
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton irradiation of DNA nucleosides in the gas phase†
Jean-Christophe
Poully
*a,
Jordan
Miles
b,
Simone
De Camillis
b,
Amine
Cassimi
a and
Jason B.
Greenwood
b
aCIMAP (UMR 6252 CEA, Université de Caen, ENSICAEN, CNRS), Boulevard Becquerel, BP 5133, 14070 CAEN Cedex 5, France. E-mail: poully@ganil.fr; Fax: +33 23145 2557; Tel: +33 23145 4442
bCentre for Plasma Physics, School of Mathematics and Physics, Queen's University Belfast, Northern Ireland, UK
First published on 11th February 2015
Abstract
The four DNA nucleosides guanosine, adenosine, cytidine and thymidine have been produced in the gas phase by a laser thermal desorption source, and irradiated by a beam of protons with 5 keV kinetic energy. The molecular ions as well as energetic neutrals formed have been analyzed by mass spectrometry in order to shed light on the ionization and fragmentation processes triggered by proton collision. A range of 8–20 eV has been estimated for the binding energy of the electron captured by the proton. Glycosidic bond cleavage between the base and sugar has been observed with a high probability for all nucleosides, resulting in predominantly intact base ions for guanosine, adenosine, and cytidine but not for thymidine where intact sugar ions are dominant. This behavior is influenced by the ionization energies of the nucleobases (G < A < C < T), which seems to determine the localization of the charge following the initial ionization. This charge transfer process can also be inferred from the production of protonated base ions, which have a similar dependence on the base ionization potential, although the base proton affinity might also play a role. Other dissociation pathways have also been identified, including further fragmentation of the base and sugar moieties for thymidine and guanosine, respectively, and partial breakup of the sugar ring without glycosidic bond cleavage mainly for adenosine and cytidine. These results show that charge localization following ionization by proton irradiation is important in determining dissociation channels of isolated nucleosides, which could in turn influence direct radiation damage in DNA.
Introduction
Biologically-relevant molecular systems such as DNA, RNA, proteins and lipids can be damaged by ionizing radiation such as photons, electrons or ions. Following these ionization events, which occur on femtosecond or sub-femtosecond timescales, in order to characterize the fundamental processes occurring up to a millisecond later, experimental and theoretical studies at the molecular scale are needed. Gas-phase investigations of the basic biomolecular building blocks have been valuable for determining their unique intrinsic structural and dynamic properties and facilitated comparison with theory, which is more tractable for isolated molecules. The effect of the local environment can then be investigated systematically by binding a given number of molecules like water to the biomolecule of interest. Besides, early work using ovens as a source of gas-phase molecules could only focus on small systems to avoid thermal decomposition. As a result, a range of biological building blocks such as amino acids,1–9 nucleobases10–16 but also sugars,17–19 have been studied. Investigations on molecules composed of several of these building blocks, for instance neutral nucleosides and nucleotides, are much rarer,20–23 though the use of electrospray ionization, laser desorption and ion traps has enabled a range of protonated and deprotonated DNA species to be investigated.24–30 Very recently, experiments involving collisions between O6+ ions and nucleosides have been performed at the GANIL facility (Caen, France).31 However, irradiation of isolated neutral nucleosides by a proton beam has never been reported, to the best of our knowledge, despite the interest in unraveling the molecular basis of proton therapy.Early studies on nucleosides used electron-impact20,32 or multi-photon ionization21 coupled to a mass spectrometer, and demonstrated the power of these techniques in extracting fragmentation mechanisms and structural molecular information. A key finding is the very high probability of glycosidic bond cleavage, separating nucleosides into their base and sugar parts. This also appeared in a more recent work on thymidine and uridine,22 which showed that thermal decomposition of thymidine occurred above 147 °C, about 10 °C more than the conditions used by Levola et al.33 for a VUV photoionization study. These two groups also identified the main fragments and reported their appearance energies. Further work has been done by Itälä et al.34 using synchrotron radiation, where they compare valence- and core-ionization of thymidine, and find that the latter leads to double ionization and extensive fragmentation into small ionic species.
This report presents the first mass spectra obtained by proton collision on the four DNA nucleosides (guanosine, adenosine, cytidine and thymidine). Protons at 5 keV have the relevant kinetic energy of secondary particles created in the track of MeV hadrons in biological matter, and have been predicted to exhibit a higher linear energy transfer than the primary beam after the Bragg peak.35 We use these results, in conjunction with existing data, to deduce the ionization, charge transfer and fragmentation mechanisms arising from these interactions. In particular, we compare our spectra with those from the Japanese SDBS database, coming from electron impact ionization at 75 eV. These electrons have a velocity comparable to 5 keV protons. However, the collision processes into play are different, ionization with electrons and electron capture with protons. It is thus interesting to check whether fragmentation is sensitive or not to the primary process.
Experimental set-up
All experiments were performed with a set-up composed of a proton source, a thermal desorption source of gas-phase neutral molecules, and the KEIRA Time-of-Flight (ToF) mass spectrometer. The proton beam was produced from an Electron Cyclotron Resonance (ECR) source that has been described elsewhere.36 The proton beam kinetic energy was 5 keV and had a continuous beam current of typically 0.4 μA. The beam was pulsed at 4 kHz, for a duration of 2 μs. Two alignment apertures (3 mm in diameter) were located before and after the extraction region of the mass spectrometer, to define the beam size and to position it. The KEIRA spectrometer has been previously described in detail37 and has the advantage that it can be operated in a high resolution trapping mode.38 For the present experiment, only the ToF mode was used as too few ions were generated per ion pulse. Gas-phase molecules were produced by a laser thermal heating source using the set-up presented in reference.39 A 100 mW, continuous wave 532 nm laser was employed to heat the nucleoside samples, which were deposited on a 10 μm stainless steel foil mounted onto the ToF repeller plate. The laser heated the reverse side of the foil to sublimate the sample which travelled 5 mm before being intersected by the proton beam. For adenosine and cytidine, some additional heating was provided by a halogen lamp.Molecular ions produced from the proton–molecule interaction were extracted by pulsing the ToF repeller and extraction plates from ground potential up to a maximum of 5 kV, 0.7 μs after the end of the proton pulse. The nucleoside ions were accelerated by the extraction field, collimated by two Einzel lenses and detected by a channel electron multiplier. Additional plates located just in front of the detector could also be used to reflect the ions in order to detect energetic neutrals formed by further delayed dissociation of nucleoside ions. Thymine (>99%), adenosine (>99%), and guanosine (>98%) were purchased from Sigma-Aldrich, cytidine (>99%) and 2′-deoxy-D-thymidine (>98%) from Carbosynth. These chemicals were used without any further purification.
Results and discussion
Comparison between thymine and thymidine
In order to test the experimental set-up, we chose the DNA nucleobase thymine, which has been widely studied by mass spectrometry with a range of ionization techniques,11,40–47 and notably by Tabet et al. after proton collision at 80 keV.48 The mass spectrum obtained is shown in Fig. 1(a), together with the one from the Japanese SDBS database (electron ionization at 75 eV).49 The same fragments can be found in both spectra, but some notable differences are nevertheless observed. First, the peaks in our spectrum are broader, and thus not fully resolved. Secondly, fragment ions are more intense in comparison to the parent ion at m/z = 126 amu. Fragments are even more abundant in the spectrum obtained by Tabet et al. Interestingly, our spectrum for protons at 5 keV is very similar to the one published by de Souza et al.50 obtained after electron impact at 1 keV. This suggests that internal energy deposition increases with the beam's kinetic energy, regardless of the nature of the incident particle. The strong intact thymine radical cation signal as well as the global similarity of our spectrum with those in the literature indicate that thermal decomposition is not a factor in our measurements. | ||
| Fig. 1 Mass spectra of gas-phase thymine (a) and thymidine (b) after collision with 5 keV protons (top) and 75 eV electrons (bottom, from the SDBS database). Thymine, protonated thymine, thymidine and deoxyribose cations are noted T+, [T + H]+, Th+ and [dR–OH]+, respectively. The notation of fragments in (b) follows that of Fig. 4. | ||
Thymidine is the DNA nucleoside composed of the thymine base linked to a deoxyribose molecule via a glycosidic bond. The spectrum obtained from the interaction with protons at 5 keV is presented in Fig. 1(b), which is compared to the SDBS spectrum (electrons at 75 eV). The intact thymidine cation is clearly observed at 242 amu, and noted as Th+. The most intense peak at 117 amu is due to the intact sugar cation (denoted as [dR–OH]+) formed by breakage of the glycosidic bond. In our spectrum, the resolution does not allow us to resolve peaks due to protonated thymine (m/z = 127 amu, [T + H]+) and to its radical cation (m/z = 126 amu, T+) but careful calibration of the mass spectrum, using ionization of Xe gas, gives a peak centered at 126.5 amu, showing that both ions are produced in similar proportions. This observation is consistent with other studies.22,33,34 The weak features around 150–155 amu are greater than the mass of the thymine and deoxyribose moieties, and are due to fragmentation of deoxyribose without glycosidic bond cleavage.34 Since the intact thymidine cation peak is also weak, we can conclude that the glycosidic bond has a very high probability of being broken after ionization of isolated thymidine, as was found in previous studies.22,33,34
Recently, Levola et al.33 performed a temperature study of thymidine sublimated from an effusion cell, and showed that the mass spectrum after UV ionization at 10 eV changes dramatically above 135 °C due to thermal decomposition. Above 135 °C the intact thymidine cation disappeared and the 98 amu fragment was the most significant peak, while 117 amu dominated at the lower temperature. The fact that we did not observe an abundant 98 amu ion and instead detected the most intense peak at 117 amu along with an intact thymidine peak, suggests that there was very little thermal decomposition of our sample due to laser heating, if any. This was further confirmed by additional experiments performed in the same conditions, but replacing the proton beam with UV laser pulses (267 nm, 130 fs, 5 × 1011 W cm−2), which provides a much softer ionization method via resonant 1 + 1 photon absorption: the spectrum obtained is shown in Fig. 2.
 | ||
| Fig. 2 Mass spectrum of gas-phase thymidine ionized by a UV femtosecond laser (267 nm, 130 fs, 5 × 1011 W cm−2). | ||
It is interesting to note that S. Maclot31 has shown that after single ionization by absorption of one 50 eV photon, the fragments observed strongly depend on the binding energy (BE) of the ejected electron. In particular, the survival rate of the intact thymidine cation is about 50% for removal of electrons with a BE close to 8 eV, around the UV photoionization threshold energy of thymidine.33 When the BE is scanned up to 16 eV, this survival rate drops rapidly while fragments increase in intensity and decrease in size. Double ionization is expected to start around 20 eV, according to experimental values for molecules such as quinoline (23 eV) or pyrrole (24 eV),51 but is not expected to be significant in the present study since protons at the much higher energy of 80 keV have been shown to induce very little double ionization of uracil52 and thymine.48 This indicates that in our measurements, the estimated range of electron binding energies is between 8 and 20 eV.
Comparing the mass spectra of thymidine and thymine, allows us to probe the role of the deoxyribose moiety on the ionization and fragmentation of the nucleoside. Such a comparison is shown in Fig. 3(a), where both spectra are presented in the m/z = 20–140 amu range, the mass of thymine being 126 amu (the blue dashed line in the figure). All the peaks contained in the thymine spectrum are found in the thymidine one (except the weak peak at 26 amu, attributed to C2H2+ by Jochims et al.42). Additional features found for thymidine but not thymine, are indicated by red dashed lines at 31, 45, 57, 69, 73, 81, 99, 110 and 117 amu (with the exception of 32 amu, which can also come from the residual gas). All these ions have been recently assigned by Itälä et al.34 to be due to the intact sugar (117 amu) and its fragments, with the exception of 81 and 110 amu. They did not assign the peak at 81 amu and attributed the peak at 110 amu to loss of O from the thymine radical cation, but this was not observed in previous studies on thymine ionization.14,40,42,48 On the other hand, peaks at 110 and 81 amu are abundant in low-energy Collision-Induced Dissociation (CID) spectra of protonated thymine in the MassBank database.53 We thus propose to assign them to loss of NH3 and (H2O + CO) from protonated thymine, assuming that its fragmentation is similar to that of protonated uracil.54 This would indicate that protonated thymine formed after proton irradiation of thymidine has enough internal energy to fragment. It is noted that 81 amu is also the mass of a fragment of deoxyribose, as reported by Ptasińska et al.,19 but in such conditions that thermal decomposition could not be ruled out.
 | ||
| Fig. 3 (a) Mass spectra of gas-phase thymidine (top) and thymine (bottom) after collision with 5 keV protons, in the thymine fragmentation region. The mass of cations corresponding to peaks appearing only in the thymidine spectrum are written in red, and the position of the thymine radical cation (126 amu) is indicated by blue dashes. (b) Mass spectra of thymidine fragment cations formed by prompt dissociation (ns timescale; top) and undergoing delayed fragmentation (μs timescale; bottom). | ||
The spectrum obtained from the detection of only energetic neutrals formed by fragmentation of molecular ions after the acceleration region is shown in Fig. 3(b). Since these neutrals are travelling at the same velocity as the initially accelerated ion, they are detected at a similar ToF. The ToF of each neutral is slightly longer due to the ions being accelerated by a further Einzel lens in the field-free region, although for the neutral spectrum in Fig. 3(b) this has been accounted for using SIMION simulations.55 Therefore a peak at a specific mass in the delayed fragmentation spectrum indicates the mass of the ion that is fragmenting, not the mass of the neutral daughter fragment. To the best of our knowledge, such delayed fragmentation on the microsecond timescale has been reported for adenine after collision with He+ and He56 as well as Ar8+,57 for isolated bromouracil and cytosine clusters ionized by multiply-charged ions,58,59 but not for nucleoside radical cations.
In our spectrum, some of the intact sugar ions (117 amu) are seen to undergo delayed fragmentation but the most intense feature corresponds to the peak at 99 amu, assigned to [dR–OH–H2O]+ in previous studies.22,46 The peak at 73 amu is also due to further breakup of a sugar fragment. The peak at 81 amu could be derived from either a sugar or base fragment but given neutrals from breakup of the base ion (126 amu) are barely visible, this delayed fragmentation is also likely to originate from sugar ions. The preponderance of sugar fragments in both the ion and neutral spectra is consistent with the fact that following ionization, the charge is localized on the deoxyribose rather than the nucleobase. This leads to the intact sugar ion or fragment thereof being produced, which may then undergo further breakup via statistical fragmentation on a microsecond timescale.
Cytidine, adenosine and guanosine
We have also performed collision experiments between protons at 5 keV and the three other DNA nucleosides: cytidine, adenosine and guanosine, the results being shown in Fig. 4. Their molecular masses are 243, 267 and 283 amu, respectively, and our spectra are compared with those from the SDBS database. For all these molecules our proton impact results generate similar fragmentation patterns as electron impact, with a few exceptions. In adenosine and guanosine, the small fragments (20–30 amu) are more significant in our spectra. In guanosine the fragment at 28 amu, which could be a base49 or sugar fragment,60 or come from the residual gas, is very strong in our spectrum, while the fragment at 57 amu due to a sugar fragment dominates the SDBS spectrum. | ||
| Fig. 4 Mass spectra of gas-phase (a) cytidine, (b) adenosine and (c) guanosine after collision with 5 keV protons (top) and low-energy electrons (bottom, from the SDBS database). Cytidine, cytosine and protonated cytosine cations are denoted Cy+, C+ and [C + H]+, respectively. Adenosine, adenine and protonated adenine cations are denoted Ad+, A+ and [A + H]+, respectively. Guanosine and guanine radical cations are denoted Gu+ and G+, respectively. (d) is a magnification of (c), to allow fragments heavier than the base to be clearly seen. Their notation follows that of Fig. 4. | ||
For these nucleosides, we performed the same laser ionization as for thymidine (see Fig. S1–S3 of the ESI†). We found that adenosine radical cation is produced fully intact, thus ruling out any thermal decomposition. In the cases of cytidine and guanosine, a strong peak for the intact molecular ion is observed, but fragment ions also appear. For the former, the most intense one is protonated cytosine, which cannot be due to formation of the nucleobase by thermal decomposition prior to proton impact.61 The other fragment peaks are also at masses greater than the base and are weaker features in the proton spectrum. For the latter, in which the guanine radical cation dominates, we cannot rule out thermal decomposition.
A list of the main ions and their assignments for each of our nucleoside spectra ranked from most to least abundant is given in Table 1. It is evident that for all the nucleosides, the breakage of the glycosidic bond to form base or sugar ions (and/or their fragments) has a high probability. However, comparing the relative yields there are some striking differences when a different base is substituted into the nucleoside. While production of intact base ions dominates the guanosine, cytidine and adenosine spectra, the largest peak in the thymidine spectrum is the intact sugar ion, and it has substantial peaks due to smaller sugar fragments (cf.Fig. 3, 4 and Table 1). In the guanosine spectrum there are also strong peaks at lower mass but these are mainly due to fragmentation of the base. In the adenosine and cytidine spectra, substantial yields are also obtained due to breakup of the sugar ring without cleavage of the glycosidic bond (labeled Sn in Fig. 1 and 4), but these ions are very minor for thymidine and guanosine. The proposed origin of these ions is shown in the schematic in Fig. 5.
| Thymidine | Cytidine | Adenosine | Guanosine | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| m/z (amu) | Assignment/origin | Intensity | m/z (amu) | Assignment/origin | Intensity | m/z (amu) | Assignment/origin | Intensity | m/z (amu) | Assignment/origin | Intensity |
| 117 | [dR–OH]+ | ++++ | 111, 112 | C+, [C + H]+ | ++++ | 135, 136 | A+, [A + H]+ | ++++ | 151 | G+ | ++++ |
| 99 | Sugar | +++ | 140 | S2 | +++ | 164 | S2 | +++ | 28 | Base/sugar/RG | ++++ |
| 73 | Sugar | +++ | 151 | S1 | ++ | 178 | S1 | ++ | 43, 44 | Base/sugar | +++ |
| 45 | Sugar | +++ | 243 | Cy+ | + | 108 | Base | ++ | 57 | Sugar | +++ |
| 43 | Base/sugar | +++ | 213 | S3 | + | 31 | Sugar | ++ | 109, 110 | Base | ++ |
| 126, 127 | T+, [T + H]+ | +++ | 178 | S6 | + | 29 | Base/sugar | ++ | 53–55 | Base | ++ |
| 31 | Sugar | ++ | 170 | S5 | + | 267 | Ad+ | + | 134, 135 | Base | ++ |
| 110 | Base | ++ | 154 | S1 | + | 237 | S3 | + | 71, 73 | Sugar | ++ |
| 71 | Base | ++ | 94, 95 | Base | + | 148 | S4 | + | 69 | Base | ++ |
| 55 | Base | ++ | 85, 86 | Sugar | + | 119 | Base | + | 60 | Sugar | ++ |
| 32 | Base/RG | ++ | 83 | Base | + | 81 | Base/sugar | + | 31 | Sugar | ++ |
| 28 | Base/sugar/RG | ++ | 81 | Sugar | + | 73 | Sugar | + | 283 | Gu+ | + |
| 252 | Th+ | + | 73 | Sugar | + | 66–70 | Base | + | 265 | [Gu–H2O]+ | + |
| 153 | S2 | + | 66–69 | Base | + | 60, 61 | Sugar | + | 194 | S1 | + |
| 150 | S1 | + | 60, 61 | Sugar | + | 45 | Sugar | + | 178 | S2 | + |
| 82, 83 | Base | + | 57 | Sugar | + | 38–42 | Base | + | 164 | S4 | + |
| 81 | Base/sugar | + | 52–56 | Base | + | 28 | Base/sugar/RG | + | 161 | B | + |
| 70 | Base/sugar | + | 45 | Sugar | + | 96, 97 | Base | + | |||
| 57 | Sugar | + | 43, 44 | Base/sugar | + | 85, 86 | Sugar | + | |||
| 52–54 | Base | + | 38–42 | Base | + | 80–83 | Base | + | |||
| 44 | Base/sugar | + | 32 | RG | + | 38–42 | Base | + | |||
| 38–42 | Base | + | 31 | Sugar | + | 29 | Base/sugar | + | |||
| 30 | Base | + | 29 | Base/sugar | + | ||||||
| 29 | Base/sugar | + | 28 | Base/sugar/RG | + | ||||||
| 27 | Base/sugar | + | 27 | Base | + | ||||||
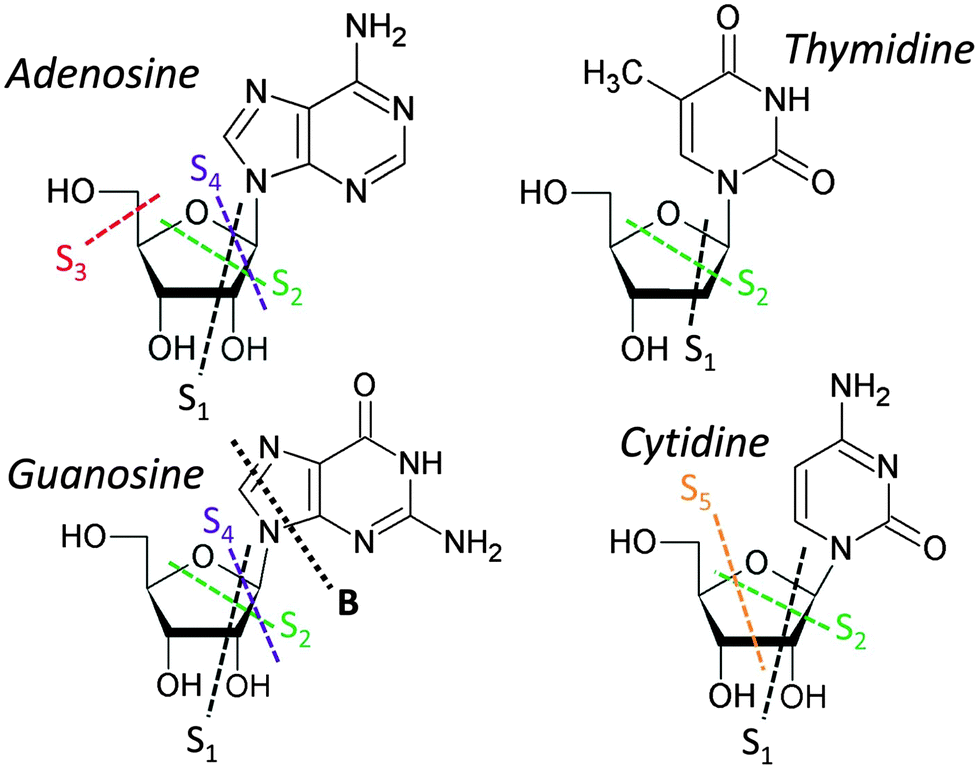 | ||
| Fig. 5 Chemical drawings of the four nucleosides studied, showing the proposed dissociations giving the observed main fragments that are heavier than the base. | ||
The relative yield of the radical base cation to the protonated base also changes significantly for each nucleoside. The protonated base is generally very significant in all our spectra (particularly for thymidine and cytidine) except for guanosine where it is virtually absent. It might be surmised that the low yield in guanosine can be attributed to thermal decomposition into the base and sugar components, followed by evaporation and ionization of guanine. However, Feketová et al.62 published a CID spectrum of the guanosine radical cation formed by means of electrospray ionization (thermal decomposition can thus be ruled out) where no protonated guanine was observed either. For cytidine and adenosine, known CID fragments of protonated bases63,64 are detected, due to loss of ammonia and H2O (the latter only for cytidine). This is consistent with the formation of vibrationally-excited protonated bases, as seen for thymidine in the previous section.
Discussion
An explanation for the striking variation in behavior we have observed for the nucleosides can be found in the relative electron binding energies of the highest occupied molecular orbitals of the nucleobase and sugar groups within the nucleosides. Table 2 gives a summary of data for the ionization energies of the nucleobases and nucleosides. A large number of studies using a variety of techniques have been used to measure these values and no one value can be quoted due to differences in definition (adiabatic/vertical ionization, appearance energies) and the range of conformers/tautomers which might be present. However, it is clear that the ionization energies for the RNA and DNA bases as well as for nucleosides are ordered as follows: G < A < C < T < U (Uracil). Meanwhile, a recent study of the isolated deoxyribose sugar has significantly revised the adiabatic ionization energy from 10.5 eV obtained in previous studies down to 8.8 eV (for theory) or 9.1 eV (experiment),17 which is similar to that of thymine (8.9 eV).65 These small differences in ionization energies may have an influence on the initial ionization event, particularly for an exothermic electron capture process. However, as was discussed in the "Comparison between thymine and thymidine" section, 5 keV protons are expected to remove electrons with binding energies up to 20 eV, which opens up removal of electrons in many valence orbitals. Following the sudden removal of the electron (a 5 keV proton takes 3 fs to travel a distance of 0.3 nm), the difference in the energies of base and sugar HOMOs is important, as this will strongly influence the final destination of the positive hole generated. Such intramolecular charge transfer is expected to be much faster than any subsequent dissociation dynamics; recently shown to be less than 5 fs in an amino acid.66,67 Therefore, as the nucleobase ionization energy reduces, it is more likely to accept the charge irrespective of the initial ionization site. It is already known in DNA that the base with the lowest ionization energy, guanine, acts as a charge sink to form guanine radicals, which subsequently influences damage to the DNA.68–70| Nucleobases | Nucleosides | ||
|---|---|---|---|
| Ionization energy | Proton affinity | Ionization energy | |
| U | 9.5 ± 0.1 | 9.04 | 9.0 ± 0.1 |
| T | 9.2 ± 0.2 | 9.13 | 8.7 ± 0.1 |
| C | 8.75 ± 0.25 | 9.85 | 8.6 ± 0.1 |
| A | 8.6 ± 0.3 | 9.77 | 8.4 ± 0.1 |
| G | 8.0 ± 0.2 | 9.95 | 8.0 ± 0.1 |
Strong localization of the charge on the base is evident in our guanosine spectrum since intact or fragment ions from the base are dominant, even if some of the guanine cation might be due to thermal decomposition (see previous section). In contrast thymidine, which contains the nucleobase with the highest ionization energy, is the only nucleoside for which the intact sugar is observed along with numerous sugar fragments, while production of base ions is uncharacteristically weak. This is consistent with the charge strongly localized on the sugar. For the intermediate cases of adenosine and cytidine, the yield of base ions signifies charge residing on the base, but the presence of Sn ions (base + part sugar) suggests that in some cases the charge may be more de-localized prior to dissociation. It is important to notice that the intact sugar peak for thymidine could also be due to the nature of the sugar (deoxyribose), which is different from the other nucleosides (ribose). However, Biemann and McCloskey20 have shown that uridine and deoxyuridine also give an intense, intact sugar peak, which supports our hypothesis that charge is increasingly localized on the sugar for higher base ionization potentials.
There is also a strong dependence on the ratio of the yield of protonated to radical base ions with the base ionization energy. For our results, as the ionization energy of the base increases this ratio changes from very low in guanosine to about one in thymidine, with the exception of cytidine which has a particularly high yield of protonated base cations. A similar trend can be found for the electron impact results, which continues if uridine is also considered (uracil has an even higher ionization energy).20,71 To form the radical base cation, the glycosidic bond must be broken in conjunction with a H atom transfer when the charge is on the base, or a proton transfer if the charge is on the sugar. Meanwhile, to form the protonated base there could be double H atom transfer if the charge is on the base,34 or H atom and proton transfer if the charge is initially on the sugar.29 Therefore, while it is evident that the base ionization energy is strongly influencing the protonated to radical ratio, the proton affinity of the base may also be playing a role. The higher proton affinity of cytosine compared to thymine (see Table 2) may explain why the cytidine ratio is out of sequence. This would suggest that prior to or during fragmentation, the radical base cation is formed by localization of the charge on the base followed by H atom transfer, while the protonated base is formed by proton and H atom transfer from the sugar. However, from our data alone we cannot be certain that these processes are dominating. To get a definitive answer would require sophisticated molecular dynamics simulations or ultrafast pump–probe laser experiments.
Conclusions
Overall, our results show that oxidation of nucleosides via proton impact results predominantly in breakage of the glycosidic bond between the sugar and base. However, the ultimate destination of this charge is strongly influenced by the local electron binding energies of the deoxyribose and base groups which determine any ultrafast charge transfer processes. In guanosine, the nucleobase acts as a sink for the charge which leads to strong fragmentation of the base. In contrast, for thymidine, localization of the charge on the other side of the glycosidic bond produces substantial disintegration of the sugar which could be a source of strand breaks in vivo. In cytidine and adenosine, significant fragmentation of the sugar ring is also present but without cleavage of the glycosidic bond.These results provide valuable insight into the mechanisms which lead to DNA radiation damage. In the near future, experiments involving highly-charged ions in the MeV range are planned to probe the effect of the kinetic energy and the ion charge state on ionization and fragmentation of nucleosides in the gas phase. These studies will be directly relevant to proton and heavy ion therapies, as the linear energy transfer is highest at these energies and corresponds to how ions interact with DNA in tumor cells at the maximum of the Bragg peak.
Acknowledgements
This work was supported by the Leverhulme Trust (research grant RPG-2012-735) and the Northern Ireland Department of Employment and Learning. J.-C. Poully acknowledges the European COST action MP 1002 Nano-IBCT for funding.Notes and references
- H. Farrokhpour, F. Fathi and A. Naves De Brito, J. Phys. Chem. A, 2012, 116, 7004–7015 CrossRef CAS PubMed.
- M. Capron, S. Diaz-Tendero, S. Maclot, A. Domaracka, E. Lattouf, A. Lawicki, R. Maisonny, J.-Y. Chesnel, A. Mery, J.-C. Poully, J. Rangama, L. Adoui, F. Martin, M. Alcami, P. Rousseau and B. A. Huber, Chem. – Eur. J., 2012, 18, 9321–9332 CrossRef CAS PubMed.
- W. Zhang, V. Carravetta, O. Plekan, V. Feyer, R. Richter, M. Coreno and K. C. Prince, J. Chem. Phys., 2009, 131, 035103 CrossRef PubMed.
- V. Bernigaud, B. Manil, L. Adoui, J. Y. Chesnel, J. Rangama, B. A. Huber, F. Alvarado, S. Bari, R. Hoekstra, J. Postma and T. Schlatholter, Xxvi International Conference On Photonic, Electronic And Atomic Collisions, 2009, vol. 194 Search PubMed.
- O. Plekan, V. Feyer, R. Richter, M. Coreno, M. de Simone, K. C. Prince and V. Carravetta, J. Electron Spectrosc. Relat. Phenom., 2007, 155, 47–53 CrossRef CAS PubMed.
- M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem., 2007, 58, 585–612 CrossRef CAS PubMed.
- M. J. Polce and C. Wesdemiotis, J. Am. Soc. Mass Spectrom., 1999, 10, 1241–1247 CrossRef CAS.
- U. Boesl, J. Phys. Chem., 1991, 95, 2949–2962 CrossRef CAS.
- I. Ipolyi, P. Cicman, S. Denifl, V. Matejčík, P. Mach, J. Urban, P. Scheier, T. D. Märk and S. Matejčík, Int. J. Mass Spectrom., 2006, 252, 228–233 CrossRef CAS PubMed.
- B. Barc, M. Ryszka, J. Spurrell, M. Dampc, P. Limao-Vieira, R. Parajuli, N. J. Mason and S. Eden, J. Chem. Phys., 2013, 139, 244311 CrossRef CAS PubMed.
- K. B. Bravaya, O. Kostko, S. Dolgikh, A. Landau, M. Ahmed and A. I. Krylov, J. Phys. Chem. A, 2010, 114, 12305–12317 CrossRef CAS PubMed.
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 7562–7568 CrossRef CAS PubMed.
- N. J. Kim, G. Jeong, Y. S. Kim, J. Sung and S. K. Kim, J. Chem. Phys., 2000, 113, 10051–10055 CrossRef CAS PubMed.
- T. Schlathölter, F. Alvarado, S. Bari, A. Lecointre, R. Hoekstra, V. Bernigaud, B. Manil, J. Rangama and B. Huber, ChemPhysChem, 2006, 7, 2339–2345 CrossRef PubMed.
- A. B. Trofimov, J. Schirmer, V. B. Kobychev, A. W. Potts, D. M. P. Holland and L. Karlsson, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, 305–329 CrossRef CAS.
- S. Wee, R. A. J. O'Hair and W. D. McFadyen, Rapid Commun. Mass Spectrom., 2005, 19, 1797–1805 CrossRef CAS PubMed.
- D. Ghosh, A. Golan, L. K. Takahashi, A. I. Krylov and M. Ahmed, J. Phys. Chem. Lett., 2012, 3, 97–101 CrossRef CAS.
- J.-W. Shin, F. Dong, M. E. Grisham, J. J. Rocca and E. R. Bernstein, Chem. Phys. Lett., 2014, 506, 161–166 CrossRef PubMed.
- S. Ptasińska, S. Denifl, P. Scheier and T. D. Märk, J. Chem. Phys., 2004, 120, 8505–8511 CrossRef PubMed.
- K. Biemann and J. A. McCloskey, J. Am. Chem. Soc., 1962, 84, 2005 CrossRef CAS.
- L. Li and D. M. Lubman, Int. J. Mass Spectrom. Ion Processes, 1989, 88, 197 CrossRef CAS.
- S. Ptasińska, P. Candori, S. Denifl, S. Yoon, V. Grill, P. Scheier and T. D. Märk, Chem. Phys. Lett., 2005, 409, 270–276 CrossRef PubMed.
- H. Asami, K. Yagi, M. Ohba, S.-h. Urashima and H. Saigusa, Chem. Phys., 2013, 419, 84–89 CrossRef CAS PubMed.
- H. D. Flosadottir, B. Omarsson, I. Bald and O. Ingolfsson, Eur. Phys. J. D, 2012, 66, 13 CrossRef.
- H. D. Flosadottir, H. Jonsson, S. T. Sigurdsson and O. Ingolfsson, Phys. Chem. Chem. Phys., 2011, 13, 15283–15290 RSC.
- V. Gabelica, T. Tabarin, R. Antoine, F. Rosu, I. Compagnon, M. Broyer, E. De Pauw and P. Dugourd, Anal. Chem., 2006, 78, 6564–6572 CrossRef CAS PubMed.
- Y. Gao and S. A. McLuckey, Rapid Commun. Mass Spectrom., 2014, 27, 249–257 CrossRef PubMed.
- J. C. Marcum, S. H. Kaufman and J. M. Weber, Int. J. Mass Spectrom., 2011, 303, 129–136 CrossRef CAS PubMed.
- O. Gonzalez-Magana, M. Tiemens, G. Reitsma, L. Boschman, M. Door, S. Bari, P. O. Lahaie, J. R. Wagner, M. A. Huels, R. Hoekstra and T. Schlatholter, Phys. Rev. A: At., Mol., Opt. Phys., 2014, 87, 032702 CrossRef.
- B. Liu, P. Hvelplund, S. B. Nielsen and S. Tomita, Int. J. Mass Spectrom., 2003, 230, 19 CrossRef CAS.
- S. Maclot, PhD thesis, 2014.
- S. J. Shaw, D. M. Desiderio, K. Tsuboyama and J. A. McCloskey, J. Am. Chem. Soc., 1970, 92, 2510 CrossRef CAS.
- H. Levola, K. Kooser, E. Rachlew, E. Nommiste and E. Kukk, Int. J. Mass Spectrom., 2013, 353, 7–11 CrossRef CAS PubMed.
- E. Itälä, M. A. Huels, E. Rachlew, K. Kooser, T. Hägerth and E. Kukk, J. Phys. B: At., Mol. Opt. Phys., 2013, 46, 215102 CrossRef.
- C. Grassberger and H. Paganetti, Phys. Med. Biol., 2011, 56, 6677–6691 CrossRef CAS PubMed.
- R. Trassl, P. Hathiramani, F. Broetz, J. B. Greenwood, R. W. McCullough, M. Schlapp and E. Salzborn, Phys. Scr., 1997, T73, 380–381 CrossRef CAS.
- O. Kelly, M. J. Duffy, R. B. King, L. Belshaw, I. D. Williams, J. Sa, C. R. Calvert and J. B. Greenwood, Analyst, 2012, 137, 64 RSC.
- J. B. Greenwood, O. Kelly, C. R. Calvert, M. J. Duffy, R. B. King, L. Belshaw, L. Graham, J. D. Alexander, I. D. Williams, W. A. Bryan, I. C. E. Turcu, C. M. Cacho and E. Springate, Rev. Sci. Instrum., 2011, 82, 043103 CrossRef CAS PubMed.
- C. R. Calvert, L. Belshaw, M. J. Duffy, O. Kelly, R. B. King, A. G. Smyth, T. J. Kelly, J. T. Costello, D. J. Timson, W. A. Bryan, T. Kierspel, P. Rice, I. C. E. Turcu, C. M. Cacho, E. Springate, I. D. Williams and J. B. Greenwood, Phys. Chem. Chem. Phys., 2012, 14, 6282–6290 RSC.
- J. d. Vries, R. Hoekstra, R. Morgenstern and T. Schlathölter, Eur. Phys. J. D, 2003, 24, 161–164 CrossRef.
- I. I. Shafranyosh, M. I. Sukhoviya, M. I. Shafranyosh and L. L. Shimon, Tech. Phys., 2008, 53, 1536–1540 CrossRef CAS.
- H. W. Jochims, M. Schwell, H. Baumgärtel and S. Leach, Chem. Phys., 2005, 314, 263–282 CrossRef CAS PubMed.
- K. W. Choi, J. H. Lee and S. K. Kim, J. Am. Chem. Soc., 2005, 127, 15674–15675 CrossRef CAS PubMed.
- M. Schneider, R. Maksimenka, F. J. Buback, T. Kitsopoulos, L. R. Lago and I. Fischer, Phys. Chem. Chem. Phys., 2006, 8, 3017–3021 RSC.
- P. J. M. van der Burgt, Eur. Phys. J. D, 2014, 68, 135 CrossRef.
- E. Itälä, D. T. Ha, K. Kooser, E. Rachlew, M. A. Huels and E. Kukk, J. Chem. Phys., 2013, 133, 154316 CrossRef PubMed.
- T. Schlathölter, R. Hoekstra and R. Morgenstern, Int. J. Mass Spectrom., 2004, 233, 173 CrossRef PubMed.
- J. Tabet, S. Eden, S. Feil, H. Abdoul-Carime, B. Farizon, M. Farizon, S. Ouaskit and T. D. Maerk, Int. J. Mass Spectrom., 2010, 292, 53–63 CrossRef CAS PubMed.
- National Institute of Advanced Industrial Science and Technology, SDBSWeb: http://sdbs.riodb.aist.go.jp, 2014.
- G. G. B. de Souza, L. H. Coutinho, C. Nunez, R. Bernini, R. B. Castilho and A. F. Lago, J. Phys.: Conf. Ser., 2007, 88, 012005 CrossRef.
- R. D. Molloy, A. Danielsson, L. Karlsson and J. H. D. Eland, Chem. Phys., 2007, 335, 49 CrossRef CAS PubMed.
- J. Tabet, S. Eden, S. Feil, H. Abdoul-Carime, B. Farizon, M. Farizon, S. Ouaskit and T. D. Märk, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 81, 012711 CrossRef.
- H. Horai, M. Arita, S. Kanaya, Y. Nihei, T. Ikeda, K. Suwa, Y. Ojima, K. Tanaka, S. Tanaka, K. Aoshima, Y. Oda, Y. Kakazu, M. Kusano, T. Tohge, F. Matsuda, Y. Sawada, M. Y. Hirai, H. Nakanishi, K. Ikeda, N. Akimoto, T. Maoka, H. Takahashi, T. Ara, N. Sakurai, H. Suzuki, D. Shibata, S. Neumann, T. Iida, K. Tanaka, K. Funatsu, F. Matsuura, T. Soga, R. Taguchi, K. Saito and T. Nishioka, J. Mass Spectrom., 2010, 45, 703 CrossRef CAS PubMed.
- L. Sadr-Arani, P. Mignon, H. Chermette and T. Douki, Chem. Phys. Lett., 2014, 605–606, 108 CrossRef CAS PubMed.
- D. A. Dahl, SIMION 3D, 2000 Search PubMed.
- F. Alvarado, S. Bari, R. Hoekstra and T. Schlatholter, J. Chem. Phys., 2007, 127, 034301 CrossRef PubMed.
- S. Martin, R. Bredy, A. R. Allouche, J. Bernard, A. Salmoun, B. Li and L. Chen, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 77, 062513 CrossRef.
- R. Delaunay, J.-P. Champeaux, S. Maclot, M. Capron, A. Domaracka, A. Mery, B. Manil, L. Adoui, P. Rousseau, P. Moretto-Capelle and B. A. Huber, Eur. Phys. J. D, 2014, 68, 162 CrossRef.
- V. Vizcaino, J. C. Poully, J. Y. Chesnel, A. Domaracka, S. Maclot, A. Méry, J. Rangama, P. Rousseau, L. Adoui and B. A. Huber, Int. J. Mass Spectrom., 2014, 365–366, 181 CrossRef CAS PubMed.
- J.-W. Shin and E. R. Bernstein, J. Chem. Phys., 2014, 140, 044330 CrossRef PubMed.
- S. J. DeLuca and K. J. Voorhees, J. Anal. Appl. Pyrolysis, 1993, 24, 211 CrossRef CAS.
- L. Feketeová, E. Yuriev, J. D. Orbell, G. N. Khairallah and R. A. J. O'Hair, Int. J. Mass Spectrom., 2011, 304, 74 CrossRef PubMed.
- C. Yao, F. Turecek, M. J. Polce and C. Wesdemiotis, Int. J. Mass Spectrom., 2007, 265, 106 CrossRef CAS PubMed.
- C. C. Nelson and J. A. McCloskey, J. Am. Chem. Soc., 1992, 114, 3661 CrossRef CAS.
- K.-W. Choi, J.-H. Lee and S. K. Kim, J. Am. Chem. Soc., 2005, 127, 15674 CrossRef CAS PubMed.
- F. Calegari, D. Ayuso, A. Trabattoni, L. Belshaw, S. De Camillis, S. Anumula, F. Frassetto, L. Poletto, A. Palacios, P. Decleva, J. B. Greenwood, F. Martín and M. Nisoli, Science, 2014, 346, 336–339 CrossRef CAS PubMed.
- L. Belshaw, F. Calegari, M. J. Duffy, A. Trabattoni, L. Poletto, M. Nisoli and J. B. Greenwood, J. Phys. Chem. Lett., 2012, 3, 3751 CrossRef CAS.
- T. Melvin, S. Botchway, A. W. Parker and P. Oneill, J. Chem. Soc., Chem. Commun., 1995, 653–654 RSC.
- L. P. Candeias and S. Steenken, J. Am. Chem. Soc., 1993, 115, 2437–2440 CrossRef CAS.
- A. W. Parker, C. Y. Lin, M. W. George, M. Towrie and M. K. Kuimova, J. Phys. Chem. B, 2010, 114, 3660 CrossRef CAS PubMed.
- NIST, in NIST Standard Reference Database, ed. P. J. L. a. W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD 20899, 2005, vol. 2005 Search PubMed.
- C. Yu, T. J. O'Donnell and P. R. LeBreton, J. Phys. Chem., 1981, 85, 3851 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05303f |
| This journal is © the Owner Societies 2015 |