
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemisorptive enantioselectivity of chiral epoxides on tartaric-acid modified Pd(111): three-point bonding
Mausumi
Mahapatra
and
Wilfred T.
Tysoe
*
Department of Chemistry and Laboratory for Surface Studies, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. E-mail: wtt@uwm.edu; Fax: +1-414-229-5036; Tel: +1-414-229-5222
First published on 8th January 2015
Abstract
The chemisorption of two chiral molecules, propylene oxide and glycidol, is studied on tartaric-acid modified Pd(111) surfaces by using temperature-programmed desorption to measure adsorbate coverage. It is found that R-glycidol shows preferential enantioselective chemisorption on (S,S)-tartaric acid modified Pd(111) surfaces, while propylene oxide does not adsorb enantioselectively. The enantioselectivity of glycidol depends on the tartaric acid coverage, and is exhibited for low tartaric acid coverages indicating that the bitartrate phase is responsible for the chiral recognition. The lack of enantioselectivity when using propylene oxide as a chiral probe implies that the enantiospecific interaction between glycidol and bitartate species is due to hydrogen-bonding interactions of the –OH group of glycidol. Scanning tunneling microscopy images were collected for tartaric acid adsorbed on Pd(111) under the same experimental conditions as used for enantioselective experiments. When tartaric acid is dosed at room temperature and immediately cooled to 100 K for imaging, individual bitartrate molecules were found. Density functional theory (DFT) calculations show that bitartrate binds to Pd(111) through its carboxylate groups and the –OH groups are oriented along the long axis of the bitartrate molecule. An enantiospecific interaction is found between glycidol and bitartate species where R-glycidol binds more strongly than S-glycidol to (S,S)-bitartate species by simultaneously forming hydrogen bonds with both the hydroxyl and carboxylate groups, thereby providing three-point bonding.
Introduction
Pharmaceuticals must generally be manufactured in their enantiopure form and are often synthesized using homogeneous-phase catalysts, thus requiring subsequent purification steps.1 This could be avoided by using heterogeneous-phase catalysts, but only very few examples have been explored; the hydrogenation of α-keto esters has been performed using supported Pt catalysts modified by cinchona alkaloids,2–6 while the hydrogenation of β-keto esters has been carried out over supported Ni catalysts modified by α-hydroxy acids (e.g. tartaric acid) and amino acids.6–11 The above reactions have been observed under catalytic conditions by chirally modifying supported catalysts by adsorbing a chiral modifier from the solution phase, which shows a strong dependence on temperature, pH, modifier concentration and modification time. Due to the complexity associated with the real catalytic systems, the way in which such chiral modifiers operate in the heterogeneous phase is not well understood. As a consequence, the surface chemistry of tartaric acid has been explored on a number of transition-metal surfaces,8,9,12–27 where several tartaric-acid derived species have been detected on these surfaces, including molecular tartaric acid, and mono- and bitartrate species, where the appearance of various species depends on the nature of the metal and the adsorbate coverage. In particular, on Pd(111), bitartrate species predominate at low coverages and monotartrate species at higher coverage when dosed at 300 K, and the presence of biacidic species is found at lower temperatures.28 Chiral modifiers such as amino acids on Pd(111) form discrete chiral assemblies which consist of ordered structures comprising tetramers or dimer rows potentially providing a chiral template in which several modifiers act in concert to provide an enantiospecific adsorption site.29 On the other hand, 1-naphthylethylamine, which is chemically similar to cinchonidine, does not self-assemble on palladium, suggesting that it acts as a one-to-one modifier.30–34While such studies provide detailed information on the local and extended chiral structures on the surface, with the exception of studies of co-adsorbed tartaric acid and methylacetoacetate on nickel,15 they provide little information on the interactions that lead to enantioselectivity. Such interactions can be explored using chiral probe molecules (here, R- and S-propylene oxide (PO) or glycidol) on chirally modified surfaces.35,36 For example, such experiments showed differences in the coverages of R- or S-PO on R- or S-2-butanol modified Palladium surfaces37 due to hydrogen-bonding interactions between the probe and the modifier.38 PO provides an ideal chiral probe since it adsorbs reversibly on Pd(111) thereby allowing coverages to be measured without interference from decomposition products.39 However, PO only allows hydrogen-bonding interactions with the epoxide oxygen to be probed. Thus, R- or S-glycidol, where the methyl group is replaced by a CH2–OH group,40 has been suggested as a more versatile chiral probe since it is capable of additional hydrogen-bonding interactions through the hydroxyl group. This is particularly important when using tartaric acid since its hydroxyl groups, which provide potential hydrogen-bonding loci, are located relatively far from the surface in all of its adsorbed forms.
The goal of this work is to measure the enantioselectivity of the tartaric acid-modified Pd(111) surfaces under UHV conditions and to identify the nature of the enantioselective tartaric acid-derived species. In this work, two different chiral probes, PO and glycidol, are used to measure the enantioselectivity of the tartaric-acid modified Pd(111) surface by using temperature programmed desorption (TPD). The enantioselectivity is measured for various coverages of the modifier to identify the nature of the tartaric acid-derived species and the active sites which are responsible for the chiral recognition. This work is supplemented by scanning tunneling microscopy (STM) experiments to explore the surface structures formed on Pd(111) as well as first-principles density functional theory (DFT) calculations to understand the chiral interactions. It is found that the bitartrate form of tartaric acid imparts enantioselectivity to glycidol, but not PO. DFT calculations of the interaction between glycidol and bitartrate indicate that glycidol adsorbs onto the surface through the epoxide oxygen, in a similar manner to PO,39 and hydrogen-bonding interactions occur between the hydroxyl groups of glycidol and bitartrate and the oxygen of COO−, and therefore, along with binding of the epoxide oxygen to the surface, it forms three bonding interactions that determine the enantioselectivity, in accord with the traditional three-point bonding rule.41,42
Experimental
Experiments were carried out in two different ultrahigh vacuum (UHV) chambers operating at base pressures of ∼1 × 10−10 Torr following bakeout.43 The Pd(111) substrate was cleaned using a standard procedure consisting of cycles of argon ion sputtering and annealing in 3 × 10−8 Torr of oxygen at 1000 K where the sample cleanliness was judged either using Auger spectroscopy or TPD after dosing with oxygen, where the absence of CO desorption indicated that the sample was carbon free.STM images of the tartaric acid-covered surfaces were acquired at a sample temperature of ∼120 K using an electrochemically etched tip made from recrystallized tungsten wire. Experiments were performed using a scanning tunneling microscope (RHK UHV350) housed in an UHV chamber operating at a base pressure below 2 × 10−10 Torr as described elsewhere.44
R- and S-PO (Aldrich, 99%) and R- and S-glycidol (Aldrich, 96%) were dosed onto the sample via a variable leak valve through a dosing tube directed towards the sample. (S,S)-tartaric acid (Aldrich, 99%) was dosed onto the sample by using a home-built Knudsen source. The tartaric acid source was repeatedly outgassed at ∼380 K to remove contaminants, primarily water. It was finally outgassed overnight at this temperature prior to dosing into the sample.
Theoretical methods
Density functional theory (DFT) calculations were performed using the projector augmented wave (PAW) method45,46 using the Vienna ab initio simulation package, VASP.47–49 The exchange–correlation potential was described using the generalized gradient approximation (GGA) of Perdew, Burke and Ernzerhof.50 A cutoff of 400 eV was used for the planewave basis set, and the wavefunctions and electron density were converged to within 1 × 10−5 eV. The first Brillouin zone was sampled with a 4 × 4 × 1 Γ-centered k-point mesh. Hydrogen bonding interactions are reasonably well reproduced (within ∼4 kJ mol−1) using this functional, although the accuracy deteriorates as the hydrogen bonds deviate from linearity.51 Geometric relaxations were considered to be converged when the force was less than 0.02 eV Å−1 on all unrestricted atoms.Results
TPD experiments are performed to measure the enantioselectivity of tartaric acid modified-Pd(111) by using PO as a chiral probe. The Pd(111) surface is dosed with a certain coverage of (S,S)-tartaric acid at ∼300 K, following which the sample is cooled to ∼150 K to dose a monolayer of one enantiomer of PO. TPD experiments are performed afterwards by monitoring 44 (tartaric acid) and 58 (PO) amu. The surface is then cleaned and the same experiment repeated for the same coverage of tartaric acid, but using the opposite enantiomer of PO. Similar experiments are carried out for various doses of tartaric acid. For the same coverage of the modifier (in this case, (S,S)-tartaric acid) the ratio between the coverage of the two enantiomers of the probe (in this case PO) is denoted the enantioselectivity ratio, Re.Fig. 1A shows the TPD results plotted as a function of tartaric acid dosing time in seconds, where the black and red curves correspond to experiments with R- and S-PO, respectively. The curves of tartaric acid are essentially identical indicating that the modifier coverages during the enantioselectivity measurements were identical. Fig. 1B plots the corresponding desorption profiles of PO (58 amu). The bottom profile in Fig. 1B is for the two enantiomers of PO adsorbed on clean Pd(111) and are identical, confirming the reproducibility of the experiment. However, as the tartaric acid coverage increases, while there is a decrease in the PO coverage due to site blocking, again the desorption profiles of R- and S-PO are identical; no enantioselectivity was observed when using PO as a chiral probe.
 | ||
| Fig. 1 TPD profiles collected following the adsorption of (S,S)-tartaric acid and a monolayer of R- or S-propylene oxide on Pd(111), as a function of (S,S)-tartaric acid exposure in seconds. The spectra which correspond to R- or S-propylene oxide are color coded by black and red respectively. The results were collected by using a heating rate of ∼3 K s−1 and by monitoring at (A) 44 amu and (B) 58 amu. | ||
Similar experiments are carried out when using glycidol as a chiral probe. The surface chemistry of glycidol on Pd(111) indicates that glycidol desorbs molecularly at ∼200 K and undergoes some decomposition reaction at higher temperature. However, only a maximum of ∼8% of glycidol decomposes during the desorption sweep on Pd(111).40 While the extent of S-glycidol decomposition is higher than for PO, it is sufficiently low to enable TPD experiments to be used to measure its coverage on chirally modified surfaces.
Fig. 2 shows the enantioselective chemisorption of glycidol on (S,S)-tartaric-acid modified Pd(111). For these experiments, glycidol is dosed at ∼200 K to avoid populating the multilayer. Fig. 2A shows the desorption profiles of both enantiomers of glycidol (at 29 amu) on clean Pd(111), which are identical, again demonstrating the reproducibility of the experiment. Fig. 2B to H compare the desorption profiles of both glycidol enantiomers from tartaric-acid-modified Pd(111). The bottom spectra, color coded in black and red, are the desorption profiles of R- and S-glycidol, respectively, while the top two spectra are due to tartaric acid. This indicates that identical coverages of tartaric acid were used for each experiment and the feature increases with tartaric acid dose. For tartaric acid doses between 35 and 45 s (Fig. 2D to F), there is clearly a discernible preferential adsorption of R-glycidol on (S,S)-tartaric-acid modified surfaces. For higher doses (Fig. 2G and H), the spectra of the two enantiomers of glycidol overlap, indicating no enantioselectivity.
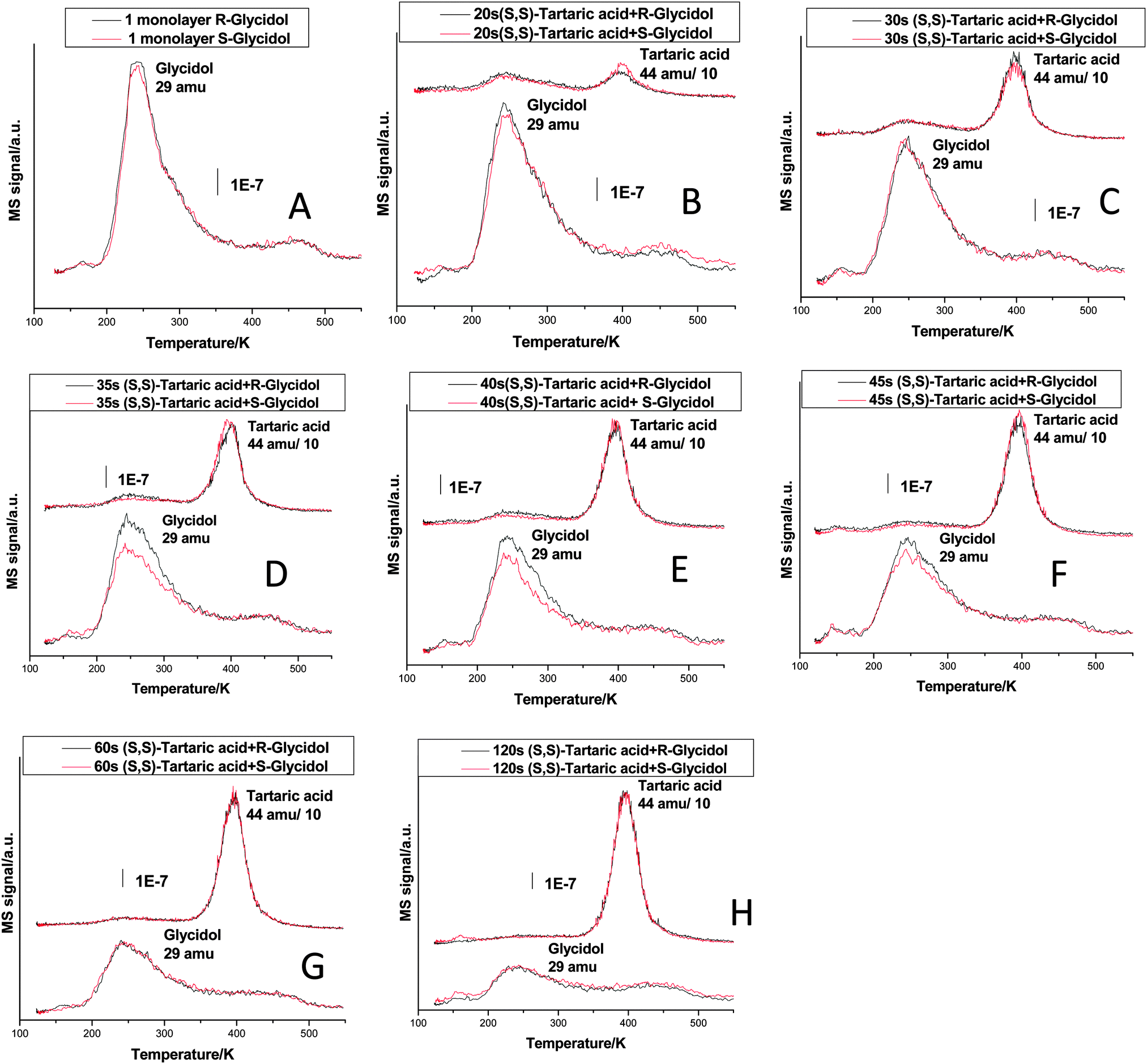 | ||
| Fig. 2 TPD profiles collected following the adsorption of (S,S)-tartaric acid and a monolayer of R- or S-glycidol on Pd(111), as a function of (S,S)-tartaric acid exposure in seconds. The spectra which correspond to R- or S-glycidol are color coded by black and red respectively. The results were collected by using a heating rate of ∼3 K s−1 and by monitoring 44 amu and 29 amu. A shows the 29 amu (glycidol) desorption profile on clean Pd(111). In B–H, the tartaric acid dosing time is increased from 20 s to 120 s. The bottom two spectra in B–H show 29 amu (glycidol) and the top two spectra show the corresponding 44 amu (tartaric acid) desorption profile. | ||
The resulting value of the enantioselective ratio (Re, defined as Re = ΘR(S,S)/ΘS(S,S) = ΘS(R,R)/ΘR(R,R)), where Θ is the probe molecule saturation coverage, the superscripts (S or R) are the chiralities of the probe and the subscripts ((S,S) or (R,R)) are those of the modifier of glycidol on tartaric acid-modified Pd(111) surfaces, is shown in Fig. 3, showing a maximum value of ∼1.3 for a (S,S)-tartaric acid dose of ∼40 s.
 | ||
| Fig. 3 Plot of the enantioselectivity ratio Re for glycidol as a function of tartaric acid coverage. Re is 1 at tartaric acid exposure of up to 25 s and reaches a maximum value of ∼1.29 at an exposure of ∼40 s and again decreases to unity for higher doses. | ||
STM images were collected for tartaric acid on Pd(111) in order to explore the distribution on the surface. An image of a relatively low coverage of tartaric acid dosed at ∼300 K and then cooled to ∼120 K, at which temperature the image was collected, is shown in Fig. 4. This mimics the conditions under which enantioselectivity was measured with glycidol as a chiral probe molecule (Fig. 2). This shows elliptical structures assigned to the presence of bitartrate species on the surface. There is no evidence for the formation of ordered surface structures on Pd(111) suggesting that they do not form templating structures. Line profiles of the elliptical structures suggest that they are ∼3 Å across and ∼6 Å long.
 | ||
| Fig. 4 STM images of (S,S)-tartaric acid dosed onto a Pd(111) surface at ∼300 K and then imaged at 120 K to mimic the conditions under which the enantioselectivity measurements were made (see Fig. 1 and 2). Vb = 0.9 V, It = 380 pA. | ||
Discussion
Propylene oxide adsorbs and desorbs from the surface without any decomposition, thereby providing an ideal chiral probe on Pd(111).39 Glycidol desorbs mostly molecularly from Pd(111) with a minor extent of decomposition40 and has been used to explore chiral recognition in the gas phase.52,53 The thermal stability and decomposition pathways of tartaric acid on Pd(111) has also been explored.28 It has been found that it adsorbs as bitartrate species at low coverages, and monotartrate species are formed at higher coverage. STM images of a low coverage of tartaric acid show the presence of individual bitartrate species and no template formation was observed.No enantioselectivity was observed for PO on tartaric-acid modified surfaces, whereas glycidol shows modest enantioselectivity over a narrow range of the tartaric acid coverages (Fig. 3). Based on previous studies, the surface contains predominantly bitartrate species in the coverage range over which enantioselectivity is detected. The observation that PO does not adsorb enantioselectively, while glycidol does, indicates that hydrogen-bonding interaction occur between the OH groups of glycidol and bitartrate species. The STM image shows that tartaric acid does not form ordered structures, implying that there is a one to one interaction between the modifier and the probe molecule.
First-principles DFT calculations were carried out to explore possible enantioselective interactions between R-glycidol and (S,S)-bitartrate species. The most stable calculated structure of bitartrate species on Pd(111) is depicted in Fig. 5, showing atop adsorption of the carboxylate oxygen atoms with the hydroxyl groups oriented along the long axis of the bitartrate species in accord with previous studies on other surfaces.54–57 The size of the bitartrate species is in accord with the size of the image seen by STM (Fig. 4). The molecule is somewhat strained to accommodate the hexagonal (111) substrate, and the adsorption energy, calculated from the difference between the energies of the structure shown in Fig. 5 and the sum of the energies of clean Pd(111) and an isolated bitartrate molecule, is ∼43 kJ mol−1.
 | ||
| Fig. 5 Depiction of the most stable structure of (S,S)-bitartrate species on Pd(111) obtained using DFT calculations showing A, the top view and B, the side view. | ||
Similar calculations were carried out for glycidol on Pd(111), where the resulting most stable structure is shown in Fig. 6. The epoxide oxygen adsorbs on a palladium atop site, as found for PO.58 However, in contrast to PO, the CH2–OH group has an additional agostic interaction with the surface resulting in an overall binding energy of ∼22 kJ mol−1.
 | ||
| Fig. 6 Depiction of the most stable structure of R-glycidol on Pd(111) obtained using DFT calculations showing A, the top view and B, the side view. | ||
In order to explore the interactions between R-glycidol and (S,S)-bitartrate species on Pd(111), a glycidol molecule was placed at various locations around the bitartrate species and the geometry was allowed to relax. The most stable structure comprising (S,S)-bitartrate species interacting with R-glycidol is depicted in Fig. 7. Here the epoxide oxygen is located at the most stable atop site, which dictates the stereochemistry of the interactions between the CH2–OH group and the bitartrate. This comprises a simultaneous hydrogen-bonding donor interaction between the CH2–OH group and an oxygen atom of the carboxylate, and a hydrogen-bonding acceptor interaction between the hydroxyl group of the bitartrate and the oxygen of the CH2–OH group. This involves a tilt of the R-glycidol molecule away from the surface to allow the hydroxyl group to interact with the carboxylate oxygen of the (S,S)-bitartrate species. This geometry is in accord with the three-point bonding rule41,42 where the bonds are indicated by dotted lines in Fig. 7. The distances between the hydrogens and oxygens are relatively close (∼2 Å), thereby facilitating such interactions. The energy gain due to this interaction, calculated from the difference in energy of the (S,S)-bitartrate and R-glycidol structures (Fig. 7) and the sum of the energies of isolated (S,S)-bitartrate species (Fig. 5) and R-glycidol (Fig. 6), is ∼−19 kJ mol−1.
 | ||
| Fig. 7 Depiction of the most stable structure of (S,S)-bitartrate + R-glycidol on Pd(111) obtained using DFT calculations showing A, the top view and B, the side view, where the interactions are indicated by dotted lines. | ||
The corresponding structure obtained from interactions between (S,S)-bitartrate and S-glycidol is shown in Fig. 8. Glycidol still adsorbs on the atop palladium site, which then controls the stereochemistry of the subsequent intermolecular hydrogen-bonding interactions since the glycidol binding energy of ∼22 kJ mol−1 is larger than the energies of typical hydrogen-bonding interactions.59S-glycidol still has a hydrogen-bonding donor interaction between the CH2–OH group and the (S,S)-bitartrate carboxylate oxygen. However, the structural constraints now imposed by the S-enantiomer of the bitartrate mean that the distance between its hydrogen and the CH2–OH oxygen is 2.93 Å which is much larger than in the most stable structure (Fig. 7) where the corresponding distance is 1.87 Å, thereby reducing the interaction energy to ∼13 kJ mol−1. Interestingly, the stabilization of R-glycidol by interaction with the (S,S)-bitartrate species is not reflected in the desorption temperature of glycidol from the surface (Fig. 2); the glycidol desorption temperature over the bitartrate coverage range where enantioselectivity is measured (in Fig. 2D to F) is identical to that on the clean surface (Fig. 2A). This suggests that the relatively weak bonding in the glycidol-bitartrate complexes allows it to dissociate below the desorption temperature during heating.
 | ||
| Fig. 8 Depiction of the most stable structure of (S,S)-bitartrate + S-glycidol on Pd(111) obtained using DFT calculations showing A, the top view and B, the side view, where the interactions are indicated by dotted lines. | ||
From the above discussion, it is clear that the enantioselective chemisorption of glycidol on the tartaric acid-modified Pd(111) surface arises due to H-bonding interaction between the modifier and the probe and those stereochemical interactions are dictated by the adsorption geometry of the probe molecule. The enantioselectivity measurement is highly dependent on tartaric acid coverage. Therefore it is possible that the nature of the surface and the reaction environment such as temperature and the nature of the solvent will have a strong influence on the surface structure not captured by experiments carried out in ultrahigh vacuum.
Conclusions
Glycidol adsorbs enantioselectively on tartaric-acid modified Pd(111) while PO does not. The tartaric acid coverage at which enantioselectivity is measured indicates that bitartrate species provides the chiral modifier, while other tartaric-acid derived structures are not enantioselective. Under the conditions where enantioselectivity is measured (dosing at ∼300 K and cooling to ∼120 K), isolated bitartrate species form on the surface and no ordered structures are found.These results clearly indicate that the local interaction between the CH2–OH group in the heterochiral complex formed between R-glycidol and (S,S)-bitartrate is responsible for the enantioselectivity and is modeled using DFT calculations. In both glycidol enantiomers, the binding is dominated by the relatively strong adsorption of the epoxide oxygen onto the palladium atop site, and the structure is controlled by hydrogen-bond donation from the CH2–OH hydrogen in R-glycidol to an oxygen atom of the carboxylate group and by donation from the (S,S)-bitartrate hydroxyl to the oxygen of the CH2–OH group of R-glycidol. In the complex formed between R-glycidol and (S,S)-bitartrate, this structure stabilizes hydrogen bonding between a bitartrate –OH and the CH2–OH oxygen, which becomes disfavored in the complex between S-glycidol and (S,S)-bitartrate.
Thus, the three-point bonding is dominated by the strongest binding of the chiral probe (or, in the case of a reaction, the prochiral reactant) to the surface and the ability of a group (or groups) in the probe or reactant to be correctly positioned to simultaneously undergo two distinct bonding interactions with the chiral center. In the case of glycidol, these are hydrogen-bonding interactions. These energies (of a few kJ mol−1) are sufficiently large to provide significant stabilization while not being so strong to inhibit catalytic reactions.
Acknowledgements
We gratefully acknowledge support of this work by the U.S. Department of Energy, Division of Chemical Sciences, Office of Basic Energy Sciences, under grant number DE-FG02-03ER15474. We thank Professor Michael Weinert and Dr Michael Garvey for advice on carrying out density functional theory calculations.References
- H. C. Kolb and K. B. Sharpless, Asymmetric Aminohydroxylation, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, Germany, 1998, vol. 2, pp. 243–260 Search PubMed.
- Y. Orito, S. Imai and S. Niwa, Asymmetric Hydrogenation of Methyl Pyruvate Using Pt-C Catalyst Modified with Cinchonidine, J. Chem. Soc. Jpn., 1979, 1118–1120 CrossRef CAS.
- Y. Orito, S. Imai and S. Niwa, Nguyengiahung, Asymmetric Hydrogenation of Methyl Benzoylformate Using Platinum-Carbon Catalysts Modified with Cinchonidine, J. Synth. Org. Chem., Jpn., 1979, 37, 173–174 CrossRef CAS.
- Y. Orito, S. Imai and S. Niwa, Asymmetric Hydrogenation of α-Keto Esters Using a Platinum-Alumina Catalyst Modified with Cinchona Alkaloid, J. Chem. Soc. Jpn., 1980, 670–672 CrossRef CAS.
- T. Harada and Y. Izumi, Improved Modified RANEY®-Nickel Catalyst for Enantioface-Differentiating (Asymmetric) Hydrogenation of Methyl Acetoacetate, Chem. Lett., 1978, 1195–1196 CrossRef CAS.
- Y. Izumi, Modified RANEY®-Nickel (Mrni) Catalyst – Heterogeneous Enantio-Differentiating (Asymmetric) Catalyst, Adv. Catal., 1983, 32, 215–271 CAS.
- A. Hoek and W. M. H. Sachtler, Enantioselectivity of Nickel Catalysts Modified with Tartaric Acid or Nickel Tartrate Complexes, J. Catal., 1979, 58, 276–286 CrossRef CAS.
- R. Raval, Chiral Expressions at Metal Surfaces, Curr. Opin. Solid State Mater. Sci., 2003, 7, 67–74 CrossRef CAS.
- C. J. Baddeley, Fundamental Investigations of Enantioselective Heterogeneous Catalysis, Top. Catal., 2003, 25, 17–28 CrossRef CAS.
- M. A. Keane and G. Webb, The Enantioselective Hydrogenation of Methyl Acetoacetate over Supported Nickel-Catalysts.1. The Modification Procedure, J. Catal., 1992, 136, 1–15 CrossRef CAS.
- M. A. Keane, Interaction of Optically Active Tartaric Acid with a Nickel-Silica Catalyst: Role of Both the Modification and Reaction Media in Determining Enantioselectivity, Langmuir, 1997, 13, 41–50 CrossRef CAS.
- M. O. Lorenzo, S. Haq, T. Bertrams, P. Murray, R. Raval and C. J. Baddeley, Creating Chiral Surfaces for Enantioselective Heterogeneous Catalysis: R,R-Tartaric Acid on Cu(110), J. Phys. Chem. B, 1999, 103, 10661–10669 CrossRef CAS.
- M. O. Lorenzo, C. J. Baddeley, C. Muryn and R. Raval, Extended Surface Chirality from Supramolecular Assemblies of Adsorbed Chiral Molecules, Nature, 2000, 404, 376–378 CrossRef CAS PubMed.
- V. Humblot, S. Haq, C. Muryn, W. A. Hofer and R. Raval, From Local Adsorption Stresses to Chiral Surfaces: (R,R)-Tartaric Acid on Ni(110), J. Am. Chem. Soc., 2002, 124, 503–510 CrossRef CAS PubMed.
- T. E. Jones and C. J. Baddeley, Direct Stm Evidence of a Surface Interaction between Chiral Modifier and Pro-Chiral Reagent: Methylacetoacetate on R,R-Tartaric Acid Modified Ni{111}, Surf. Sci., 2002, 519, 237–249 CrossRef CAS.
- T. E. Jones and C. J. Baddeley, A Rairs, STM and TPD Study of the Ni{111}/R,R-Tartaric Acid System: Modelling the Chiral Modification of Ni Nanoparticles, Surf. Sci., 2002, 513, 453–467 CrossRef CAS.
- M. O. Lorenzo, V. Humblot, P. Murray, C. J. Baddeley, S. Haq and R. Raval, Chemical Transformations, Molecular Transport, and Kinetic Barriers in Creating the Chiral Phase of (R,R)-Tartaric Acid on Cu(110), J. Catal., 2002, 205, 123–134 CrossRef CAS.
- W. A. Hofer, V. Humblot and R. Raval, Conveying Chirality onto the Electronic Structure of Achiral Metals: (R,R)-Tartaric Acid on Nickel, Surf. Sci., 2004, 554, 141–149 CrossRef CAS PubMed.
- V. Humblot, S. Haq, C. Muryn and R. Raval, (R,R)-Tartaric Acid on Ni(110): The Dynamic Nature of Chiral Adsorption Motifs, J. Catal., 2004, 228, 130–140 CrossRef CAS PubMed.
- V. Humblot, M. O. Lorenzo, C. J. Baddeley, S. Haq and R. Raval, Local and Global Chirality at Surfaces: Succinic Acid Versus Tartaric Acid on Cu(110), J. Am. Chem. Soc., 2004, 126, 6460–6469 CrossRef CAS PubMed.
- T. E. Jones and C. J. Baddeley, An Investigation of the Adsorption of (R,R)-Tartaric Acid on Oxidised Ni{111} Surfaces, J. Mol. Catal. A: Chem., 2004, 216, 223–231 CrossRef CAS PubMed.
- T. E. Jones, T. C. Q. Noakes, P. Bailey and C. J. Baddeley, Adsorbate-Induced Segregation in the Ni{111}/Au/(R,R)-Tartaric Acid System, J. Phys. Chem. B, 2004, 108, 4759–4766 CrossRef CAS.
- M. Parschau, T. Kampen and K. H. Ernst, Homochirality in Monolayers of Achiral Meso Tartaric Acid, Chem. Phys. Lett., 2005, 407, 433–437 CrossRef CAS PubMed.
- K. H. Ernst, Supramolecular Surface Chirality, in Supramolecular Chirality, 2006, vol. 265, pp. 209–252 Search PubMed.
- T. E. Jones and C. J. Baddeley, Influence of Modification Conditions on the Interaction of Methylacetoacetate with (R,R)-Tartaric Acid-Modified Ni{111}, J. Phys. Chem. C, 2007, 111, 17558–17563 CAS.
- K. H. Ernst, Amplification of Chirality in Two-Dimensional Molecular Lattices, Curr. Opin. Colloid Interface Sci., 2008, 13, 54–59 CrossRef CAS PubMed.
- A. J. Gellman, Y. Huang, Y. Feng, Y. V. Pushkarev, B. Holsclaw and B. S. Mhatre, Superenantioselective Chiral Surface Explosions, J. Am. Chem. Soc., 2013, 135, 19208–19214 CrossRef CAS PubMed.
- M. Mahapatra and W. T. Tysoe, Structure and Decomposition Pathways of D-(–)-Tartaric Acid on Pd(111), Surf. Sci., 2014, 629, 132–138 CrossRef CAS PubMed.
- M. Mahapatra, L. Burkholder, Y. Bai, M. Garvey, J. A. Boscoboinik, C. Hirschmugl and W. T. Tysoe, Formation of Chiral Self-Assembled Structures of Amino Acids on Transition-Metal Surfaces: Alanine on Pd(111), J. Phys. Chem. C, 2014, 118, 6856–6865 CAS.
- I. Lee, Z. Ma, S. Kaneko and F. Zaera, 1-(1-Naphthyl)Ethylamine Adsorption on Platinum Surfaces: On the Mechanism of Chiral Modification in Catalysis, J. Am. Chem. Soc., 2008, 130, 14597–14604 CrossRef CAS PubMed.
- L. Burkholder, D. Stacchiola, J. A. Boscoboinik and W. T. Tysoe, Enantioselective Chemisorption on Model Chirally Modified Surfaces: 2-Butanol on Alpha-(1-Naphthyl)Ethylamine/Pd(111), J. Phys. Chem. C, 2009, 113, 13877–13885 CAS.
- J. L. Sales, V. Gargiulo, I. Lee, F. Zaera and G. Zgrablich, Monte Carlo Modeling of the Enantioselective Adsorption of Propylene Oxide on 1-(1-Naphthyl)Ethylamine-Modified Pt(111) Surfaces, Catal. Today, 2010, 158, 186–196 CrossRef CAS PubMed.
- J. A. Boscoboinik, Y. Bai, L. Burkholder and W. T. Tysoe, Structure and Distribution of S-Alpha-(1-Naphthyl)-Ethylamine on Pd(111), J. Phys. Chem. C, 2011, 115, 16488–16494 CAS.
- A. D. Gordon and F. Zaera, Adsorption of 1-(1-Naphthyl)Ethylamine from Solution onto Platinum Surfaces: Implications for the Chiral Modification of Heterogeneous Catalysts, Angew. Chem., Int. Ed., 2013, 52, 3453–3456 CrossRef CAS PubMed.
- D. Stacchiola, L. Burkholder and W. T. Tysoe, Probing Enantioselective Chemisorption in Ultrahigh Vacuum, J. Mol. Catal. A: Chem., 2004, 216, 215–221 CrossRef CAS PubMed.
- D. Stacchiola, L. Burkholder, T. Zheng, M. Weinert and W. T. Tysoe, Requirements for the Formation of a Chiral Template, J. Phys. Chem. B, 2005, 109, 851–856 CrossRef CAS PubMed.
- D. Stacchiola, L. Burkholder and W. T. Tysoe, Enantioselective Chemisorption on a Chirally Modified Surface in Ultrahigh Vacuum: Adsorption of Propylene Oxide on 2-Butoxide-Covered Palladium(111), J. Am. Chem. Soc., 2002, 124, 8984–8989 CrossRef CAS PubMed.
- F. Gao, Y. L. Wang, L. Burkholder and W. T. Tysoe, Enantioselective Chemisorption of Propylene Oxide on a 2-Butanol Modified Pd(111) Surface: The Role of Hydrogen-Bonding Interactions, J. Am. Chem. Soc., 2007, 129, 15240–15249 CrossRef CAS PubMed.
- V. Bustos, D. Linares, A. G. Rebaza, W. T. Tysoe, D. Stacchiola, L. Burkholder and G. Zgrablich, Monte Carlo Theory Analysis of Thermal Programmed Desorption of Chiral Propylene Oxide from Pd(111) Surfaces, J. Phys. Chem. C, 2009, 113, 3254–3258 CAS.
- M. Mahapatra and W. T. Tysoe, Adsorption and Reaction Pathways of a Chiral Probe Molecule, S-Glycidol on a Pd(111) Surface, Catal. Sci. Technol., 2015 10.1039/C4CY00904E.
- L. H. Easson and E. Stedman, Studies on the Relationship between Chemical Constitution and Physiological Action: Molecular Dissymmetry and Physiological Activity, Biochem. J., 1933, 27, 1257–1266 CAS.
- A. G. Ogston, Interpretation of Experiments on Metabolic Processes, Using Isotopic Tracer Elements, Nature, 1948, 162, 963 CrossRef CAS.
- F. Gao, Z. J. Li, Y. L. Wang, L. Burkholder and W. T. Tysoe, Chemistry of Alanine on Pd(111): Temperature-Programmed Desorption and X-Ray Photoelectron Spectroscopic Study, Surf. Sci., 2007, 601, 3276–3288 CrossRef CAS PubMed.
- J. A. Boscoboinik, F. C. Calaza, Z. Habeeb, D. W. Bennett, D. J. Stacchiola, M. A. Purino and W. T. Tysoe, One-Dimensional Supramolecular Surface Structures: 1,4-Diisocyanobenzene on Au(111) Surfaces, Phys. Chem. Chem. Phys., 2010, 12, 11624–11629 RSC.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. Ireta, J. Neugebauer and M. Scheffler, On the Accuracy of DFT for Describing Hydrogen Bonds: Dependence on the Bond Directionality, J. Phys. Chem. A, 2004, 108, 5692–5698 CrossRef CAS.
- N. Borho and M. A. Suhm, Glycidol Dimer: Anatomy of a Molecular Handshake, Phys. Chem. Chem. Phys., 2002, 4, 2721–2732 RSC.
- A. Maris, B. M. Giuliano, D. Bonazzi and W. Caminati, Molecular Recognition of Chiral Conformers: A Rotational Study of the Dimers of Glycidol, J. Am. Chem. Soc., 2008, 130, 13860–13861 CrossRef CAS PubMed.
- L. A. M. M. Barbosa and P. Sautet, Stability of Chiral Domains Produced by Adsorption of Tartaric Acid Isomers on the Cu(110) Surface: A Periodic Density Functional Theory Study, J. Am. Chem. Soc., 2001, 123, 6639–6648 CrossRef CAS PubMed.
- C. G. M. Hermse, A. P. van Bavel, A. P. J. Jansen, L. A. M. M. Barbosa, P. Sautet and R. A. van Santen, Formation of Chiral Domains for Tartaric Acid on Cu(110): A Combined Dft and Kinetic Monte Carlo Study, J. Phys. Chem. B, 2004, 108, 11035–11043 CrossRef CAS.
- J. N. James and D. S. Sholl, Density Functional Theory Studies of Dehydrogenated and Zwitterionic Glycine and Alanine on Pd and Cu Surfaces, J. Mol. Catal. A: Chem., 2008, 281, 44–48 CrossRef CAS PubMed.
- J. N. James and D. S. Sholl, Theoretical Studies of Chiral Adsorption on Solid Surfaces, Curr. Opin. Colloid Interface Sci., 2008, 13, 60–64 CrossRef CAS PubMed.
- F. Roma, G. Zgrablich, D. Stacchiola and W. T. Tysoe, Theoretical Analysis of the Coverage Dependence of Enantioselective Chemisorption on a Chirally Templated Surface, J. Chem. Phys., 2003, 118, 6030–6037 CrossRef CAS PubMed.
- G. C. Pimentel and A. L. McLellan, The Hydrogen Bond, W. H. Freeman, San Francisco, 1960 Search PubMed.
| This journal is © the Owner Societies 2015 |