
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Collisional relaxation of apocarotenals: identifying the S* state with vibrationally excited molecules in the ground electronic state S0*†
Florian
Ehlers
a,
Mirko
Scholz
b,
Jens
Schimpfhauser
c,
Jürgen
Bienert
c,
Kawon
Oum
*b and
Thomas
Lenzer
*b
aGeorg-August-Universität Göttingen, Institut für Physikalische Chemie, Tammannstr. 6, 37077 Göttingen, Germany
bUniversität Siegen, Physikalische Chemie 2, Adolf-Reichwein-Str. 2, 57076 Siegen, Germany. E-mail: lenzer@chemie.uni-siegen.de; oum@chemie.uni-siegen.de; Fax: +49 271 740 2943; Tel: +49 271 740 2803
cMax-Planck-Institut für biophysikalische Chemie, Am Faßberg 11, 37077 Göttingen, Germany
First published on 24th March 2015
Abstract
In recent work, we demonstrated that the S* signal of β-carotene observed in transient pump–supercontinuum probe absorption experiments agrees well with the independently measured steady-state difference absorption spectrum of vibrationally hot ground state molecules S0* in solution, recorded at elevated temperatures (Oum et al., Phys. Chem. Chem. Phys., 2010, 12, 8832). Here, we extend our support for this “vibrationally hot ground state model” of S* by experiments for the three terminally aldehyde-substituted carotenes β-apo-12′-carotenal, β-apo-4′-carotenal and 3′,4′-didehydro-β,ψ-caroten-16′-al (“torularhodinaldehyde”) which were investigated by ultrafast pump–supercontinuum probe spectroscopy in the range 350–770 nm. The apocarotenals feature an increasing conjugation length, resulting in a systematically shorter S1 lifetime of 192, 4.9 and 1.2 ps, respectively, in the solvent n-hexane. Consequently, for torularhodinaldehyde a large population of highly vibrationally excited molecules in the ground electronic state is quickly generated by internal conversion (IC) from S1 already within the first picosecond of relaxation. As a result, a clear S* signal is visible which exhibits the same spectral characteristics as in the aforementioned study of β-carotene: a pronounced S0 → S2 red-edge absorption and a “finger-type” structure in the S0 → S2 bleach region. The cooling process is described in a simplified way by assuming an initially formed vibrationally very hot species S0** which subsequently decays with a time constant of 3.4 ps to form a still hot S0* species which relaxes with a time constant of 10.5 ps to form S0 molecules at 298 K. β-Apo-4′-carotenal behaves in a quite similar way. Here, a single vibrationally hot S0* species is sufficient in the kinetic modeling procedure. S0* relaxes with a time constant of 12.1 ps to form cold S0. Finally, no S0* features are visible for β-apo-12′-carotenal. In that case, the S1 → S0 IC process is expected to be roughly 20 times slower than S0* relaxation. As a result, no spectral features of S0* can be found, because there is no chance that a detectable concentration of vibrationally hot molecules is accumulated.
1. Introduction
Photoexcitation of a carotene in the blue-green spectral region provides access to its optically bright second electronically excited state S2(11Bu+). The subsequent dynamics of carotenoids in solution and in light-harvesting arrangements have been interpreted in the framework of various kinetic models involving different electronic states, with several of them still being the subject of considerable debate.1,2 The simplest model for the intramolecular nonradiative relaxation of a carotenoid considers only two consecutive internal conversion (IC) steps via the S1(21Ag−) to the S0(11Ag−) state, as shown in Fig. 1(A). In this simplest case, modeling the optical response of such a kinetic system would require only three species-associated spectra (SAS) for S0, S1 and S2 and the time constants τ2 and τ1 for the two IC processes. | ||
| Fig. 1 Two kinetic schemes for the interpretation of carotenoid photophysics. Horizontal and tilted arrows denote electronic transitions between different singlet states, whereas vertical arrows denote intramolecular vibrational redistribution or vibrational cooling by collisional energy transfer to the surrounding solvent. | ||
However, the situation is indeed much more complicated: firstly, the SAS of all species Si (i = 0–2) depend on the excess energy in the respective state: “Vibrationally hot” molecules Si* show characteristic and complex spectral changes, including an increase of absorption at the red edge of the electronic band and a decrease of absorption maxima. This has been shown in a number of previous studies on molecules in the gas phase and in solution, such as azulene, anthracene and carotene derivatives.3–7 Due to collisional energy transfer (CET) to the surrounding solvent molecules, the SAS will change with a characteristic time constant τCET, typically in the range 7–15 ps for standard organic solvents.4,6,7 In addition, it has to be kept in mind that a simple single exponential decay of a transient absorption signal due to CET can only be expected if the absorption coefficient of the relaxing molecule linearly depends on its internal energy and at the same time the average energy transferred per collision 〈ΔE〉 linearly depends on internal energy.4
Secondly, even for a fixed energy in an electronic state, the way the energy is distributed among the different vibrational modes might severely affect the appearance of the SAS. For instance, IC of a carotene from S2 to S1 initially produces an excited S1 state with a non-statistical energy distribution. The energy will “randomize” over time, eventually leading to a statistical distribution (provided the electronic state lives long enough). This process is well-known as “intramolecular vibrational redistribution (IVR)”.5,8 It is accompanied by considerable spectral changes of the SAS9,10 because energy will be “reshuffled” between optically “dark” and “bright” modes.
Thirdly, the IC time constants τi (i = 1, 2) may also vary with internal energy in the respective state Si. For instance, it is well known from molecular beam experiments that IC rate constants increase with excess energy, e.g. by two or more orders of magnitude in the case of azulene in the S2 and toluene in the S1 state, when their excess energy is raised from 0 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1.11,12
000 cm−1.11,12
A complete description of such a kinetic system is complicated, because the SAS of each species Si smoothly changes with its internal energy (as probably also does its lifetime). We recently introduced a simplified kinetic treatment where such spectral changes are modeled by using time-dependent SAS. This way, IVR in S1 and collisional relaxation of the hot ground state S0* of β-carotene derivatives was successfully described.6,7
Alternatively, following an Occam's razor approach, one may resort to a simplified kinetic scheme with a sufficient number of species (with static SAS) and time constants to describe the essentials of the dynamics, leading us to the scheme in Fig. 1(B): after photoexcitation to S2, a “hot” S1** species9,10 is quickly formed by IC (typically τ2 < 200 fs).1 S1** experiences fast IVR forming an isoenergetic S1* species with a different spectrum. For β-carotene derivatives in n-hexane, one expects a time constant τIVR,1 in the range 400–600 fs.7 Collisional relaxation τCET,1 is expected to be much slower than IVR, as already mentioned, in the 7–15 ps range. Therefore, the sequential relaxation scheme within S1 should be accurate enough.
The relaxation processes within S1 compete with electronic relaxation to the ground electronic state by IC. As noted above, IC of the isoenergetic S1** and S1* species is possibly faster than for the relaxed S1 species, because of their higher internal energy. Nevertheless, due to the fact that energy dependent τ1 values are not yet experimentally available for carotenoids, we assume here the same IC time constant τ1 for all three S1 species. IC accesses a vibrationally “hot” S0 species, denoted as S0**. Especially for short lifetimes τ1 of the S1 state, description of the spectral development of the hot ground state by a single spectrum might not be sufficient, and therefore we also consider here a slightly cooled-down species S0*. Note that the spectral shape of the hot ground state species in reality smoothly varies with excess energy.4,5 Relaxation of a carotenoid by collisional energy transfer to the solvent is then described by two collisional cooling time constants τCET,0a and τCET,0b, respectively. The scheme in Fig. 1(B) will serve as the basis for describing the carotenoid systems studied here.
It should be noted that additional spectral features have been observed which have been assigned to different intermediates formed during the electronic relaxation of carotenoids. For instance, the S* signal was observed which is still under debate.2,6,7,13 Others and we identify this S* signal as the vibrationally hot ground electronic state (S0**/S0*) discussed above,6,7,14–18 Alternatively, S* has been either described as an electronically excited state,18–24 different S0 conformers,25,26 or the vibrationally hot S1 state.27
In our previous investigations of β-carotene and 13,13′-diphenyl-β-carotene, we presented strong evidence that the S* spectral features found in broadband transient absorption experiments arise from the characteristic absorption of vibrationally “hot” carotene molecules in S0 which are generated by IC from the S1 state, i.e. the S0**/S0* species in Fig. 1(B).6,7 These species showed the typical “hot band” absorption on the red edge of the S0 → S2 absorption band and a characteristic “finger-type” structure in the S0 → S2 ground-state bleach (GSB). Both result from the subtraction of the “cold” (298 K) GSB spectrum from the transient “hot” spectra of the S0**/S0* molecules. This interpretation was strongly supported by the result of steady-state temperature-dependent absorption spectra, which nicely reproduced the difference spectra in the transient absorption experiments, including the “hot band” absorption and the “finger-type” structure.6
Here, we provide additional strong support for this mechanism and the relaxation scheme in Fig. 1(B) by a systematic study of S0**/S0* spectral features for three all-trans apocarotenals featuring increasing conjugation lengths (Fig. 2). This way, the S1 lifetime and thus the time constant τ1 for generating S0**/S0* population via IC from S1 can be precisely varied in wide ranges.1,28 On the other hand, it is a well-known fact that time constants of collisional relaxation τCET in solution do not strongly depend on the type of the vibrationally excited molecule (e.g. 10–13 ps in the case of azulene in n-alkanes).4,6,7 Therefore one should be able to “tune” the intensity and spectral shape of the S0* state spectral features by variation of the conjugation length of the apocarotenal. As will be shown below, we indeed observe exactly such a behavior in the apocarotenal systems and therefore believe that the spectral features of S* arise from vibrationally hot molecules in the ground electronic state.
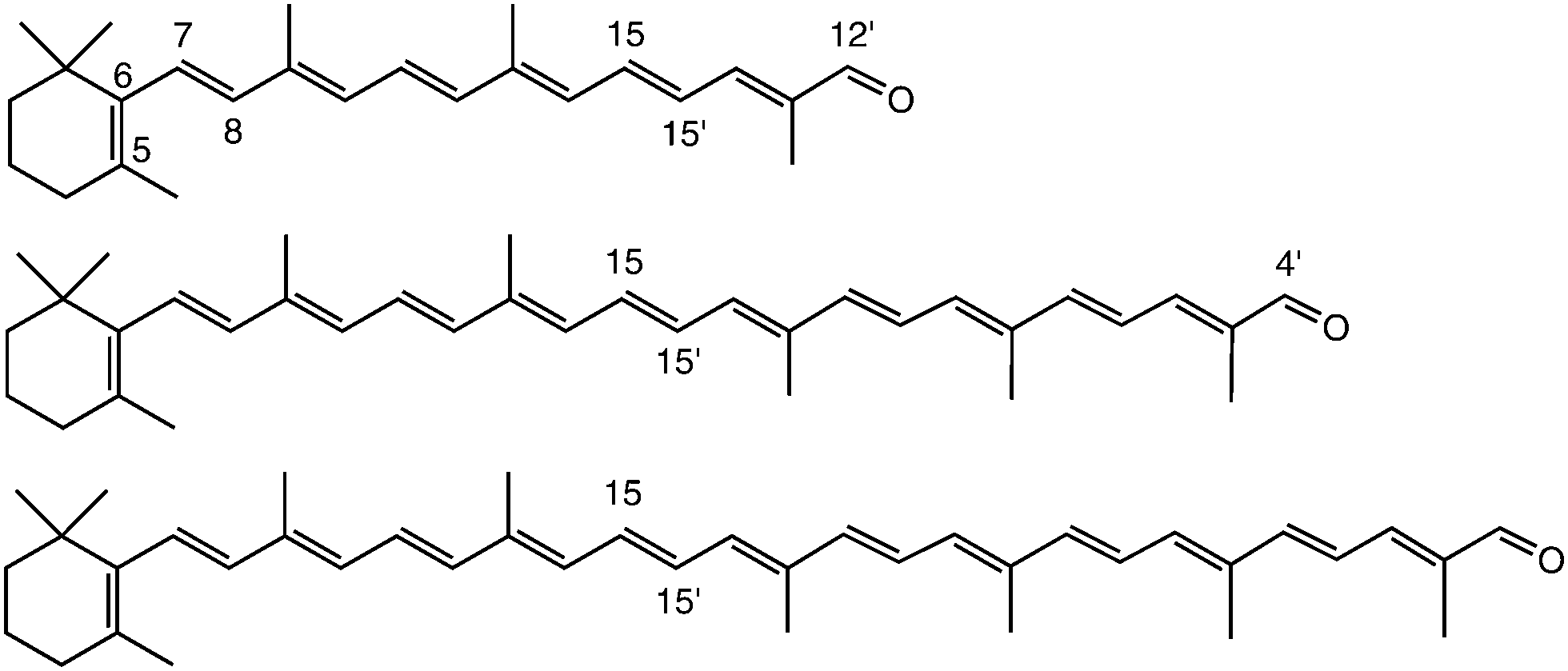 | ||
| Fig. 2 Chemical structure of β-apo-12′-carotenal (C25H34O, top), β-apo-4′-carotenal (C35H46O, middle) and torularhodinaldehyde (C40H52O, bottom). | ||
2. Experimental
2.1 Chemicals
The structure of the three all-trans apocarotenals β-apo-12′-carotenal (C25 aldehyde), β-apo-4′-carotenal (C35 aldehyde) and 3′,4′-didehydro-β,ψ-caroten-16′-al (C40 aldehyde, “torularhodinaldehyde”) are shown in Fig. 2. β-Apo-12′-carotenal was a generous gift from BASF SE (Dr Hansgeorg Ernst) and used as received (purity > 97%). The other two apocarotenals were synthesized by repeated extension of the polyene chain via Wittig reaction of β-apo-8′-carotenal (C30 aldehyde) and a C5 phosphonium salt (both also generous gifts by Dr Hansgeorg Ernst from BASF SE). Details of the synthesis and purification procedures can be found in the ESI.† All experiments were performed in the solvent n-hexane (Merck Uvasol).2.2 Pump–supercontinuum probe (PSCP) spectroscopy
Ultrafast broadband transient absorption experiments were carried out using our setup for pump–supercontinuum probe (PSCP) spectroscopy.7,29–32 The apocarotenal sample was excited by a NOPA (λpump = 490 or 500 nm, beam diameter ca. 250 μm, energy ca. 1.0–1.5 μJ pulse−1) and probed by a supercontinuum (350–770 nm, beam diameter ca. 150 μm), with the relative polarization set at magic angle. The pump–probe intensity cross-correlation time was ca. 90 fs, with a time zero accuracy of ca. 10 fs. To avoid any influence of photoinduced sample degradation, 10–15 mL of an apocarotenal solution in n-hexane was circulated through a flow cell (path length 400 μm, window thickness 200 μm), so that the sample volume was exchanged after each pump–probe experiment (repetition frequency 920 Hz). Solutions with an OD (at maximum, 400 μm path length) of 0.333 (β-apo-12′-carotenal), 0.174 (β-apo-4′-carotenal) and 0.752 (torularhodinaldehyde) were employed. For these conditions, the transient absorption signals were in the linear regime, as confirmed in separate experiments. Steady-state absorption spectra of the sample solutions were recorded on a Varian Cary 5000 dual-beam spectrometer.2.3 Global analysis procedure
The PSCP spectra were chirp-corrected by using the transient response of the pure n-hexane solvent. The spectra were then subjected to kinetic modeling. Each data set, which consisted of 512 kinetic traces, was analyzed globally using the kinetic schemes discussed below. Each species-associated spectrum was represented by a sufficiently large number of Gaussian functions, and only the known S0 spectrum was kept fixed. During the fitting procedure, all parameters (position, width, height of the Gaussian functions and time constants of the kinetic model) were optimized simultaneously to arrive at the best fit, which also considered the experimentally determined time resolution by a convolution procedure.3. Results and discussion
3.1 Steady-state absorption spectra
Fig. 3 shows steady-state absorption spectra of the three apocarotenals in n-hexane solution at 298.15 K. Upon extension of the polyene chain, there is a clear red-shift of the S0 → S2 band of the apocarotenals, suggesting a reduction of the S0–S2 energy gap. At the same time, the structure of the electronic band becomes more pronounced. The latter can be explained by a reduced influence of the terminal β-ionone ring on the absorption properties of the carotenes, as suggested by Christensen et al.:33 according to their “distribution of conformers” model, conformations featuring different torsional angles C5–C6–C7–C8 (Fig. 2) of the β-ionone ring with respect to the rest of the polyene system, result in different “effective conjugation lengths”, and thus a distribution of transition energies. The impact of this effect will be much weaker for a larger number of conjugated double bonds, thus the spectra will show more structure and appear more resolved.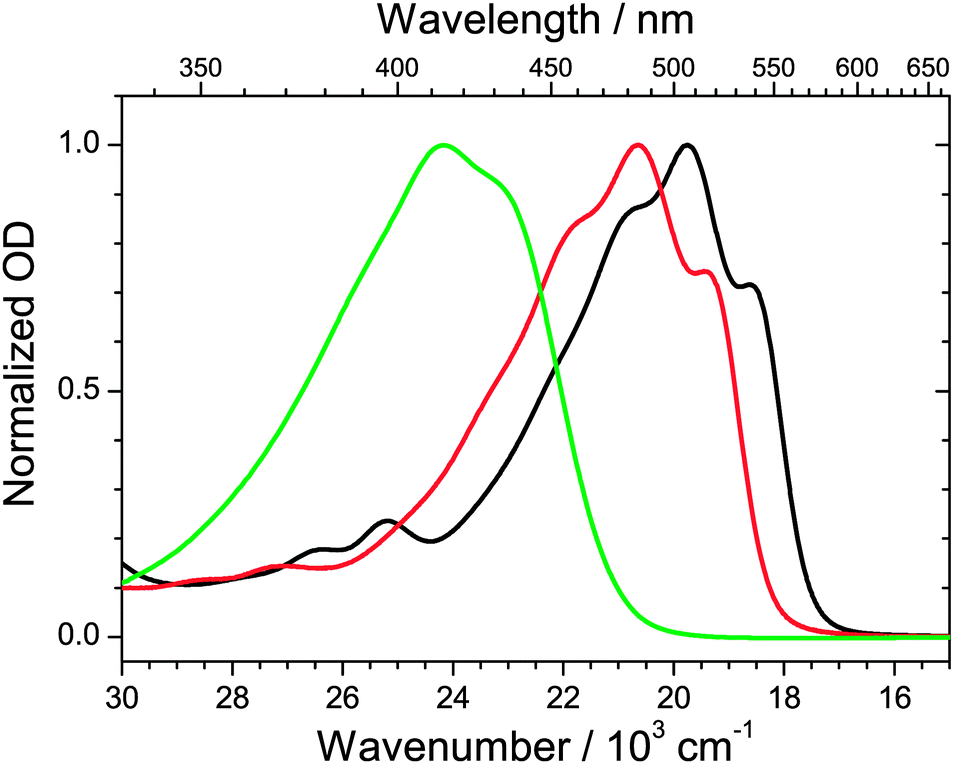 | ||
| Fig. 3 Steady-state absorption spectra of β-apo-12′-carotenal (green), β-apo-4′-carotenal (red) and torularhodinaldehyde (black) in n-hexane at 298.15 K. | ||
3.2 Ultrafast dynamics of torularhodinaldehyde
We commence with the PSCP transient absorption data for torularhodinaldehyde in n-hexane, shown in Fig. 4. The carotene was excited at 500 nm (S0 → S2 band). The top panel shows the dynamics around t = 0. We observe a pronounced bleach of the ground electronic state centered at 510 nm. In addition, absorption features appear in the 570–770 nm range. We assign these to S2 → Sn excited state absorption (ESA), in agreement with our earlier PSCP experiments on other carotene systems.6,7,31 The wavelength range below 450 nm appears to be spectrally silent, however, based on the inverted steady-state absorption spectrum, shown in the bottom panel as a dotted orange line, it is clear that there must be superimposed broad and unstructured S2 → Sn ESA in this wavelength range compensating the bleach.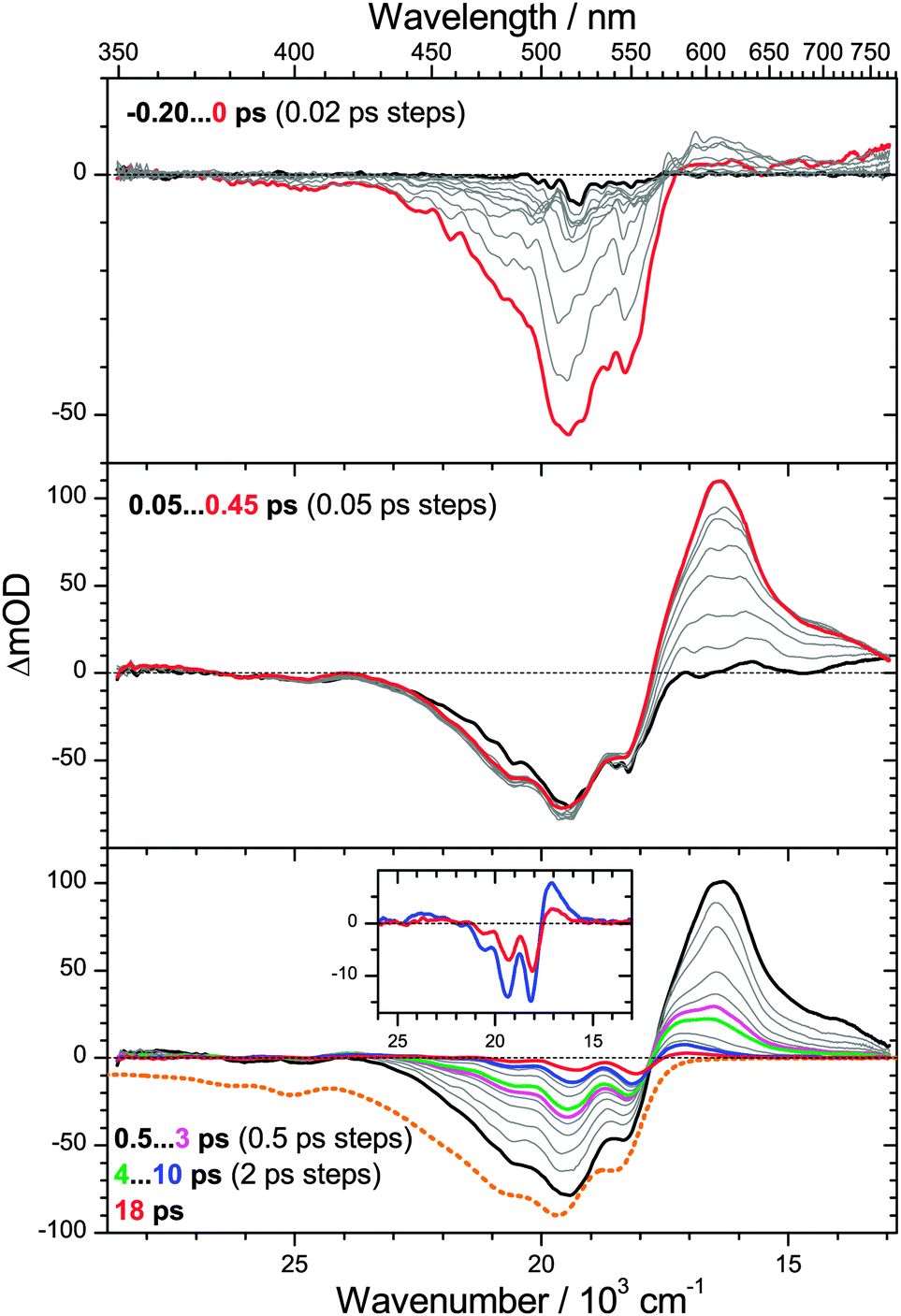 | ||
| Fig. 4 Transient PSCP absorption spectra of torularhodinaldehyde in n-hexane for excitation at 500 nm: (upper panel) −0.2–0 ps with 20 fs steps; (middle panel) 0.05–0.45 ps with 50 fs steps; (lower panel) 0.5–3 ps and 4–10 ps with 0.5 ps and 2 ps steps, respectively, and 18 ps. Selected transient spectra are shown as thick colored lines for guidance. For comparison, the inverted and scaled steady-state absorption spectrum is shown in the lower panel as an orange dotted line. The inset in the bottom panel shows a magnification of the spectra at 10 and 18 ps. | ||
In the middle panel, the spectral development up to 0.45 ps is depicted. One observes the rise of a strong S1 → Sn ESA band above 560 nm, as a result of S2 → S1 internal conversion (IC) with a time constant τ2 = 200 fs, obtained from a global analysis procedure. The value is reasonable, compared with our earlier values found for β-carotene derivatives (160 fs), yet it could be also shorter, as the spectral dynamics could be “contaminated” by fast relaxation processes of the nascent S1** state due to IVR. While there is a slight narrowing of the S1 ESA band, a separate time constant τIVR,1 cannot be reliably determined. The region below 450 nm remains spectrally silent. This suggests that there is an S1 → Sn ESA band which must be similarly unstructured and weak as in the case of the S2 state.
The bottom panel shows the dynamics on longer timescales. Starting from 0.5 ps (black line), the absorption band decays in a non-uniform way: the peak centered at 600 nm quickly disappears, and what remains is a relatively broad absorption in the range 550 to 650 nm, see e.g. the green spectrum at 4 ps. We assign this process to IC from S1 with a time constant τ1 = 1.2 ps. In addition, the peaks in the bleach region are more and more sharpening. Based on our previous findings for β-carotene derivatives,6,7 we assign the remaining spectral features to the almost pure difference spectrum of vibrationally hot S0 molecules which are generated by the fast IC process. The shape of this spectrum is explained as follows: first, a spectral hallmark of such species is a “hot-band” absorption (resulting from absorption of the population in the “Boltzmann tail”) which should appear on the red edge of the room-temperature electronic spectrum (compare Fig. 3). Indeed, a pronounced absorption “bump” is located between 570 and 650 nm, see also the inset in the bottom panel of Fig. 4 for the spectra at 10 ps (blue line) and 18 ps (red line). The structured bleach in the 470–570 nm region results from another typical spectral property of vibrationally hot molecules, namely a decrease of spectral maxima and increase of spectral minima compared to room temperature spectra, see e.g. previous temperature-dependent gas-phase studies of the electronic spectra of azulene and anthracene by Schwarzer and co-workers5 and our previous temperature dependent study of β-carotene steady-state absorption spectra in solution.6 Subtraction of the room-temperature steady-state absorption spectrum from a transient spectrum will therefore result in a pronounced undulatory structure in the bleach region. This is also clearly seen in the inset of the bottom panel in Fig. 4 for the spectra at 10 and 18 ps. The “modulation depth” of the maxima and minima in the two transient spectra is much stronger than for the room temperature spectrum (orange dotted line in the bottom panel).
Its short S1 lifetime makes torularhodinaldehyde a textbook example for studying S0* collisional energy transfer of carotenoids in solution. As shown in more detail below, collisional relaxation in the ground electronic state can be modeled best by employing two “hot” species S0** and S0* in the global kinetic analysis, relaxing with time constants of 3.4 and 10.5 ps, respectively. The second time constant agrees well with our previous findings for β-carotene derivatives in the same solvent: 11.9 ps for β-carotene6 and 10.4 ps for 13,13′-diphenyl-β-carotene.7 In the latter two cases, the S0* state features were weaker, because of the seven times larger S1 lifetime (τ1 = 8.7 and 9.2 ps, respectively), and the correspondingly smaller S0* transient absorption features. The cooling time constant of S0* of torularhodinaldehyde also compares well with vibrational relaxation time constants of smaller organic molecules, such as azulene, where one finds τCET,0 = 10–13 ps for a series of alkanes.4
3.3 Photoinduced dynamics of β-apo-4′-carotenal
To further test the aforementioned generation and cooling mechanism of S0* molecules, we examined the photoinduced dynamics of β-apo-4′-carotenal in n-hexane. This carotenoid possesses the same type of chromophore as torularhodinaldehyde, yet the conjugated polyene system is slightly shorter. This should lead to a longer S1 lifetime, as already suggested by results from our previous single-wavelength pump–probe studies in various solvents.28PSCP spectra are shown in Fig. 5. In the top panel, the early-time dynamics around t = 0 are similar to those for torularhodinaldehyde, with pronounced S0 → S2 bleach features centered at 500 nm and weak S2 → Sn ESA above 600 nm. In the middle panel, the formation of S1 → Sn ESA is observed, with a time constant τ2 = 180 fs, again very similar to torularhodinaldehyde. However, we note that the peak of the bleach and the S1 ESA are shifted to the blue by ca. 840 cm−1 and 570 cm−1, respectively, a result of the shorter conjugation length of β-apo-4′-carotenal.
 | ||
| Fig. 5 Transient PSCP absorption spectra of β-apo-4′-carotenal in n-hexane for excitation at 500 nm: (upper panel) −0.2–0 ps with 20 fs steps; (middle panel) 0.05–0.50 ps with 50 fs steps; (lower panel) 1–8 ps with 1 ps steps and 10, 14 and 18 ps. Selected transient spectra are shown as thick colored lines for guidance. For comparison, the inverted and scaled steady-state absorption spectrum is shown in the lower panel as an orange dotted line. The inset in the bottom panel shows a magnification of the spectra at 10 and 18 ps. | ||
The bottom panel depicts the behavior on longer time scales. The decay time of the S1 ESA band (peak at 590 nm) is τ1 = 4.9 ps. The time constant of the S1 → S0* IC process is thus by a factor of four larger than for torularhodinaldehyde. This becomes particularly clear in the inset, where the blue spectrum at 10 ps still shows a remainder of the S1 ESA peak at 590 nm (in contrast to Fig. 4) overlapped by the characteristic signature of the vibrationally hot S0* molecules formed by the IC process. The “S0* part” of the transient spectrum at 10 ps shows the same fingerprints as in Fig. 4: a “hot band” absorption at 560 nm and a bleach which is much more structured than the ground state bleach of cold S0 molecules. Importantly, the blue-shift of the S0* state difference spectra (840 cm−1) is identical to the blue-shift of the S0 → S2 steady-state absorption spectra, in agreement with the assignment to a hot ground-state species. The decay time of the S0* state feature obtained from global spectral analysis is τCET,0 = 12.1 ps, in line with the S0* collisional cooling time of torularhodinaldehyde.
3.4 Ultrafast dynamics of β-apo-12′-carotenal
So far, we have presented transient spectra for two apocarotenals where τ1 < τCET,0, i.e. the S0* species are formed faster than they are cooled by collisions. As a result, pronounced transient spectral features of S0* were clearly identified. We now turn to an example, where τ1 ≫ τCET,0. In such a case, cooling of S0* molecules by collisions with solvent molecules is much faster than their generation from S1. Thus, a detectable concentration of vibrationally hot S0* species in the ground electronic state is never accumulated and spectral features of S0* should be absent.Experimentally, such conditions are established by further shortening the polyene chain of an apocarotenal, which largely increases τ1,28,34,35 whereas the time constant for collisional energy transfer τCET is much less sensitive to such a structural modification, as demonstrated by the results for torularhodinaldehyde and β-apo-4′-carotenal in this paper and previous experiments on a range of organic molecules.4,36 Here, we therefore choose β-apo-12′-carotenal which features a rather short conjugation length. In our previous preliminary studies on this molecule we found a lifetime τ1 of ca. 200 ps,34,35 therefore one expects τ1 ≈ 20·τCET,0 in this case.
PSCP spectra of β-apo-12′-carotenal in n-hexane (λpump = 490 nm) are presented in Fig. 6. The behavior at early times in the top panel is shown for the same delay times as in Fig. 4 and 5. Because of the much shorter conjugation length, the pronounced S0 → S2 ground state bleach appears in the range 360–470 nm, in agreement with the steady-state absorption spectrum (Fig. 3). We note that the center peak at 430 nm is due to anti-Stokes Raman scattering of the C–H stretching modes of n-hexane and only present during temporal overlap of the pump and probe pulses. Above 470 nm, one observes S2 → Sn ESA features, again blue-shifted compared to torularhodinaldehyde and β-apo-4′-carotenal.
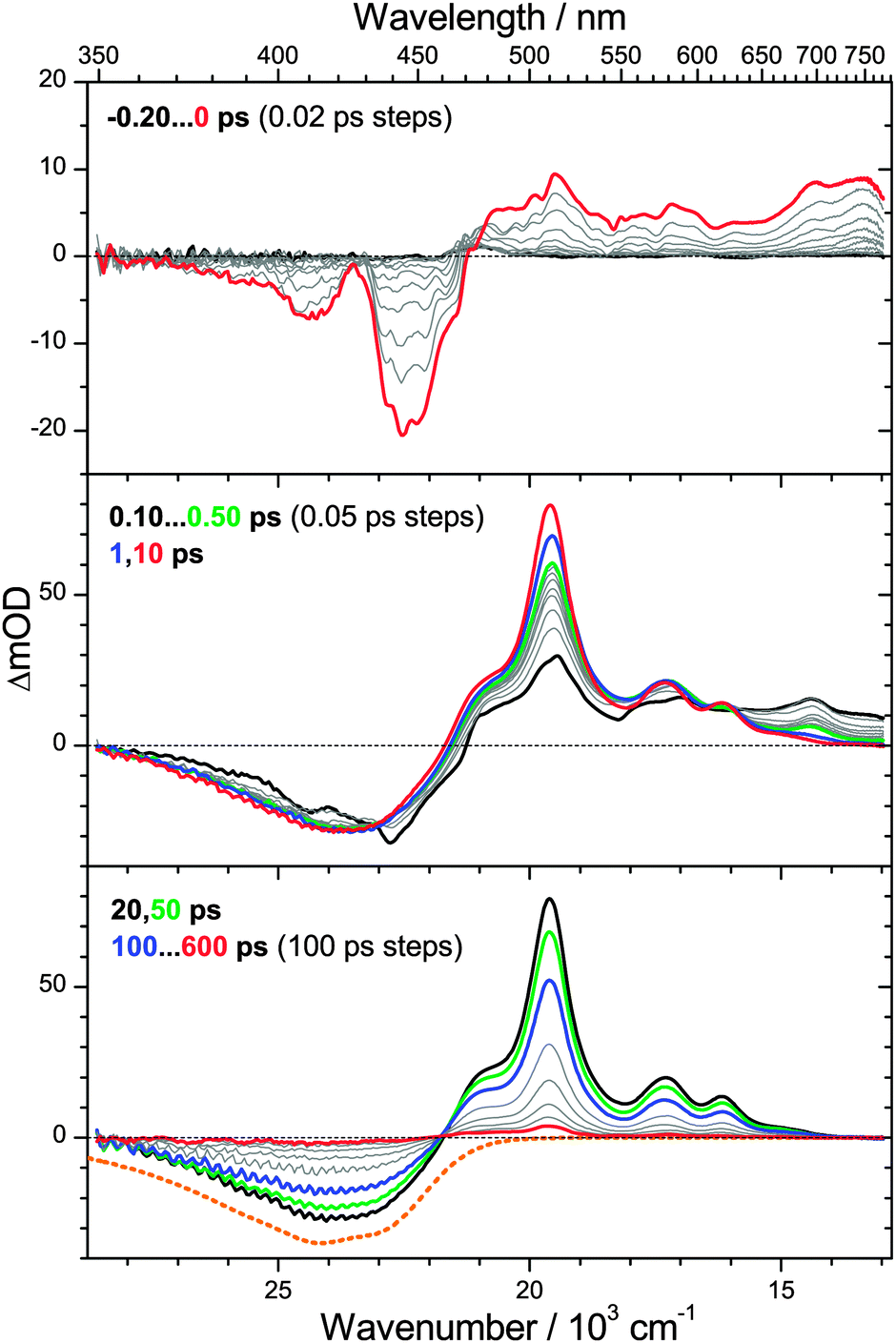 | ||
| Fig. 6 Transient PSCP absorption spectra of β-apo-12′-carotenal in n-hexane for excitation at 490 nm: (upper panel) −0.2–0 ps with 20 fs steps; (middle panel) 0.10–0.50 ps with 50 fs steps, 1 ps and 10 ps; (lower panel) 20 ps and 50 ps, 100–600 ps with 100 ps steps. Selected transient spectra are shown as thick colored lines for guidance. For comparison, the inverted and scaled steady-state absorption spectrum is shown in the lower panel as an orange dotted line. | ||
In the middle panel one finds that the S2 → Sn ESA quickly disappears due to ultrafast IC (τ2 = 105 fs) and is replaced by a pronounced S1 → Sn ESA band. The S1 state undergoes further internal relaxation, as indicated by the green, blue and red lines for delay times of 0.50, 1 and 10 ps, respectively. The ESA band becomes sharper, and especially the peak at 510 nm rises. We assign these dynamics to two processes: fast IVR of the initially prepared S1** species with a time constant τIVR,1 = 0.44 ps to form S1*. The value is in very good agreement with values previously obtained by us for IVR timescales of β-carotene derivatives.6,7 The second, slower, process is collisional deactivation of S1* by the n-hexane solvent with a time constant of τCET,1 = 8.3 ps. Note that the latter process is analogous to the S0* → S0 relaxation discussed above, however the initial vibrational excess energy of the S1* molecules is of course lower and therefore the “hot band” dynamics in the S1 state is less pronounced. Still it is clearly detectable for β-apo-12′-carotenal because its S1 lifetime is much larger than for torularhodinaldehyde and β-apo-4′-carotenal.
In the bottom panel, one follows the slow S1 → S0 IC process from the internally almost relaxed S1 state with a time constant of 192 ps. The S1 → Sn ESA band decays and at the same time the ground state bleach is filled up. There is an isosbestic point at 460 nm and, in contrast to the other two apocarotenals, we do not find indications for spectral features of S0*.
3.5 Simplified kinetic schemes for apocarotenals in n-hexane
The transient PSCP spectra of the three apocarotenal systems were analyzed using simplified forms of the general scheme in Fig. 1(B). In the case of torularhodinaldehyde and β-apo-4′-carotenal we could not observe clear spectral indications of IVR of S1**, except for a very minor narrowing of the S1 band, which did not allow us to extract a reliable value for τIVR,1. Also, because of the short IC lifetime τ1, spectral indications for the collisional relaxation of S1* are of course not present, and therefore a single S1 species is sufficient for modeling the dynamics.We simulate the results for torularhodinaldehyde employing the simplified sequential scheme in Fig. 7(A). Because of the very short S1 lifetime of 1.2 ps, a substantial population of hot ground state molecules with initially high excess energy is quickly generated. Two vibrationally hot species S0** and S0* are employed to describe the complex change of the hot ground state spectrum resulting from vibrational cooling by solvent collisions. The corresponding SAS are shown in Fig. 8(A). Characteristic spectral changes due to collisional energy transfer are evident in the S0**, S0* and S0 spectra (green, red and blue): the pronounced hot band at ca. 620 nm in the S0** spectrum disappears upon cooling and the spectrum narrows, leading to S0* which then further cools down to 298 K (S0). Time constants of 3.4 and 10.5 ps are found for this collisional cooling process. Such biexponential dynamics is not unusual and expected if the absorption coefficient of S0 does not linearly depend on excess energy.4 The oscillator strength of the S0 species is approximately conserved, as expected for a CET process. Note also that the spectral development is comparable to β-carotene derivatives, as previously reported by us in ref. 6 and 7. A similar spectral behavior of vibrationally hot ground state molecules is also observed for other organic molecules, compare e.g. the temperature-dependent absorption spectra for azulene and anthracene reported in Fig. 3 and 4 of ref. 5. This experimental evidence therefore supports our assignment of the S* spectral features2 to vibrationally hot ground state molecules. Kinetic traces for three selected wavelengths are included in Fig. 9. They show the superposition of the fast S1 decay dynamics with the collisional relaxation of the hot S0**/S0* species resulting in the curved shape and the slow final decay of the transients. Global fit parameters are summarized in Table 1.
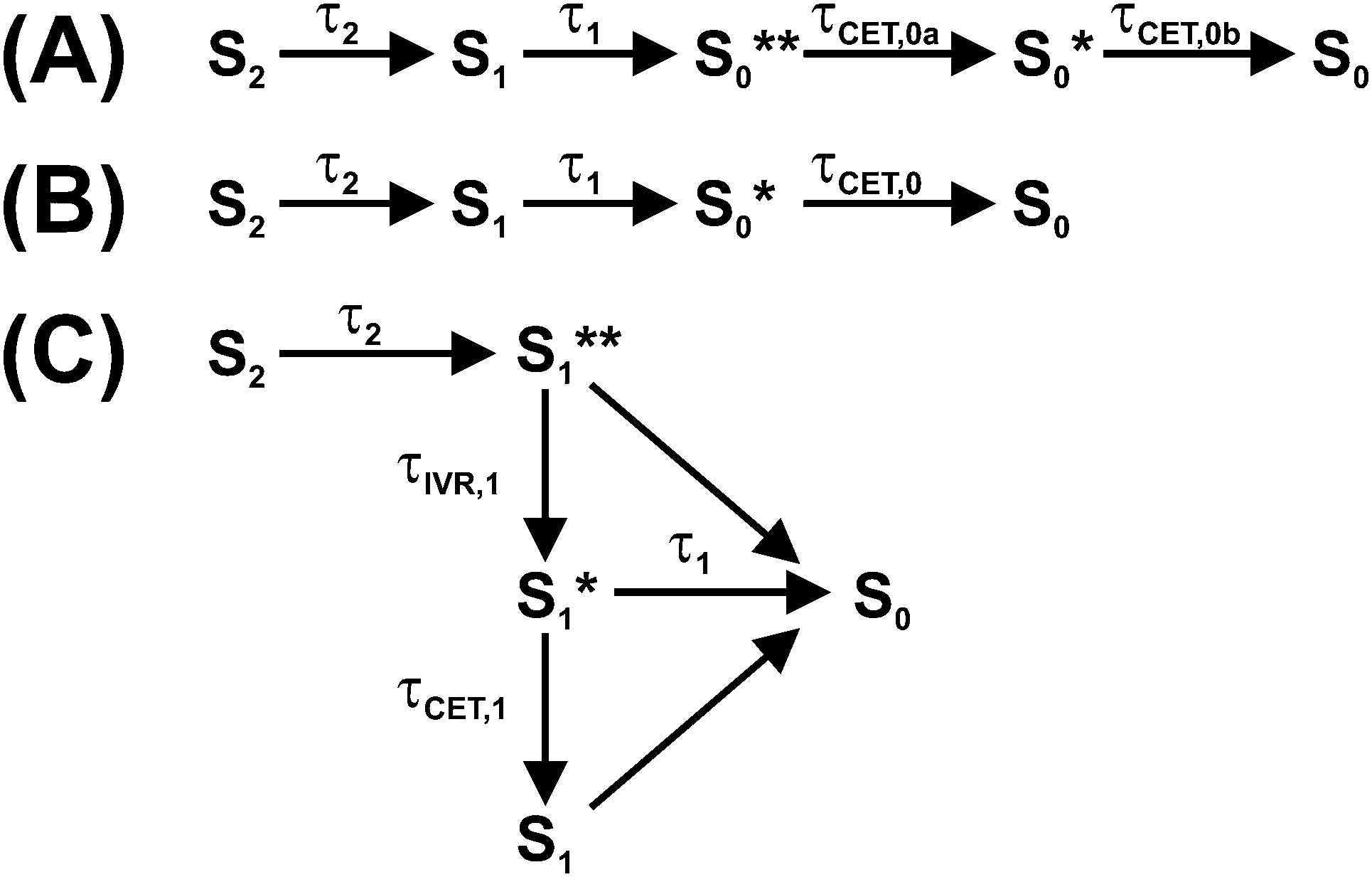 | ||
| Fig. 7 Simplified kinetic schemes based on the general mechanism in Fig. 1(B) for interpreting the photophysics of the three apocarotenals in n-hexane. Relaxation schemes for (A) torularhodinaldehyde, (B) β-apo-4′-carotenal and (C) β-apo-12′-carotenal. | ||
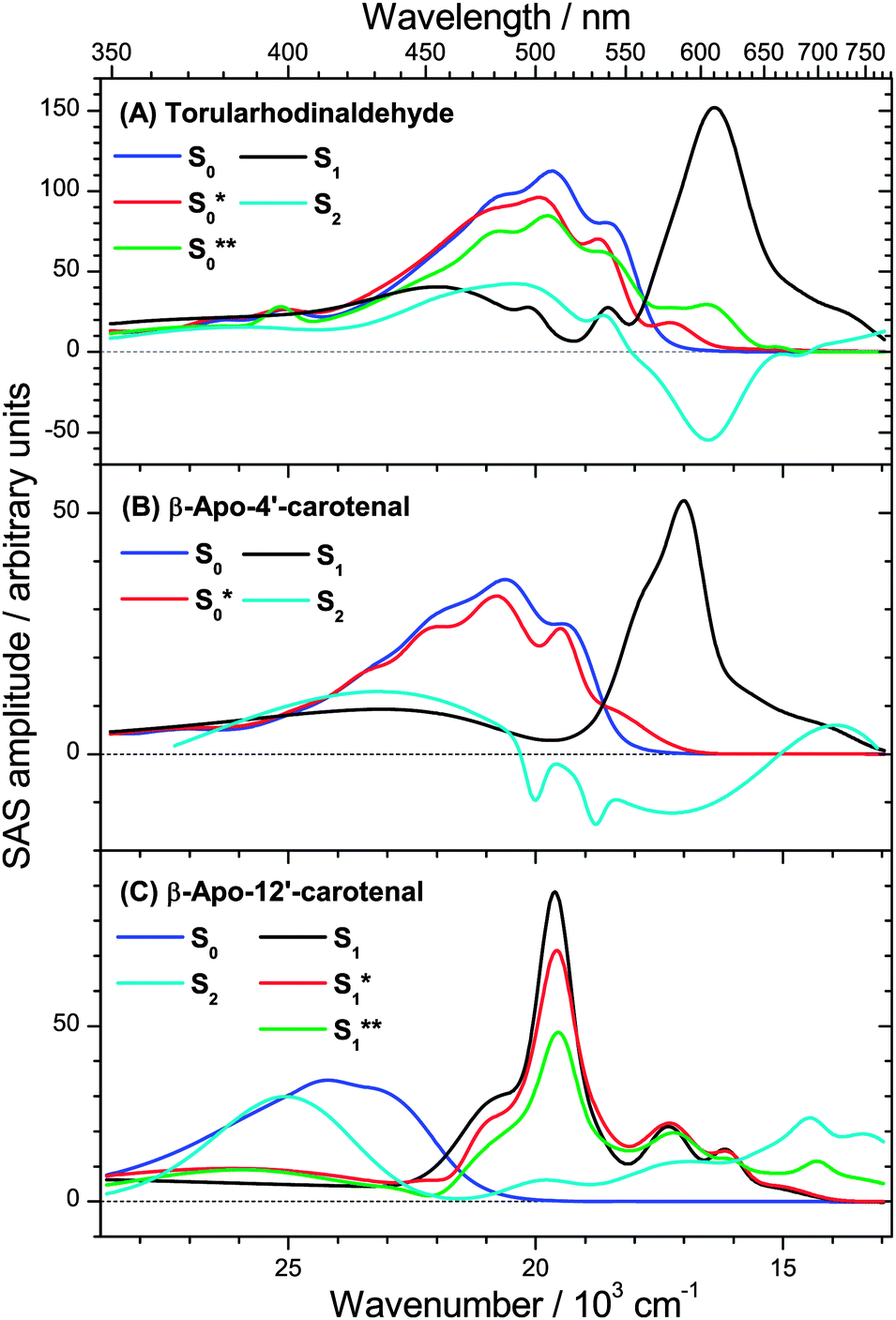 | ||
| Fig. 8 Species-associated spectra (SAS) in n-hexane of (A) torularhodinaldehyde, (B) β-apo-4′-carotenal and (C) β-apo-12′-carotenal. | ||
 | ||
| Fig. 9 Kinetic traces for torularhodinaldehyde in n-hexane at (A) λprobe = 501 nm, (B) λprobe = 557 nm and (C) λprobe = 585 nm. | ||
| Carotenal | λ pump (nm) |
τ
CCa (fs) |
τ
2b (fs) |
τ
IVR,1 and τCET,1c (ps) |
τ
1d (ps) |
τ
CET,0a and τCET,0be (ps) |
|---|---|---|---|---|---|---|
| a Pump–probe intensity cross-correlation of the experiment. b Internal conversion (IC) time constant for S2 → S1, initially populating a hot S1 state (S1**), which is not internally equilibrated. c Time constant for narrowing of the S1 → Sn ESA band due to IVR (intramolecular vibrational redistribution; fast process) and CET (collisional energy transfer; slow process), not clearly assignable for torularhodinaldehyde and β-apo-4′-carotenal because of the short S1 lifetime. d Internal conversion (IC) time constant populating the vibrationally hot ground electronic state. e Time constants for collisional energy transfer in the ground electronic state. Two time constants are required for torularhodinaldehyde, whereas for β-apo-4′-carotenal one time constant suffices. In the case of β-apo-12′-carotenal, IC from S1 is very slow: thus the concentration of S0* is very low and its contribution to the PSCP spectra too low to be detected. | ||||||
| Torularhodinaldehyde | 500 | 90 | 200 | — | 1.2 | 3.4, 10.5 |
| β-Apo-4′-carotenal | 500 | 90 | 180 | — | 4.9 | 12.1 |
| β-Apo-12′-carotenal | 490 | 90 | 105 | 0.44, 8.3 | 192 | — |
Our interpretation is further confirmed for β-apo-4′-carotenal. SAS can be found in Fig. 8(B). Because of the longer S1 lifetime of τ1 = 4.9 ps and the therefore slower production of hot ground state molecules, collisional cooling (τCET = 12.1 ps) competes more efficiently. As a result, the PSCP spectra can be fully modeled by considering the simplified kinetic scheme given in Fig. 7(B), including only a single (cooler) hot ground state species S0*. Note that the resulting SAS for S0* is very similar to the slightly cooled S0* species of torularhodinaldehyde. Three selected kinetic traces are shown in Fig. 10. Here, the longer lifetime of S1 is evident (4.9 vs. 1.2 ps for torularhodinaldehyde), and therefore the S1 decay is not as clearly separated from the cooling process of S0* as in the case of torularhodinaldehyde. A summary of global fit parameters is included in Table 1.
 | ||
| Fig. 10 Kinetic traces for β-apo-4′carotenal in n-hexane at (A) λprobe = 471 nm, (B) λprobe = 529 nm and (C) λprobe = 565 nm. | ||
In the case of β-apo-12′-carotenal, IC from the S1 state is very slow: τ1 ≫ τIVR,1, τCET,1. As a result, IVR and CET in S1 should be clearly observable. At the same time, this also means that τ1 ≫ τCET,0. Therefore, the steady-state approximation for S0* holds, and thus the concentration of S0* will be very small at all times. One therefore does not expect to see any S0* spectral features and thus the simplified scheme in Fig. 7(C) is applied. The resulting SAS are depicted in Fig. 8(C). Characteristic spectral changes accompany the relaxation within the S1 state: the initially formed S1** species shows a broad spectrum (green line) extending to the upper wavelength limit of our spectral observation window, including a small but distinct peak at 700 nm. The main peak at 510 nm is already visible but it is small and broad, as is the rest of the spectrum. Due to fast IVR, an S1* species is quickly formed (τIVR,1 = 440 fs), which is internally equilibrated. This species exhibits a narrower spectrum and a steeper main peak (red line). S1* then relaxes further by CET to the solvent to form the room temperature S1 species (black line), with the aforementioned time constant τCET = 8.3 ps. Because of very slow IC to S0 (τ1 = 192 ps) we do not find spectral indications for an S0* species which due to its extremely low concentration is below the detection limit.
Three representative kinetic traces are depicted in Fig. 11. CET in S1 is apparent in the 501 nm transient as a characteristic round shape in the 2–30 ps range. Here the steepening of the main peak of the S1 ESA band due to CET is superimposed by slow population loss due to IC to S0. Kinetic parameters obtained from the global analysis are again summarized in Table 1.
 | ||
| Fig. 11 Kinetic traces for β-apo-12′carotenal in n-hexane at (A) λprobe = 405 nm, (B) λprobe = 501 nm and (C) λprobe = 577 nm. | ||
The impact of vibrationally highly excited S0 and S1 species for the different apocarotenals is finally highlighted in Fig. 12, where PSCP spectra (open circles) and global analysis results (lines) are shown for the fixed delay time of 10 ps.
 | ||
| Fig. 12 PSCP spectra (circles) at 10 ps in n-hexane for (A) torularhodinaldehyde, (B) β-apo-4′carotenal and (C) β-apo-12′-carotenal, including total fit and contributions from individual species. | ||
For torularhodinaldehyde (Fig. 12(A)), the S1 population has already completely decayed (flat magenta line), and the structured spectrum is a result of the superposition of hot ground state absorption (mainly S0* (red), but also S0**(green)) and the bleach of room-temperature S0 molecules (blue).
For β-apo-4′-carotenal (Fig. 12(B)), the resulting spectrum is a superposition of remaining S1 absorption (magenta), hot ground state absorption of S0* (red) and the bleach of cold S0 molecules (blue). The S1 and S0* contributions are responsible for the characteristic broad double-peak structure in the range 560–590 nm, which we similarly observed previously for β-carotene and 13,13′-diphenyl-β-carotene,6,7 which feature only a slightly longer S1 lifetime (ca. 9 ps).
In the case of β-apo-12′-carotenal (Fig. 12(C)), the spectrum at 10 ps contains only ESA contributions from the S1 and S1* species (CET in the first electronically excited state is still ongoing), which is superimposed by the bleach of cold S0 molecules (blue). Because of the long S1 lifetime (192 ps) the concentration of S0* molecules is negligible, and therefore spectral features of S0* are not visible at all.
4. Conclusions
We have demonstrated that three apocarotenals with very different S1 lifetimes can be described by simplified versions of the relaxation model in Fig. 1(B), considering only three electronic species S2, S1 and S0 and additional “distortions” of their spectra due to internal vibrational excess energy or incomplete IVR (Fig. 7). These “spectral distortions” were captured in a simplified kinetic model by introducing vibrationally “hot” electronic species S0**, S0* and S1* and a not internally equilibrated S1** species. The visibility of these species in transient spectra depends on the specific value of the S1 lifetime τ1, e.g. if it is much larger or much smaller than typical time constants for collisional relaxation (τCET ≈ 10 ps). The spectral properties of the “hot” species found in our global analysis are completely consistent with experimental data in the literature for vibrationally hot organic molecules, e.g. showing a characteristic hot band absorption tail reaching by 1000–2000 cm−1 toward the red spectral range.4,5 Moreover, in our previous study of β-carotene, the difference absorption spectrum of S0* was independently measured in separate temperature-dependent steady-state absorption experiments and found to be very similar to the S* spectral features in the transient absorption spectra.6 The same characteristic spectral signatures are found in the current study for torularhodinaldehyde and β-apo-4′-carotenal, see Fig. 12(A) and (B), and this striking similarity is further highlighted in the comparison shown in Fig. S1 of the ESI.† We also believe that this model is applicable to a wide range of carotenoids. As one example, we already demonstrated in two previous studies that the photoinduced processes of two β-carotene derivatives are well described in the same way.6,7 As another example, clear S0*-like spectral signatures were found by Gillbro and co-workers for the long-chain dodecapreno-β-carotene which features a very short S1 lifetime of τ1 = 0.5 ps.14 Its spectral features are very similar to those of torularhodinaldehyde.Also, various observations from previous experimental transient absorption studies of carotenoids can be traced back to vibrationally hot ground state molecules: for instance, Papagiannakis et al.37 and Savolainen et al.38 found that the excitation intensity modified the intensity ratio between S* and S1 absorption signals for carotenoids bound to natural and artificial light-harvesting complexes. In both cases, the S*/S1 absorption ratio increased when the pump energy was increased and entered the nonlinear regime. Such a behavior can be traced back to the increasing influence of a two-photon excited population with high initial excess energy. After IC, such carotenoid molecules will enter S0 with an excess energy much higher than for an initially one-photon excited population. The S0** spectrum of these molecules with high internal temperature will be considerably distorted giving rise to much stronger hot band absorption features than those of the one-photon excited S0* population.
In addition, Billsten et al. observed that the intensity of the S* signal of zeaxanthin was correlated with the amount of initial excess energy:39 the S* absorption features became more pronounced when decreasing the excitation wavelength (485, 400 and 266 nm). This can be explained in the same way as above. For higher initial excitation energies, zeaxanthin will enter S0 with much higher excess energy which gives rise to more pronounced S* absorption features.
We therefore believe that extensions of our kinetic model will be only required in special cases: for instance, systems such as β-apo-12′-carotenal are known to exhibit significant charge transfer character in polar solvents, including the appearance of substantial stimulated emission and a drastic reduction of the S1 lifetime:28,34,35,40,41 the first electronically excited state is then better termed “S1/ICT”,42–44 but the basic model of three electronic states survives. Because formation of the S1/ICT state leads to a substantial change in dipole moment, solvation dynamics will induce a transient Stokes shift of the S1/ICT stimulated emission and possibly also excited state absorption features, as already demonstrated by us previously.34 In the simplest approach, solvation dynamics can be handled e.g. by introducing an additional S1 species in the models of Fig. 1(B) and 7 or by introducing a time-dependence for the S1 spectrum, as demonstrated by us previously.6,7 Short-chain apocarotenals, such as all-trans-retinal (= all-trans-β-apo-15-carotenal), might constitute another special class. In that case triplet formation (via an intermediate nπ* state) and trans–cis isomerization were proposed as additional channels.41,45
Acknowledgements
We thank H. Ernst from BASF SE for his valuable advice and for providing β-apo-12′-carotenal for the PSCP experiments, and β-apo-8′-carotenal and additional reagents as precursors for our synthesis of β-apo-4′-carotenal and torularhodinaldehyde. We also thank J. Troe, D. Schwarzer, K. Luther, J. Schroeder, A. M. Wodtke (Georg August University Göttingen, Germany), as well as N. P. Ernsting and J. L. Pérez Lustres (Humboldt University Berlin, Germany) for their continuous support and advice.Notes and references
- T. Polívka and V. Sundström, Chem. Rev., 2004, 104, 2021 CrossRef PubMed.
- T. Polívka and V. Sundström, Chem. Phys. Lett., 2009, 477, 1 CrossRef PubMed.
- L. Brouwer, H. Hippler, L. Lindemann and J. Troe, J. Phys. Chem., 1985, 89, 4608 CrossRef CAS.
- D. Schwarzer, J. Troe, M. Votsmeier and M. Zerezke, J. Chem. Phys., 1996, 105, 3121 CrossRef CAS PubMed.
- D. Schwarzer, C. Hanisch, P. Kutne and J. Troe, J. Phys. Chem. A, 2002, 106, 8019 CrossRef CAS.
- T. Lenzer, F. Ehlers, M. Scholz, R. Oswald and K. Oum, Phys. Chem. Chem. Phys., 2010, 12, 8832 RSC.
- K. Golibrzuch, F. Ehlers, M. Scholz, R. Oswald, T. Lenzer, K. Oum, H. Kim and S. Koo, Phys. Chem. Chem. Phys., 2011, 13, 6340 RSC.
- D. J. Nesbitt and R. W. Field, J. Phys. Chem., 1996, 100, 12735 CrossRef CAS.
- H. H. Billsten, D. Zigmantas, V. Sundström and T. Polívka, Chem. Phys. Lett., 2002, 355, 465 CrossRef.
- F. L. de Weerd, I. H. M. van Stokkum and R. van Grondelle, Chem. Phys. Lett., 2002, 354, 38 CrossRef CAS.
- U. Hold, T. Lenzer, K. Luther and A. C. Symonds, J. Chem. Phys., 2003, 119, 11192 CrossRef CAS PubMed.
- H. Frerichs, T. Lenzer, K. Luther and D. Schwarzer, Phys. Chem. Chem. Phys., 2005, 7, 620 RSC.
- P. Chabéra, M. Fuciman, P. Hríbek and T. Polívka, Phys. Chem. Chem. Phys., 2009, 11, 8795 RSC.
- P. O. Andersson and T. Gillbro, J. Chem. Phys., 1995, 103, 2509 CrossRef CAS PubMed.
- W. Wohlleben, T. Buckup, H. Hashimoto, R. J. Cogdell, J. L. Herek and M. Motzkus, J. Phys. Chem. B, 2004, 108, 3320 CrossRef CAS.
- T. Buckup, J. Savolainen, W. Wohlleben, J. L. Herek, H. Hashimoto, R. R. B. Correia and M. Motzkus, J. Chem. Phys., 2006, 125, 194505 CrossRef PubMed.
- V. Namboodiri, M. Namboodiri, G. Flachenecker and A. Materny, J. Chem. Phys., 2010, 133, 054503 CrossRef CAS PubMed.
- D. Kosumi, S. Maruta, T. Horibe, Y. Nagaoka, R. Fujii, M. Sugisaki, R. J. Cogdell and H. Hashimoto, J. Chem. Phys., 2012, 137, 064505 CrossRef PubMed.
- C. C. Gradinaru, J. T. M. Kennis, E. Papagiannakis, I. H. M. van Stokkum, R. J. Cogdell, G. R. Fleming, R. A. Niederman and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2364 CrossRef CAS PubMed.
- D. M. Niedzwiedzki, J. O. Sullivan, T. Polívka, R. R. Birge and H. A. Frank, J. Phys. Chem. B, 2006, 110, 22872 CrossRef CAS PubMed.
- D. Niedzwiedzki, J. F. Koscielecki, H. Cong, J. O. Sullivan, G. N. Gibson, R. R. Birge and H. A. Frank, J. Phys. Chem. B, 2007, 111, 5984 CrossRef CAS PubMed.
- H. Cong, D. M. Niedzwiedzki, G. N. Gibson and H. A. Frank, J. Phys. Chem. B, 2008, 112, 3558 CrossRef CAS PubMed.
- N. Christensson, F. Milota, A. Nemeth, J. Sperling, H. F. Kauffmann, T. Pullerits and J. Hauer, J. Phys. Chem. B, 2009, 113, 16409 CrossRef CAS PubMed.
- A. E. Jailaubekov, S.-H. Song, M. Vengris, R. J. Cogdell and D. S. Larsen, Chem. Phys. Lett., 2010, 487, 101 CrossRef CAS PubMed.
- J. Hauer, M. Maiuri, D. Viola, V. Lukes, S. Henry, A. M. Carey, R. J. Cogdell, G. Cerullo and D. Polli, J. Phys. Chem. A, 2013, 117, 6303 CrossRef CAS PubMed.
- V. Lukeš, N. Christensson, F. Milota, H. F. Kauffmann and J. Hauer, Chem. Phys. Lett., 2011, 506, 122 CrossRef PubMed.
- E. E. Ostroumov, M. G. Müller, M. Reus and A. R. Holzwarth, J. Phys. Chem. A, 2011, 115, 3698 CrossRef CAS PubMed.
- F. Ehlers, D. A. Wild, T. Lenzer and K. Oum, J. Phys. Chem. A, 2007, 111, 2257 CrossRef CAS PubMed.
- K. Oum, P. W. Lohse, O. Flender, J. R. Klein, M. Scholz, T. Lenzer, J. Du and T. Oekermann, Phys. Chem. Chem. Phys., 2012, 14, 15429 RSC.
- P. W. Lohse, J. Kuhnt, S. I. Druzhinin, M. Scholz, M. Ekimova, T. Oekermann, T. Lenzer and K. Oum, Phys. Chem. Chem. Phys., 2011, 13, 19632 RSC.
- T. Lenzer, S. Schubert, F. Ehlers, P. W. Lohse, M. Scholz and K. Oum, Arch. Biochem. Biophys., 2009, 483, 213 CrossRef CAS PubMed.
- A. L. Dobryakov, S. A. Kovalenko, A. Weigel, J. L. Pérez Lustres, J. Lange, A. Müller and N. P. Ernsting, Rev. Sci. Instrum., 2010, 81, 113106 CrossRef CAS PubMed.
- R. L. Christensen, M. Goyette, L. Gallagher, J. Duncan, B. DeCoster, J. Lugtenburg, F. J. Jansen and I. van der Hoef, J. Phys. Chem. A, 1999, 103, 2399 CrossRef CAS.
- K. Oum, P. W. Lohse, F. Ehlers, M. Scholz, M. Kopczynski and T. Lenzer, Angew. Chem., Int. Ed., 2010, 49, 2230 CrossRef CAS PubMed.
- M. Kopczynski, F. Ehlers, T. Lenzer and K. Oum, J. Phys. Chem. A, 2007, 111, 5370 CrossRef CAS PubMed.
- S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, J. Chem. Phys., 2001, 115, 3256 CrossRef CAS PubMed.
- E. Papagiannakis, I. H. M. van Stokkum, M. Vengris, R. J. Cogdell, R. van Grondelle and D. S. Larsen, J. Phys. Chem. B, 2006, 110, 5727 CrossRef CAS PubMed.
- J. Savolainen, T. Buckup, J. Hauer, A. Jafarpour, C. Serrat, M. Motzkus and J. L. Herek, Chem. Phys., 2009, 357, 181 CrossRef CAS PubMed.
- H. H. Billsten, J. Pan, S. Sinha, T. Pascher, V. Sundström and T. Polívka, J. Phys. Chem. A, 2005, 109, 6852 CrossRef CAS PubMed.
- F. Ehlers, T. Lenzer and K. Oum, J. Phys. Chem. B, 2008, 112, 16690 CrossRef CAS.
- T. Polívka, S. Kaligotla, P. Chábera and H. A. Frank, Phys. Chem. Chem. Phys., 2011, 13, 10787 RSC.
- J. A. Bautista, R. E. Connors, B. B. Raju, R. G. Hiller, F. P. Sharples, D. Gosztola, M. R. Wasielewski and H. A. Frank, J. Phys. Chem. A, 1999, 103, 8751 CrossRef CAS.
- D. Zigmantas, T. Polívka, R. G. Hiller, A. Yartsev and V. Sundström, J. Phys. Chem. A, 2001, 105, 10296 CrossRef CAS.
- D. Zigmantas, R. G. Hiller, A. Yartsev, V. Sundström and T. Polívka, J. Phys. Chem. B, 2003, 107, 5339 CrossRef CAS.
- S. Yamaguchi and H. Hamaguchi, J. Chem. Phys., 1998, 109, 1397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of β-apo-4′-carotenal and torularhodinaldehyde; Fig. S1 featuring a comparison of S* signals for β-carotene, torularhodinaldehyde and β-apo-4′-carotenal. See DOI: 10.1039/c4cp05600k |
| This journal is © the Owner Societies 2015 |