CAS PubMed; (b) T. Bauert, L. Zoppi, G. Koller, J. S. Siegel, K. K. Baldridge and K.-H. Ernst, Quadruple Anionic Buckybowls by Solid-State Chemistry of Corannulene and Cesium, J. Am. Chem. Soc., 2013, 135, 12857–12860 CrossRef CAS PubMed.
- V. M. Silkin, J. Zhao, F. Guinea, E. V. Chulkov, P. M. Echenique and H. Petek, Image potential states in graphene, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 121408 CrossRef.
- (a) P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef; (b) W. Kohn and L. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- A. J. Cohen, P. Mori-Sánchez and W. Yang, Challenges for Density Functional Theory, Chem. Rev., 2012, 112, 289–320 CrossRef CAS PubMed.
- G. Onida, L. Reining and A. Rubio, Electronic excitations: density-functional versus many-body Green's-function approaches, Rev. Mod. Phys., 2002, 74, 601–659 CrossRef CAS.
- (a) L. Hedin, New Method for Calculating the One-Particle Green's Function with Application to the Electron-Gas Problem, Phys. Rev., 1965, 139, A796–A823 CrossRef; (b) L. Hedin and S. Lundqvist, Effects of Electron-Electron and Electron-Phonon Interactions on the One-electron States in Solids, in Solid State Physics: Advances in research and Applications, ed. H. Ehrenreich, F. Seitz and D. Turnbull, Academic, New York, 1969, vol. 23 Search PubMed.
- N. Marom, F. Caruso, X. Ren, O. T. Hofmann, T. Korzdorfer, J. R. Chelikowsky, A. Rubio, M. Scheffler and P. Rinke, Benchmark of GW methods for azabenzenes, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 245127 CrossRef.
- T. Körzdörfer and N. Marom, Strategy for finding a reliable starting point for G0W0 demonstrated for molecules, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 041110 CrossRef.
- (a) J. B. Neaton, M. S. Hybertsen and S. G. Louie, Renormalization of Molecular Electronic Levels at Metal-Molecule Interfaces, Phys. Rev. Lett., 2006, 97, 216405 CrossRef CAS; (b) J. M. Garcia-Lastra, C. Rostgaard, A. Rubio and K. S. Thygesen, Polarization-induced renormalization of molecular levels at metallic and semiconducting surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245427 CrossRef.
- K. Kaasbjerg and K. S. Thygesen, Benchmarking GW against exact diagonalization for semiempirical models, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085102 CrossRef.
- (a) X. Blase, C. Attaccalite and V. Olevano, First-principles GW calculations for fullerenes, porphyrins, phtalocyanine, and other molecules of interest for organic photovoltaic applications, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115103 CrossRef; (b) T. Körzdörfer, R. M. Parrish, N. Marom, J. S. Sears, C. D. Sherrill and J.-L. Brédas, Assessment of the performance of tuned range-separated hybrid density functionals in predicting accurate quasiparticle spectra, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 205110 CrossRef; (c) F. Bruneval and M. A. L. Marques, Benchmarking the Starting Points of the GW Approximation for Molecules, J. Chem. Theory Comput., 2013, 9, 324–329 CrossRef CAS.
- M. Nooijen and R. J. Bartlett, Equation of motion coupled cluster method for electron attachment, J. Chem. Phys., 1995, 102, 3629–3646 CrossRef CAS PubMed.
- S. J. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, M. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus and S. T. Elbert, General atomic and molecular electronic structure system, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. D. Corso, S. D. Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- L. Martin-Samos and G. Bussi, SaX: an open source package for electronic-structure and optical-properties calculations in the GW approximation, Comput. Phys. Commun., 2009, 180, 1416–1425 CrossRef CAS PubMed.
- J. P. Perdew and A. Zunger, Self-interaction correction to density functional approximations for many-electron systems, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 5048–5079 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional, J. Chem. Phys., 1999, 110, 5029–5035 CrossRef CAS PubMed.
- M. A. Petrukhina and L. T. Scott, Fragments of Fullerenes and Carbon Nanotubes: Designed Synthesis, Unusual Reactions, and Coordination Chemistry, Wiley, 2011 Search PubMed.
- V. K. Voora, L. S. Cederbaum and K. D. Jordan, Existence of a Correlation Bound s-Type Anion State of C60, J. Phys. Chem. Lett., 2013, 4, 849–853 CrossRef CAS.
- (a) T. Huang, J. Zhao, M. Feng, H. Petek, S. Yang and L. Dunsch, Superatom orbitals of Sc3N@C80 and their intermolecular hybridization on Cu(110)-(2 × 1)-O surface, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085434 CrossRef; (b) M. Feng, Y. Shi, C. I. Lin, J. Zhao, F. Liu, S. Yang and H. Petek, Energy stabilization of the s-symmetry superatom molecular orbital by endohedral doping of C82 fullerene with a lanthanum atom, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 075417 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05776g |
| This journal is © the Owner Societies 2015 |