
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe shape of ibuprofen in the gas phase†
Thomas
Betz
abc,
Sabrina
Zinn
abc and
Melanie
Schnell
*abc
aMax Planck Institute for the Structure and Dynamics of Matter, Luruper Chaussee 149, D-22761 Hamburg, Germany. E-mail: melanie.schnell@mpsd.mpg.de
bThe Hamburg Centre for Ultrafast Imaging, Luruper Chaussee 149, D-22761 Hamburg, Germany
cCenter for Free-Electron Laser Science, Notkestrasse 85, D-22607 Hamburg, Germany
First published on 6th January 2015
Abstract
Ibuprofen's pain-relieving properties arise from its ability to physically block the active site of an enzyme, thus making its structural and conformational properties highly interesting. We here present a conformer-selective high-resolution broadband rotational spectroscopy study of gas-phase ibuprofen. The interpretation of the spectroscopic results is supported by quantum-chemical calculations. We identify four low-energy conformers that differ in the structural arrangement of the isobutyl moiety with respect to the remainder of the molecule. While the isobutyl group shows high structural flexibility – resulting in distinct low-energy conformers – the propanoic acid group favors a stable arrangement.
1 Introduction
Ibuprofen (derived from iso-butyl–phenyl–propanoic-acid) is of high relevance in medical applications because of its anti-flammatory, pain-relieving effect and is today a widespread non-prescriptive drug. The World Health Organisation lists it as one of the essential drugs for a basic health-care system.1 It belongs to the class of non-steroidal anti-inflammatory drugs (NSAIDs), which inhibit the enzyme cyclooxygenase (COX) by interacting with and thus competitively blocking its active site (e.g. see ref. 2–5). Cyclooxygenase, in turn, plays a key role in the production chain of pain, inflammation and fever mediators, such as the class of prostaglandins. Studies of the enzyme–inhibitor complex in crystals revealed that the inhibiting effect of ibuprofen is achieved by inserting its carboxyl group into the hydrogen bond network of the active site of the COX enzyme and thus blocking the molecular recognition mechanisms needed for its catalyzing effect.6–9Ibuprofen ((RS)-2-(4-(2-methylpropyl)phenyl)propanoic acid) consists of an aromatic ring bridging an isobutyl and a propanoic acid group (see Fig. 1) so that a rich conformational landscape can be expected. Indeed, previous quantum-chemical calculations predict a large number of low-energy conformers.10–13 On the other hand, the carboxylic acid group can function both as hydrogen donor and acceptor, which is a main structural motif in crystalline ibuprofen and which can be important for its function as an inhibitor.14,15 Furthermore, ibuprofen is chiral, featuring a stereogenic center at the α-carbon site connecting the carboxyl group and the aromatic ring. Interestingly, only the S-enantiomer has a pharmaceutical effect.16,17
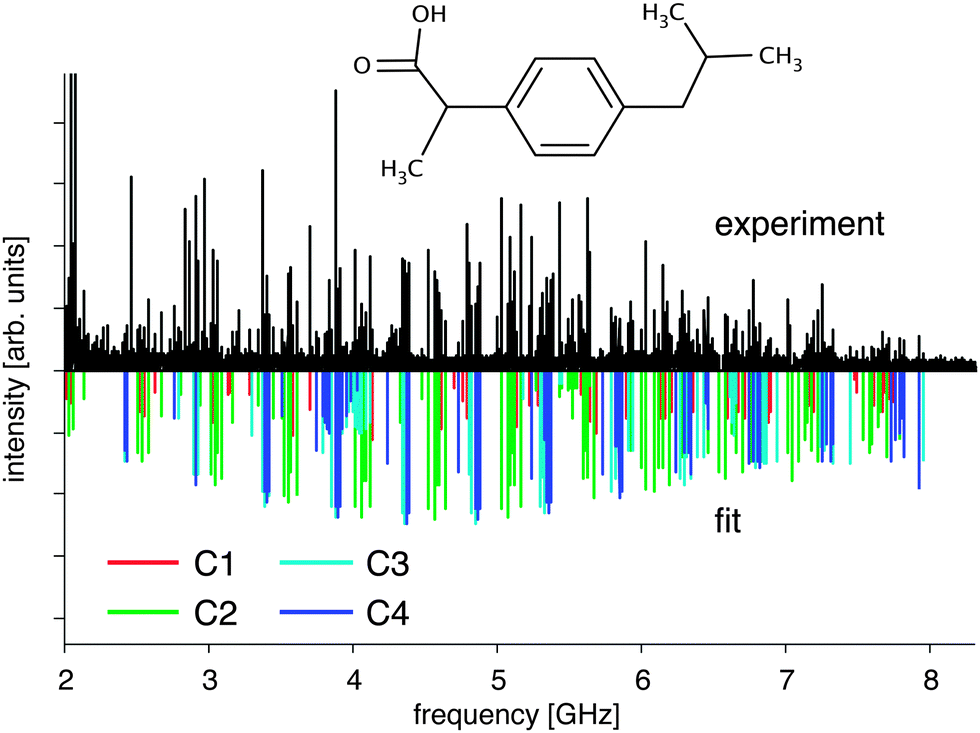 | ||
| Fig. 1 Broadband rotational spectrum of ibuprofen: the upper trace shows the experimental data obtained by chirped-pulse Fourier transform microwave spectroscopy, and the lower trace represents the result of fitting an asymmetric rotor Hamiltonian to the four lowest energy conformers C1–C4 at a temperature of 1.5 K. Known background lines have been removed. The individual spectra of the conformers are dominated by a- and c-type transitions. | ||
A detailed knowledge of the molecular structures can help to further elucidate the structure–function relationship of pharmaceutically relevant molecules such as ibuprofen, in particular the interactions and conformational changes accompanied with the molecular recognition of the inhibitors. In this respect, it is particularly interesting to compare the molecular structures of isolated, unperturbed molecules with those in an enzyme–inhibitor complex. High-resolution rotational spectroscopy has been proven to be a sensitive tool to precisely determine molecular structures of isolated molecules in the gas phase. Already subtle changes in the molecular structure, such as isotopic substitution (vide infra) and different conformational arrangements, lead to unambiguous differences in the rotational constants and thus in the rotational spectra. Precise molecular structures can be determined by analyzing the rotational spectra of isotopically substituted species. From the changes in the molecular moments of inertia due to isotopic substitution, atom positions can be experimentally determined by Kraitchman's equations, for example. Alternatively, valuable structural information as well as important benchmarking of theoretical techniques can also be achieved by a direct comparison of the experimental parameters with quantum-chemical data.
Here, the gas-phase rotational spectrum of ibuprofen in the frequency range from 2–8.5 GHz under the cold conditions of a molecular jet is reported. This study is in line with previous spectroscopic work on the structurally related, pain-relieving molecules aspirin18 and paracetamol19 that both show rich conformational flexibility. Recently, two different conformers were observed spectroscopically for ibuprofen's structural motif isobutyl benzene.20 It will be interesting to compare these findings with the different conformers of ibuprofen, which is structurally more complex. Intramolecular interactions arising from the carboxy group can become important.
2 Experimental details
Here, we employ the broadband chirped-pulse Fourier transform (CP-FTMW) microwave spectroscopy technique.21,22 Its advent eased the characterization of larger, conformationally flexible molecules, such as strawberry aldehyde (C12H14O3) for which five low-energy conformers were observed,23 and it also facilitates the present study of ibuprofen.Ibuprofen was purchased from Sigma-Aldrich (98% purity) and used without further purification. Ibuprofen is a solid at room temperature with a melting point around 76 °C. The sample was heated to 110 °C in a reservoir to generate sufficient vapor pressure and then supersonically expanded into vacuum using neon as carrier gas. The ibuprofen molecules were excited by a 1 μs long microwave pulse, which was linearly frequency chirped from 2 to 8.5 GHz. The chirp was amplified by a 300 W traveling wave tube amplifier and broadcast into the vacuum chamber using a horn antenna. 10 μs of the resulting free induction decay (FID) were recorded, starting 5 μs after the excitation pulse and then amplified and converted into a digital signal. The length of the recorded FID sets the frequency resolution to 100 kHz, whereas the full width at half maximum of observed lines was on the order of 30 kHz. 650![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 FIDs have been recorded, averaged and Fourier transformed to obtain the final rotational spectrum depicted in Fig. 1. More experimental details can be found in ref. 22 and 24.
000 FIDs have been recorded, averaged and Fourier transformed to obtain the final rotational spectrum depicted in Fig. 1. More experimental details can be found in ref. 22 and 24.
3 Results and discussion
The recorded spectrum is displayed in Fig. 1. It is very rich, and the data analysis was complicated due to overlapping contributions from the individual ibuprofen conformers. We therefore developed a simple guessing algorithm to support the spectral analysis. For this purpose the rotational spectra were predicted for various input values of the rotational constants B and C using the simulation program PGOPHER,25 and an overlap integral with the experimental data was calculated. A reasonable input parameter space was estimated by using ab initio predictions of the rotational constants. The rotational constants with maximal overlap represented very good starting values for further assignment and refinement.In total, four conformers of ibuprofen were observed with comparable transition intensities (see Fig. 2). Each spectrum was fitted using an asymmetric rotor Hamiltonian (Watson S-Reduction in Ir representation) as implemented in the program PGOPHER.25 The obtained spectroscopic parameters are listed and compared to density-functional values in Table 1. We could assign 100–150 lines per conformer, both of R- and of Q-type character, and the average error of the fits is on the order of 30 kHz. Transitions involving rotational quantum numbers J up to 15 are present in the spectrum. In accordance with the predicted dipole-moment components (Table 1), both a- and c-type transitions were observed. Centrifugal distortion constants were omitted in the fits presented in Table 1 since their magnitudes would be below 1 kHz and their inclusion only slightly changes the quality of the fits, as can be seen from results including centrifugal distortion constants DJ, DK, and DJK that are given in the ESI.†
 | ||
| Fig. 2 Part of the broadband rotational spectrum of ibuprofen (J + 1 ← J = 7 ← 6 rotational transition including K substructure), illustrating the four conformers C1–C4. | ||
| Parameter | C1 | Calc. | C2 | Calc. | C3 | Calc. | C4 | Calc. |
|---|---|---|---|---|---|---|---|---|
| A (MHz) | 1325.646(2) | 1337.0 | 1357.714(2) | 1365.5 | 1570.526(2) | 1585.4 | 1534.777(2) | 1545.7 |
| B (MHz) | 260.7134(3) | 263.3 | 261.4543(2) | 264.3 | 245.4431(2) | 248.6 | 244.8919(3) | 247.7 |
| C (MHz) | 251.1784(3) | 253.8 | 248.3089(2) | 250.8 | 239.7512(2) | 242.4 | 242.1010(3) | 244.9 |
| μ a (D) | −1.1 | −1.2 | 1.2 | 1.1 | ||||
| μ b (D) | −0.08 | −0.5 | −0.8 | 0.25 | ||||
| μ c (D) | 1.2 | 1.3 | 1.2 | 1.2 | ||||
| μ tot (D) | 1.7 | 1.9 | 1.9 | 1.7 | ||||
| E (kJ mol−1) | 0 | 0.13 | 0.12 | 0.19 | ||||
| Assigned lines | 139 | 151 | 113 | 109 | ||||
| Error (kHz) | 44 | 43 | 33 | 44 |
For conformer C3, which has a somewhat larger magnitude for μb, also a few b-type transitions were recorded (see Table 1 and Section S3 of the ESI†). There are lines left in the recorded rotational spectrum, which cannot be assigned to additional conformers of ibuprofen but most likely arise from unidentified complexes or fragments (see below). No line splitting due to internal rotation was observed. Ab initio calculations predict barrier heights of more than 13 kJ mol−1 for internal rotation of the different methyl tops, which do not result in resolvable effects in our experiments.
Unfortunately, the spectrum is too weak to detect 13C isotopologues that would allow us to determine the structure very precisely. Instead, we compare our experimentally obtained molecular parameters to the results of quantum-chemical calculations at the M06-2X/aug-cc-pVTZ level of theory as implemented in Gaussian09 to analyse the shape of the various ibuprofen conformers (see Table 1). Due to the good agreement between experiment and theory, the experimentally identified conformers can directly be assigned to the predicted four lowest energy conformers, which are presented in Fig. 3 and labeled C1–C4. All conformers show the same orientation of the propanoic acid group with respect to the phenyl ring, but differ in the arrangement of the isobutyl group. Their energies and dipole moments are very similar (see Table 1), explaining the comparable intensities in the spectrum (see Fig. 2). Further conformers, for example with a different orientation of the COOH group, were not observed in the spectrum. They are at least 1.5 kJ mol−1 higher in energy, and we assume that they are not populated under the cold conditions of our molecular jet anymore. A table of all calculated conformers, including higher energy conformers, can be found in the ESI.†
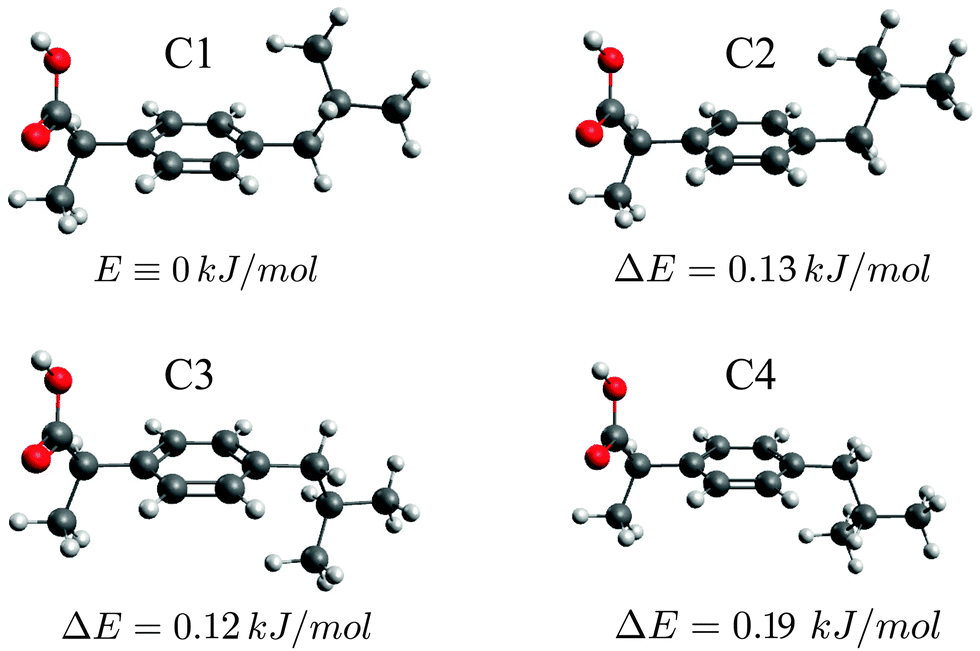 | ||
| Fig. 3 Optimized geometries and relative energies of the four experimentally observed ibuprofen conformers (M06-2X/aug-cc-pVTZ). They all show the same orientation of the propanoic acid group but differ in the arrangement of the isobutyl group with respect to the phenyl ring. | ||
The stabilization of the carboxyl group arises from the interaction of the hydrogen atom of the α-methyl group with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group via hydrogen bonding. The nature of this interaction can be further illuminated with the help of calculations. They predict that the Mulliken charge of the carbonyl oxygen is approximately 0.4 e while that of the hydroxy group as a whole is 0.04 e, so that their electrostatic interaction with the α-methyl hydrogen leads to significantly different energies (ΔE ≈ 3.7–4.2 kJ mol−1). For the related NSAID molecule aspirin, both orientations of the carboxyl group were obtained in the gas-phase rotational spectra. This difference might arise from the absence of a stabilizing group in aspirin, since the carboxyl group is directly attached to the phenyl ring.
O group via hydrogen bonding. The nature of this interaction can be further illuminated with the help of calculations. They predict that the Mulliken charge of the carbonyl oxygen is approximately 0.4 e while that of the hydroxy group as a whole is 0.04 e, so that their electrostatic interaction with the α-methyl hydrogen leads to significantly different energies (ΔE ≈ 3.7–4.2 kJ mol−1). For the related NSAID molecule aspirin, both orientations of the carboxyl group were obtained in the gas-phase rotational spectra. This difference might arise from the absence of a stabilizing group in aspirin, since the carboxyl group is directly attached to the phenyl ring.
The differences between the four conformers arise from changes in the orientation of the isobutyl group with respect to the remaining molecule. Generally, the isobutyl group can either be located on the same side as the carboxylic acid group COOH with respect to the phenyl ring (conformers C1 and C2), or on the opposite site (conformers C3 and C4). The rotational constants within the two conformational groups (C1 and C2 vs. C3 and C4) are extremely similar to each other. This similarity also becomes visible in the small spacing between the a-type transition groups shown in Fig. 2. Note that structure optimizations using the B3LYP density functional do not provide a sufficient agreement with the experimentally obtained parameters (also see ESI†). This strongly points to the presence of intramolecular dispersion interactions between one of the isobutyl methyl groups and the phenyl ring that is known as dispersion-energy donor.
In addition, the two terminal methyl groups of the branched isobutyl moiety can arrange themselves in three different ways: they can be located on the same side (conformers C2 and C4) or on the opposite side (conformers C1 and C3) as the carboxyl group with respect to an imaginary mirror plane σv of the phenyl ring that contains the center carbon atom of the propanoic acid group, or, finally, they can be oriented such that the two methyl groups would be symmetric with respect to that imaginary mirror plane. According to our calculations, this latter structure is about 1.8 kJ mol−1 higher in energy, and it is not observed in the spectrum. For isobutyl benzene, i.e., ibuprofen's structural motif without the propanoic acid group, both the asymmetric and the symmetric conformers were detected in a recent cavity-based Fourier transform microwave (FTMW) spectroscopy study.20 The transition intensities of the symmetric one were about one order of magnitude lower than of the asymmetric form. Based on their experimental data, the authors estimated an energy difference between the lower-lying asymmetric and the symmetric conformer of about 4 kJ mol−1. The energetically favored asymmetric isopropyl arrangement that we also observed for ibuprofen is very similar to the structural motif that occurs in crystalline ibuprofen,14,15 for which the symmetric arrangement was not observed.
In additional measurements, the sample was kept at the thermal decomposition temperature of ibuprofen (around 100 °C) for a longer time, and the spectra of two previously uncharacterized species started to dominate. These spectra consist mainly of a-type transitions. The determined molecular parameters and further details are given in the ESI.† We suggest that the uncharacterized species correspond to decarboxylation products, following the fragmentation behavior observed before in mass spectrometry studies.26,27 In addition, both spectra show a pronounced line splitting, which might be due to the internal rotation of the former α-methyl group that is now less sterically hindered than in ibuprofen. However, a detailed analysis is still ongoing.
4 Conclusions and outlook
We identified four energetically close conformers of ibuprofen in the gas phase via their rotational spectra. Ibuprofen shows rich conformational flexibility of the isobutyl group that might be needed for transporting the molecule to the COX enzyme and its recognition. The conformational arrangements agree to the spectroscopic findings for isobutyl benzene as well as to crystalline ibuprofen. Furthermore, the structural assignment of the conformers revealed a stabilizing interaction of the α-methyl top with the carbonyl subunit of the carboxyl group, which is present for all observed conformers. The respective arrangement of the isobutyl group with respect to the central phenyl ring seems to be stabilized by intramolecular dispersion interactions. The obtained spectroscopic parameters can now be used to study ibuprofen's enantiomers using microwave three-wave mixing.28 Our results can furthermore be used as reference structures to identify structural changes of ibuprofen upon formation of the enzyme–inhibitor complex and may serve as an excellent point of reference for further structure elucidation of other related solid drugs. In further studies, it will be intriguing to use gas-phase ibuprofen as a binding partner for molecular recognition studies, aiming at unraveling the interplay of inter- and intramolecular forces at the molecular level.References
- WHO, Model Lists of Essential Medicines, http://www.who.int/medicines/publications/essentialmedicines/en/.
- S. Offermanns and W. Rosenthal, Encyclopedic Reference of Molecular Pharmacology, Springer Verlag, edn 1, 2004 Search PubMed.
- K. Aktories, U. Förstermann, F. Hofmann and K. Starke, Allgemeine undspezielle Pharmakologie und Toxikologie, Elsevier, Munich, 11. Aufl., 2013 Search PubMed.
- F. D. Hart and E. C. Huskisson, Drugs, 1984, 27(3), 232–255 CrossRef CAS PubMed.
- G. Dannhardt and W. Kiefer, Eur. J. Med. Chem., 2001, 36(2), 109–126 CrossRef CAS.
- B. S. Selinsky, K. Gupta, C. T. Sharkey and P. J. Loll, Biochemistry, 2001, 40(17), 5172–5180 CrossRef CAS PubMed.
- A. Viegas, J. Manso, M. C. Corvo, M. M. B. Marques and E. J. Cabrita, J. Med. Chem., 2011, 54(24), 8555–8562 CrossRef CAS PubMed.
- V. Limongelli, M. Bonomi, L. Marinelli, F. L. Gervasio, A. Cavalli, E. Novellino and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(12), 5411–5416 CrossRef CAS PubMed.
- A. L. Blobaum and L. J. Marnett, J. Med. Chem., 2007, 50(7), 1425–1441 CrossRef CAS PubMed.
- A. Jubert, M. L. Legarto, N. E. Massa, L. L. Tévez and N. B. Okulik, J. Mol. Struct., 2006, 783(1–3), 34–51 CrossRef CAS PubMed.
- M. L. Vueba, M. E. Pina and L. A. E. Batista de Carvalho, J. Pharm. Sci., 2008, 97(2), 845–859 CrossRef CAS PubMed.
- Z. Fu, X. Li and K. M. Merz, J. Comput. Chem., 2011, 32(12), 2587–2597 CrossRef CAS PubMed.
- L. Liu and H. Gao, Spectrochim. Acta, Part A, 2012, 89, 201–209 CrossRef CAS PubMed.
- N. Shankland, C. Wilson, A. Florence and P. Cox, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 951–954 Search PubMed.
- L. Hansen, G. Perlovich and A. Bauer-Brandl, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, 1357–1358 Search PubMed.
- H. Hao, G. Wang and J. Sun, Drug Metab. Rev., 2005, 37(1), 215–234 CrossRef CAS.
- G.-Q. Lin, Q.-D. You and J.-F. Cheng, Chiral Drugs: Chemistry and Biological Action, John Wiley & Sons, Hoboken, New Jersey, 2011 Search PubMed.
- C. Cabezas, J. L. Alonso, J. C. López and S. Mata, Angew. Chem., Int. Ed., 2012, 51(6), 1375–1378 CrossRef CAS PubMed.
- M. Varela, C. Cabezas, J. C. López and J. L. Alonso, J. Phys. Chem. A, 2013, 117(50), 13275–13278 CrossRef CAS PubMed.
- G. Churchill, R. Hassey, K. Utzat, J. Fournier, R. Bohn and H. H. Michels, J. Phys. Chem. A, 2010, 114, 1029–1031 CrossRef CAS PubMed.
- G. G. Brown, B. C. Dian, K. O. Douglass, S. M. Geyer, S. T. Shipman and B. H. Pate, Rev. Sci. Instrum., 2008, 79(5), 053103 CrossRef PubMed.
- D. Schmitz, V. A. Shubert, T. Betz and M. Schnell, J. Mol. Spectrosc., 2012, 280, 77–84 CrossRef CAS PubMed.
- S. T. Shipman, J. L. Neill, R. D. Suenram, M. T. Muckle and B. H. Pate, J. Phys. Chem. Lett., 2011, 2(5), 443–448 CrossRef CAS.
- T. Betz, S. Zinn, J. B. Graneek and M. Schnell, J. Phys. Chem. A, 2014, 118(28), 5164–5169 CrossRef CAS PubMed.
- C. M. Western, PGOPHER, a Program for Simulating Rotational Structure, University of Bristol, http://pgopher.chm.bris.ac.uk Search PubMed.
- M. A. Zayed, M. F. Hawash, M. A. Fahmey and A. M. M. El-Gizouli, J. Therm. Anal. Calorim., 2012, 108(1), 315–322 CrossRef CAS.
- G. Caviglioli, P. Valeria, P. Brunella, C. Sergio, A. Attilia and B. Gaetano, J. Pharm. Biomed. Anal., 2002, 30(3), 499–509 CrossRef CAS.
- V. A. Shubert, D. Schmitz, D. Patterson, J. M. Doyle and M. Schnell, Angew. Chem., Int. Ed., 2014, 53, 1152–1155 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05529b |
| This journal is © the Owner Societies 2015 |