
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of protein–materials interaction by soft colloidal probe spectroscopy†
Steve
Martin
a,
Hanqing
Wang
b,
Laura
Hartmann
b,
Tilo
Pompe
a and
Stephan
Schmidt
*a
aInstitute of Biochemistry, Leipzig University, Johannisallee 21-23, 04103 Leipzig, Germany. E-mail: stephan.schmidt@uni-leipzig.de; Fax: +49 341 9736939; Tel: +49 341 9732978
bInstitute of Organic and Macromolecular Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, Düsseldorf, Germany. Fax: +49 211 8115840; Tel: +49 211 8110360
First published on 23rd December 2014
Abstract
We present a robust and fast method to quantify the adhesion energy of surface anchored proteins on material surfaces using soft colloidal particles as sensors. The results obtained from studying the adhesion of fibronectin on surfaces with different hydrophobicity were in good agreement with theoretical considerations demonstrating the feasibility of the method.
Introduction
Protein–material interactions and associated contact phenomena are important for many areas of science and technology.1–4 In particular, newly emerging areas in biomedicine and sensor technology benefit from an improved mechanistic understanding of protein interactions e.g. with implant surfaces5 or nanoparticles.6 Fundamental insights into the protein–material interactions have been gained from protein adsorption measurements in simplified model systems using various techniques including surface plasmon resonance, ellipsometry or fluorescence microscopy.7,8 These techniques basically yield the adsorbed amount of protein as well as adsorption kinetics, but also the exchange characteristics of proteins at surfaces.9–11 In addition for large surfaces (nanoparticles or nanoporous systems) calorimetric methods can be used to measure specific adsorption enthalpies of proteins.6,12 According to these studies protein adsorption is present on almost any surface and typically irreversible if large proteins with multiple adhesion sites are involved. Protein adsorption measurements are typically analyzed via models assuming adsorption–desorption equilibria (e.g. Langmuir isotherms).7,8 However, in case of quasi-irreversible adsorption of large proteins the experiments do not reach the equilibrium situation. Thus, in a strict sense these models are not valid for such experiments. Hence, adsorption enthalpies and in-depth analysis of the thermodynamics of protein–materials interaction cannot be derived within this approach. Moreover, besides studying protein adsorption phenomena, investigating the interactions of quasi-stationary protein layers is of great importance. For example, the interaction of surface anchored proteins forming the actual contact of an implant surface with tissue have to be understood in detail in order to control the long-term response of the surrounding tissue. Furthermore, strongly adsorbing proteins, like the large adhesion receptor fibronectin (FN), are known to be mobile or displaced under external cues, like displacing serum proteins or receptor forces of adherent cells.11,13Atomic force microscopy (AFM) or surface force apparatus (SFA) are suitable techniques to study contact phenomena between surface anchored proteins and materials.14–16 These methods have been used to investigate adhesion forces of proteins layers which is an important part of the rational design of biomaterial surfaces. Hence, such force-based techniques can be considered as an alternative to measuring the thermodynamics of protein surface adsorption. However, handling of SFA or AFM is rather difficult and their throughput too low in order to process a significant number of proteins and material surfaces. Therefore, in the present work, we adapted a novel screening method to study interactions of surface anchored proteins in a simplified and rapid fashion.17–19 Our method uses soft protein coated hydrogel particles, also called soft colloidal probes (SCPs), which undergo mechanical deformation when adhering to material surfaces.19,20 The mechanical deformation can be conveniently read out by reflection interference contrast microscopy (RICM) and related to the adhesion energy of the protein layer bound to the SCP (Fig. 1). The underlying theory was developed by Johnson, Kendall and Roberts (JKR model21). The JKR adhesion energy Wadh of an elastomeric particle resting on a surface can be calculated as:
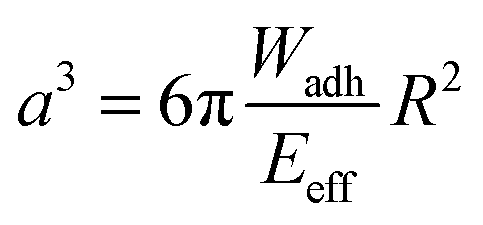 | (1) |
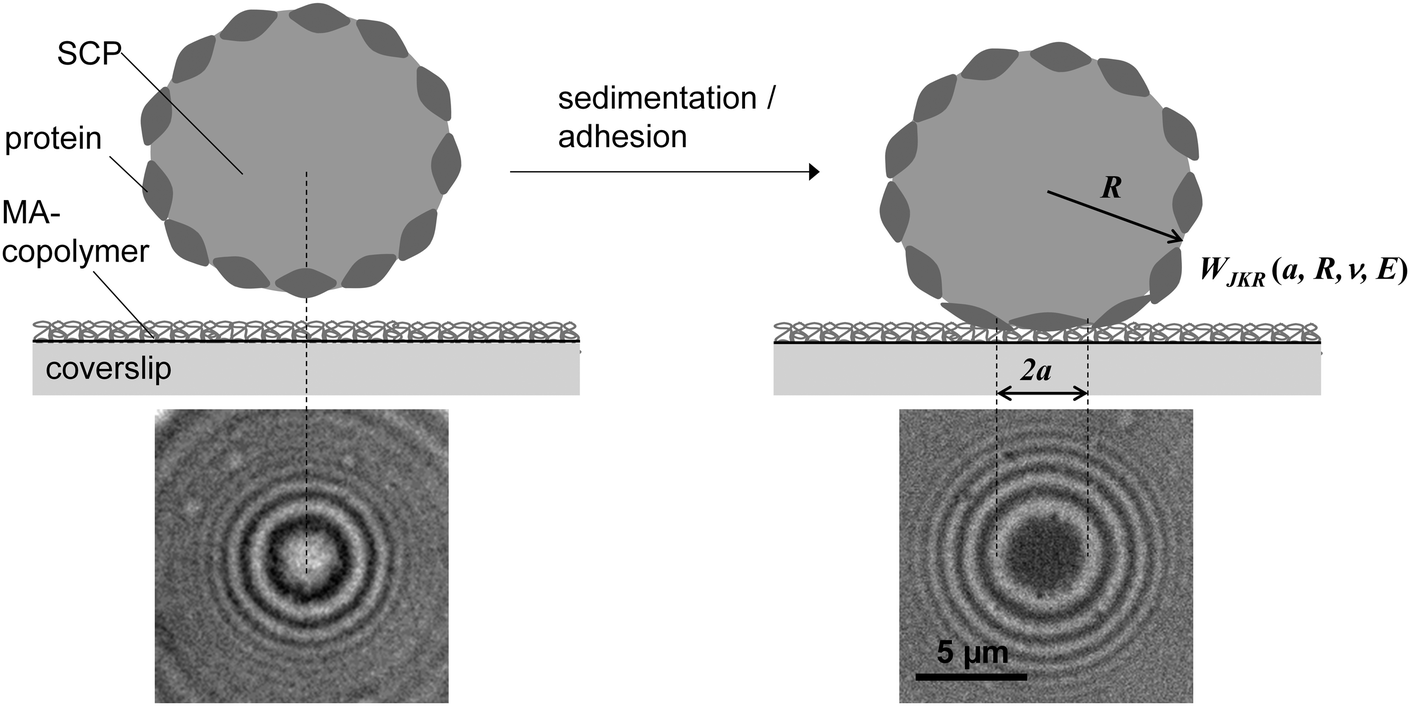 | ||
| Fig. 1 SCP adhesion measurement schematic (top): a protein coated SCP comes into contact with a maleic anhydride (MA) copolymer surface due to sedimentation (left), then the protein layer starts to adhere on the polymer surface inducing mechanical deformation of the SCP (right). The contact area of the particle can be read out via reflection interference contrast microscopy (RICM) from the central circular interference minimum (bottom). | ||
Results and discussion
We tested the SCP setup to study protein adsorption phenomena by measuring the adhesion energies of SCP bound layers of fibronectin (FN) on a series of polymer coated surfaces. The SCPs consisted of covalently crosslinked polyacrylic acid and were post-functionalized with proteins via carbodiimide chemistry (ESI,† S1 and S2). Briefly, SCPs were prepared via an inverse emulsion polymerization of acrylic acid in presence of bisacrylamide as crosslinker. Radical polymerization was thermally initiated using azo-bis(isobutyronitril) (AIBN). The carboxylic acid groups of the resulting polyacrylic acid (PAA) microgels allowed for straightforward coupling of FN using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). Note that the covalent attachment via multiple activated acrylic acid groups results in strong immobilization of FN, possibly hampering conformational changes. The protein functionalized SCPs were characterized by confocal laser scanning microscopy (ESI,† S3). The results indicate a dense monolayer packing of FN on the SCP surface (ESI,† S3 and S4), which is reasonable for the high concentration of FN in the immobilization solution (200 μg ml−1) and the high number of activated carboxy groups at the SCP surface. The calculation of adhesion energy requires elastic modulus determination of the SCPs (see eqn (1)). AFM colloidal probe indentation measurements in PBS buffer were conducted to determine the elastic modulus of the protein functionalized particles with a modified Hertzian contact model (ESI,† S5).22The polymer coated model surfaces were composed of alternating maleic anhydride (MA) copolymers where the co-monomers are varied from ethene, propene, styrene and octadecene. Accordingly, the different MA copolymers are termed PEMA, PPMA, PSMA and POMA (see Fig. 2A). The MA-copolymers surfaces were prepared on amino-silane coated glass coverslips as previously described23 (for details see ESI,† S6). Note that the polymer layer achieves densely packed dangling chains on the surface in mushroom-brush conformation with an overall thickness of some tens of nm.24 Importantly, the different copolymers lead to variations of the surface hydrophobicity. The water contact angle strongly decreases from POMA (100°), PSMA (75°), PPMA (38°) to PEMA (21°) coated surfaces.9,13 As expected, the contact areas of FN coated SCPs resting on MA-copolymer surfaces followed the trend in hydrophobicity of the polymer surfaces, as can be seen in the RICM images (Fig. 2B). A clear increase of the contact area was observed for more hydrophobic surfaces.
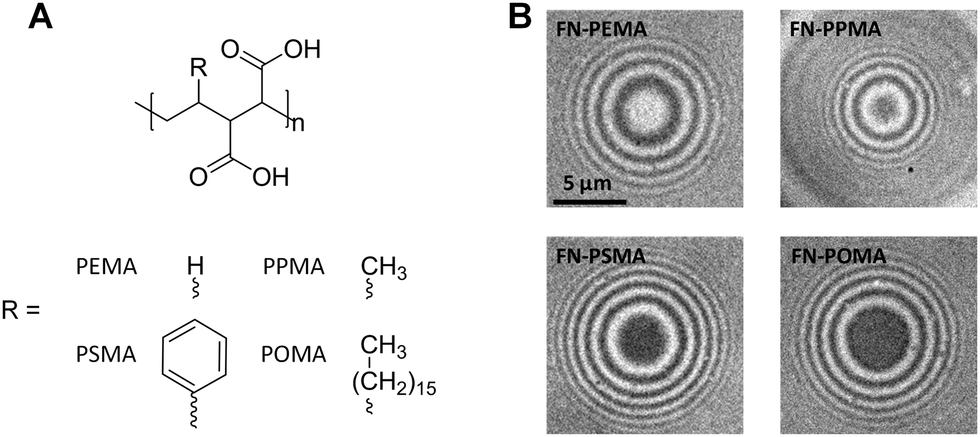 | ||
| Fig. 2 (A) Chemical structures of the hydrolyzed MA-copolymers; (B) typical RICM images of FN SCPs on the different MA-copolymer surfaces. | ||
From the RICM images the contact radii of the SCPs were evaluated in order to calculate the adhesion energy per area via the JKR approach. For this purpose the contact radii of a large number of SCPs was measured and plotted vs. the SCP radius (Fig. 3A). Using eqn (1) the data were fitted yielding the adhesion energy Wadh as single fit parameter. The comparison of the obtained adhesion energies for FN on the MA-polymer surfaces show the expected trend, more hydrophobic surfaces resulting in increased adhesion energies (Fig. 3B). Wadh is a measure of the change in free energy when the hydrated polymer and protein surface form a contact. Generally, the two main contributions to Wadh are the interaction free energy of FN and polymer at the interface and the solvation free energy (hydrophobic effect) of FN and the polymer. The solvation free energy of the polymer should lead to larger adhesion energies for more hydrophobic surfaces as less energy is required to overcome water–polymer interactions to form FN–polymer contacts. According to Young's equation, the water contact angle cos(θ) of a material surface is proportional to its solvation free energy. Interestingly, when plotting the adhesion energies versus cos(θ) of the water contact angle of MA-copolymer surfaces (Fig. 4), we found an almost linear trend. This suggests that the large Wadh values on hydrophobic MA-polymers are mostly driven by the entropic gain due to the solvation free energy (hydrophobic effect). The presence of strong entropic effects is supported by the fact that large proteins like FN have the ability to adhere via conformational changes resulting in presentation of hydrophobic sites in the contact zone, while still retaining the beneficial hydration layer facing the bulk solution. It is known that such conformational changes are less strong on polar surfaces with low contact angle; therefore Wadh is reduced in this case. Of course it could be expected that not only hydrophobic effects but also the interaction free energy of FN and polymer changes with the type of MA-copolymer layer. Alternatively it could be argued that changes in the interaction free energy plays only a minor role in this case since both the MA-polymers and FN show a negative net charge at the measurement conditions (pH 7.4) thus reducing attractive electrostatic and dipole contributions.
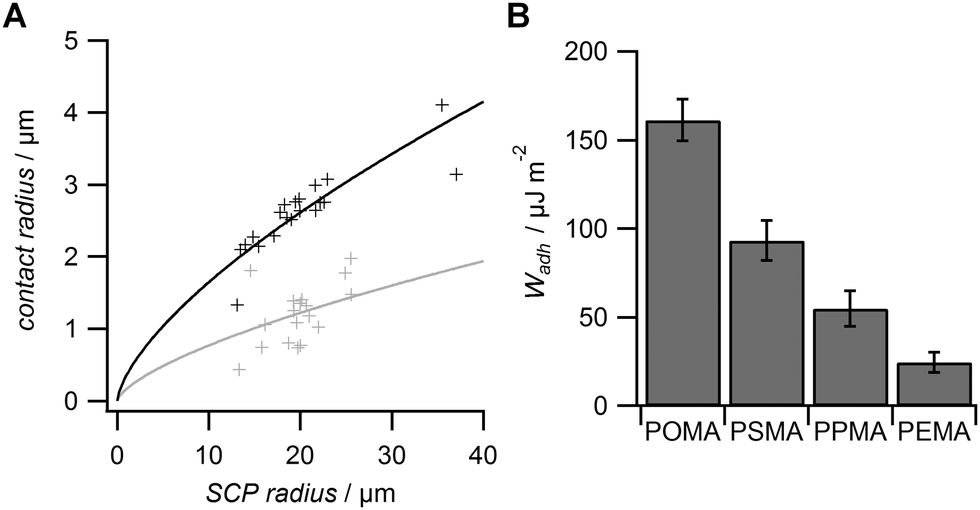 | ||
| Fig. 3 (A) Typical contact radii of FN SCPs on POMA (black) and PEMA (grey). The lines represent least square fits via the JKR approach. Data was obtained on at least three different polymer surfaces for all coatings. (B) Adhesion energy per area for FN coated SCP on different MA-copolymer surfaces. | ||
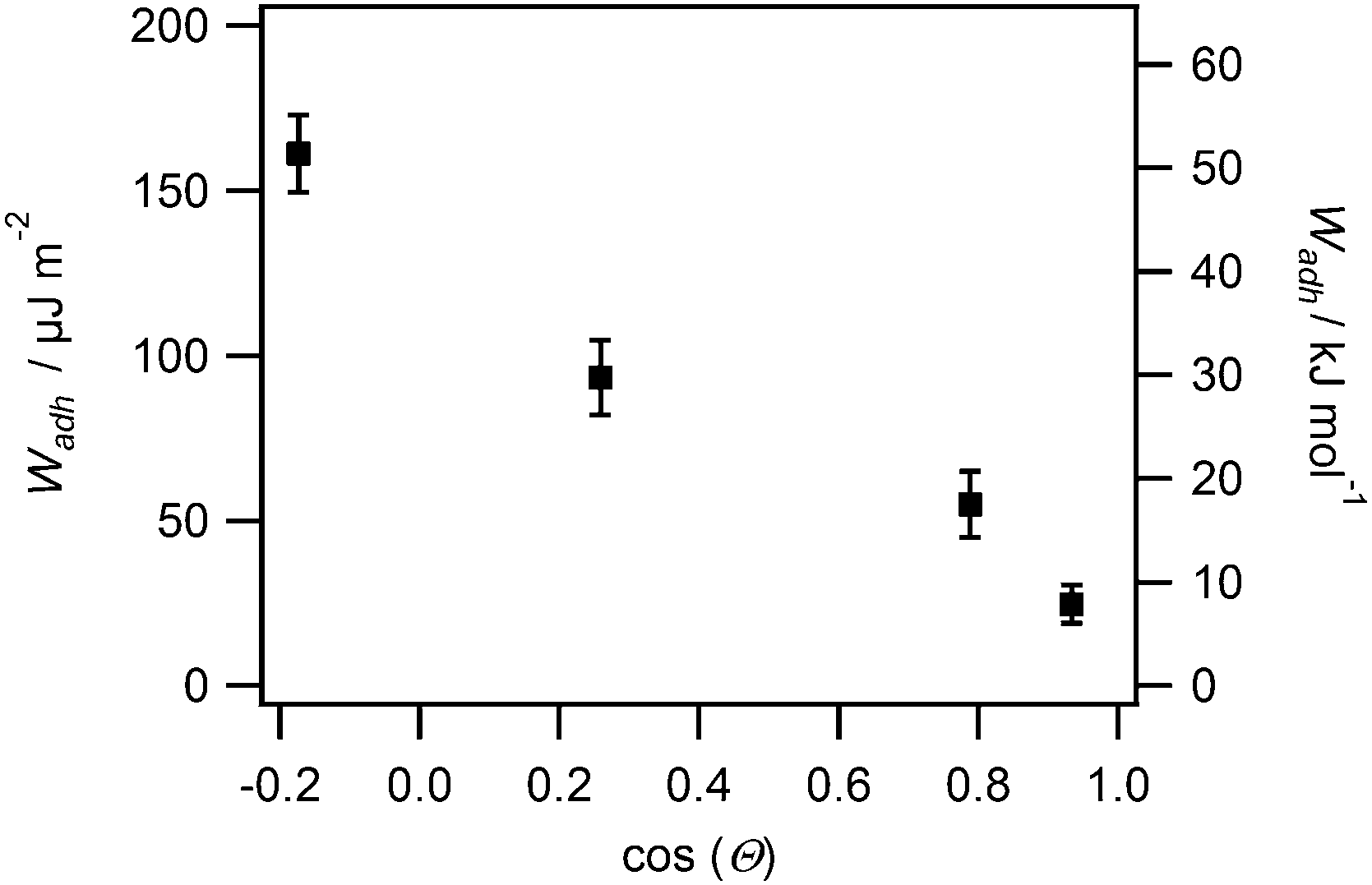 | ||
| Fig. 4 Adhesion energy per area for FN coated SCP (left axis) and estimated adhesion energy per FN molecule plotted versus cosine of the water contact angle cos(θ) of MA-copolymer surfaces as a measure of the solvation free energy of the polymer. | ||
From the adhesion energy per area and the estimated SCPs protein density (ESI,† S4) we calculated the adhesion energy per mole of surface bound protein (Fig. 4 right axis, ESI,† S7). The adhesion energy per mol decreases from 51 kJ mol−1 to 8 kJ mol−1 from the most hydrophobic surface (POMA) to the most hydrophilic surface (PEMA). These molecular interaction energies seem very low when compared to literature values. For example ITC measurements revealed almost two orders of magnitude larger adhesion energies (∼1000 kJ mol−1) for BSA adsorbing on SiO2, TiO2 or polystyrene surfaces.6,25 Also theoretical studies7 and kinetic studies on short peptides8 predicted much larger adsorption energies on hydrophobic surfaces. There are several factors which can cause the observed quantitative differences compared to adsorption measurements in solution. At first the densely packed and covalently bound proteins on the SCP cannot undergo conformational changes to the same degree as freely adsorbing proteins.4 Therefore spreading upon adhesion on the polymer surface is reduced, which results in a reduction of the molar adhesion energy compared to adsorption measurements. It is known that entropic and enthalpic contributions via such processes considerably contribute the overall protein–material interaction. Secondly, surface roughness of both interaction surfaces can generally reduce the effective contact points of adhering surfaces and thus the overall adhesion energy.26 This effect could lead to an underestimation of the molar interaction energies. However, surface roughness effects are considered negligible in the presented study. Surface roughness of SCP should be on the order of the size of the protein as the mesh size of the PAA network is on a similar length scale (5.7 nm, see ESI,† S3 and S4). The surface roughness of the MA-copolymer film is known to be of molecular length scale as well.23 Furthermore, deformation of the SCP during contact should lead to a decrease of possible roughness effects.
It is important to note that in the presented setup protein–material interaction is not only constrained by the inhibition of conformational changes of proteins but furthermore by the mechanical forces of the supporting polymeric network of the SCP. Near the edge of the contact zone the attached proteins can be considered to be under a restoring force due to the polymeric linker when the SCP forms a contact with the surface. In contrast, the central contact area is characterized by small compressive forces of the deformed SCP onto the adsorbed protein (see ESI,† S7). While this effect – impacting the quantitative data of the adhesion energy – might be assumed to be disadvantageous, it illustrates the strength of the approach at the same time. The force-based assay allows quantifying adsorption phenomena of large proteins at various surfaces and resembles interaction process occurring at cell culture substrates under physiological conditions. Therein, cells bind via surface receptors, like integrins, to adhesion ligands such as Arg–Gly–Asp motifs presented by FN adsorbed on materials surfaces and apply considerable forces in the process of cell adhesion. In that way, cells can be described as deformable objects binding to surfaces via large adhesion proteins such as FN. Such a setup is nicely modelled by SCP adhesion in our experiments. The exact stress distribution in the contact zone of cells is of course much more complex as is the case for adherent SCP (see ESI,† S7). But again, it is non-homogeneously distributed with tensile and compressive areas within a cell especially during dynamic process like cell migration. Thus, we suggest that the presented assay is able to probe similar interactions and provides meaningful results in a cell adhesion context. Interestingly, a study investigating the molecular reorganisation of the adhesion ligand FN by cell receptor force gave theoretical estimates of the protein–materials interactions in a similar order of magnitude as presented here.27 FN–substrate interactions were estimated in the range of 1–6 kT comparing to 3–20 kT found in our study.
In light of the latter discussion and the nice correspondence of adhesion energies with surface properties of the polymer layers, namely hydrophobicity, the set of FN adhesion energies obtained in this work very well explains the variations of cell adhesion behaviour on the different MA-polymer coatings from earlier studies, which is largely affected by FN–surface interactions.9,11 It was found that the FN–surface interaction can lead to different modes of traction force behaviour, which is based on the fact that cells would sense different adhesion strengths on surfaces with varying hydrophobicity.28 These results highlight the importance to directly measure the adhesion energies between protein layers and material surfaces since pure adsorption measurements do not capture the involved multivalent surface effects.
Conclusions
Overall, the developed method is capable of fast and direct measurement of the adhesion energies of protein layers with material surfaces, even for highly adhesive large proteins. Importantly, the method captures colligative adhesions of surface anchored proteins on a soft surface, which cannot be detected by classic surface science tools. For example, calculations have shown that flexible interfaces induce cooperative binding of surface anchored binding partners to a much higher degree than stiff interfaces.29 Such cooperative binding is a common feature of cell–matrix and cell–cell interactions as well as interactions on artificial material surfaces. The quantitative measurements of protein–material interaction by the SCP method with contact areas sized with a cell adhesion site characteristic and cell-like mechanical properties nicely reflects the biological context. Future studies will thus focus on the effect of specific interaction partners (e.g. matrix ligands and cell receptors), possible conformational changes of proteins in the contact zone via SCP-AFM,30,31 as well as determining the effect of protein–protein interaction during matrix assembly or protein adsorption in complex solutions.Acknowledgements
Financial support was granted by the SMWK-program 4-7531.60/30/27 and the German Research Foundation through the Emmy Noether program (HA5950/1-1) and project SCHM 2748/3-1.Notes and references
- H. Chen, L. Yuan, W. Song, Z. K. Wu and D. Li, Prog. Polym. Sci., 2008, 33, 1059–1087 CrossRef CAS PubMed.
- Y. Qian and C. Hong, Prog. Chem., 2014, 26, 1275–1284 Search PubMed.
- A. E. Rodda, L. Meagher, D. R. Nisbet and J. S. Forsythe, Prog. Polym. Sci., 2014, 39, 1312–1347 CrossRef CAS PubMed.
- Q. Wei, T. Becherer, S. Angioletti-Uberti, J. Dzubiella, C. Wischke, A. T. Neffe, A. Lendlein, M. Ballauff and R. Haag, Angew. Chem., Int. Ed., 2014, 53, 8004–8031 CrossRef CAS PubMed.
- S. L. Bellis, Biomaterials, 2011, 32, 4205–4210 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- R. A. Latour, J. Biomed. Mater. Res., Part A, 2006, 78, 843–854 CrossRef PubMed.
- Y. Wei and R. A. Latour, Langmuir, 2009, 25, 5637–5646 CrossRef CAS PubMed.
- T. Pompe, K. Keller, C. Mitdank and C. Werner, Eur. Biophys. J. Biophys. Lett., 2005, 34, 1049–1056 CrossRef CAS PubMed.
- L. Renner, T. Pompe, K. Salchert and C. Werner, Langmuir, 2005, 21, 4571–4577 CrossRef CAS.
- T. Pompe, M. Kaufmann, M. Kasimir, S. Johne, S. Glorius, L. Renner, M. Bobeth, W. Pompe and C. Werner, Biophys. J., 2011, 101, 1863–1870 CrossRef CAS PubMed.
- S. Chakraborti, P. Joshi, D. Chakravarty, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2012, 28, 11142–11152 CrossRef CAS PubMed.
- L. Renner, T. Pompe, K. Salchert and C. Werner, Langmuir, 2004, 20, 2928–2933 CrossRef CAS.
- Y. F. Dufrene and A. E. Pelling, Nanoscale, 2013, 5, 4094–4104 RSC.
- C. K. Lee, Y. M. Wang, L. S. Huang and S. M. Lin, Micron, 2007, 38, 446–461 CrossRef CAS PubMed.
- J. A. Koehler, M. Ulbricht and G. Belfort, Langmuir, 2000, 16, 10419–10427 CrossRef CAS.
- D. Pussak, D. Ponader, S. Mosca, S. V. Ruiz, L. Hartmann and S. Schmidt, Angew. Chem., Int. Ed., 2013, 52, 6084–6087 CrossRef CAS PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008–2016 CrossRef CAS PubMed.
- D. Pussak, D. Ponader, S. Mosca, T. Pompe, L. Hartmann and S. Schmidt, Langmuir, 2014, 30, 6142–6150 CrossRef CAS PubMed.
- D. Pussak, M. Behra, S. Schmidt and L. Hartmann, Soft Matter, 2012, 8, 1664–1672 RSC.
- K. L. Johnson, K. Kendall and A. D. Roberts, Proc. R. Soc. London, Ser. A, 1971, 324, 301–313 CrossRef CAS.
- M. Glaubitz, N. Medvedev, D. Pussak, L. Hartmann, S. Schmidt, C. A. Helm and M. Delcea, Soft Matter, 2014, 10, 6732–6741 RSC.
- T. Pompe, S. Zschoche, N. Herold, K. Salchert, M. F. Gouzy, C. Sperling and C. Werner, Biomacromolecules, 2003, 4, 1072–1079 CrossRef CAS PubMed.
- T. Pompe, L. Renner, M. Grimmer, N. Herold and C. Werner, Macromol. Biosci., 2005, 5, 890–895 CrossRef CAS PubMed.
- G. Baier, C. Costa, A. Zeller, D. Baumann, C. Sayer, P. H. H. Araujo, V. Mailaender, A. Musyanovych and K. Landfester, Macromol. Biosci., 2011, 11, 628–638 CrossRef CAS PubMed.
- L. Segeren, B. Siebum, F. G. Karssenberg, J. W. A. Van den Berg and G. J. Vancso, J. Adhes. Sci. Technol., 2002, 16, 793–828 CrossRef CAS PubMed.
- T. Pompe, J. Starruss, M. Bobeth and W. Pompe, Biointerphases, 2006, 1, 93–97 CrossRef CAS PubMed.
- T. Pompe, S. Glorius, T. Bischoff, I. Uhlmann, M. Kaufmann, S. Brenner and C. Werner, Biophys. J., 2009, 97, 2154–2163 CrossRef CAS PubMed.
- H. Krobath, B. Rozycki, R. Lipowsky and T. R. Weikl, Soft Matter, 2009, 5, 3354–3361 RSC.
- J. Erath, S. Schmidt and A. Fery, Soft Matter, 2010, 6, 1432–1437 RSC.
- J. Buumlnsow, J. Erath, P. M. Biesheuvel, A. Fery and W. T. S. Huck, Angew. Chem., Int. Ed., 2011, 50, 9629–9632 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SCP synthesis and functionalization, surface preparation, surface coverage, meshwidh, elastic modulus measurements, stress distribution, RICM protocols. See DOI: 10.1039/c4cp05484a |
| This journal is © the Owner Societies 2015 |