
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProspects for plasmonic hot spots in single molecule SERS towards the chemical imaging of live cells
Darya
Radziuk
* and
Helmuth
Moehwald
Max-Planck Institute of Colloids and Interfaces, Department of Interfaces, D14476 Potsdam (Golm), Germany. E-mail: darya.radziuk@mpikg.mpg.de
First published on 8th January 2015
Abstract
Single molecule surface enhanced Raman scattering (SM-SERS) is a highly local effect occurring at sharp edges, interparticle junctions and crevices or other geometries with a sharp nanoroughness of plasmonic nanostructures (“hot spots”). The emission of an individual molecule at SM-SERS conditions depends on the local enhancement field of the hot spots, as well as the binding affinity and positioning at a hot spot region. In this regard, the stability of near-field nano-optics at hot spots is critical, particularly in a biological milieu. In this perspective review, we address recent advances in the experimental and theoretical approaches for the successful development of SM-SERS. Significant progress in the understanding of the interaction between the excitation electromagnetic field and the surface plasmon modes at the metallic or metallic/dielectric interface of various curvatures are described. New knowledge on methodological strategies for positioning the analytes for SM-SERS and Raman-assisted SERS or the SERS imaging of live cells has been acquired and displayed. In the framework of the extensive development of SM-SERS as an advancing diagnostic analytical technique, the real-time SERS chemical imaging of intracellular compartments and tracing of individual analytes has been achieved. In this context, we highlight the tremendous potential of SERS chemical imaging as a future prospect in SERS and SM-SERS for the prediction and diagnosis of diseases.
Introduction
Less than forty years have passed since the initial discovery of strong Raman signals enhanced at a rough silver surface,1,2 which quickly gave rise to a new emerging research field – surface enhanced Raman spectroscopy, or in brief: SERS. SERS enhancement occurs during the interaction of an incident electromagnetic field with surface plasmon resonance at a metal surface. High SERS enhancement factors (>107–108) can be developed at the interparticle junctions of plasmonic nanostructures (‘plasmonic hot spots’), sufficient enough to enable single molecule detection. A typical single molecule SERS spectrum appears as signals randomly fluctuating with time at local positions with the enhancement on the SERS substrate. For this reason, the single molecule SERS phenomenon is controversial, and indeed there has been much debate about its existence over the years.From a fundamental point of view, it is important to understand how the average enhancement factor can contribute to each molecule under SERS conditions on the surface. In this aspect, one of the critical parameters is an analyte adsorption and positioning at the plasmonic hot spot, especially concerning the transfer of an analyte from the bulk to the SERS active surface. The adsorption efficiency directly affects the analytical SERS enhancement factors. For example, 10% adsorption efficiency means 10 times less signal at a given concentration. One molecule positioned at plasmonic hot spot can provide 10 times higher signal than one only a few nanometers away and 100 times more than SERS signal from randomly adsorbed molecules.3
What are the SERS conditions needed to ensure single molecule detection? New methods have been developed to position analytes at hot spots resulting in strong improvements in analytical enhancement factors and in the statistics of single molecule detection. Various colloidal lithography methods, self-assembly procedures and surface-functionalization chemistry techniques have been advanced in this regard. However, the transportation of analytes from the bulk solution to the SERS active surface and selective positioning at hot spots is still a critical problem especially in a biological environment.
As single molecule SERS is a highly local effect the understanding of its coupling into the plasmon modes at hot spots is important for the development of new plasmonic nanostructures and theoretical models for SERS emission.4 How the nanoparticle geometry, dimensions and sustainability contribute to the electromagnetic field enhancement at hot spots in the SERS phenomenon is not yet fully understood. What is the diffusion behaviour of analytes at the plasmonic hot spots?5
The main task of SERS is the amplification of weak Raman signals. In this regard, electromagnetic field enhancement is a dominating factor and can be well controlled if we understand the relationship between the localized surface plasmon effect and the metallic or metallic/dielectric nanostructure, i.e. the physico-chemical properties of the plasmonic hot spots. For this reason, this perspective aims to reveal the future prospects from experimental and theoretical points of scientific view for plasmonic hot spots for SERS detection of single analytes. As SERS is a diagnostic analytical tool, we aim to apply its power to real applications in biology and medicine, with a particular focus on single living cells. We believe that the recent progress described in this review will help to advance the SERS technique as a reliable routine method for the fast and facile diagnostic of the early stages of diseases.
In this review, we will describe the perspective strategies applied in the design of plasmonic nanostructures to form sustainable hot spots for the SERS detection of single molecules (Scheme 1). We will explain why and how these methods, such as colloidal lithography and self-assembly, have been significantly improved to address the problem of single molecules under SERS conditions. For single molecule detection, we need to find out the origin of SERS and its relationship with the extinction of plasmonic nanostructures and surface plasmon resonance coupling. In this context, new perspectives in the development of enhancement mechanisms, alongside developments in plasmon hybridization theory for hot spots will be examined. Furthermore, progress made in the qualitative analysis of SERS will be studied, with a focus on the theoretical methods used for computing the near-field nano-optics and electromagnetic coupling, followed by discussions on the great advances made in the spectral and spatial imaging of single molecules.
 | ||
| Scheme 1 Scheme of the main topics covered in this review. | ||
As we review the major advances in the physico-chemical properties of plasmonic nanostructures as a progressive contribution to SERS, this perspective review will focus on describing successful strategies for the SERS detection of individual molecules (e.g. ions, glucose or lipids) in single living cells (bacteria, plants, yeast, HeLa or fibroblasts) in vivo or in vitro. It is important to note, that SERS at the present stage is entering a new era in the evolution of biomedical tools at a molecular level to significantly improve the chemical imaging of single molecules in living cells. The unique combination of the spectral and spatial potential of SERS allows one to trace individual molecular interactions of living organelles in the cells in real time. This a tremendous step forward, especially in the early diagnosis of diseases and for revealing the transition mechanisms from ‘healthy’ to ‘unhealthy; states.
1. Design of plasmonic hot spots for single molecule detection
1.1. SERS requirements for plasmonic hot spots
SERS relies on an understanding of the Raman phenomenon. By definition, it is an inelastic light scattering technique, where incident photons interact with an analyte via the system's polarizability tensor, either depositing a quantum of energy into an excitation state (Stokes scattering) or acquiring a quantum of energy from an excitation state (anti-Stokes scattering). Beyond the particular polarization matrix elements, the Raman scattering rate is proportional to the electromagnetic intensity at the molecule, and the density of states available for the outgoing Raman-scattered photon. Through a combination of geometry and metal dielectric function, metal nanostructures have a frequency-dependent optical response, with contributions from plasmons.6,7 The plasmon response involves the displacement of charge density from its equilibrium position on length scales generally smaller than the excitation wavelength. As a result, local areas of high electromagnetic field increase and plasmonic hot spots are formed for Raman scattering enhancement.In this way, SERS requires a nanoroughness of crevices or interparticle junctions (<10 nm) for the plasmonic hot spots. The choice of metals for SERS is not restricted to only those that solely exhibit plasmon resonance (e.g. silver, alkali metals, gold, copper, aluminum, gallium, etc.),8 but can be extended by alloying or coupling with other materials. For example, the alloying of silver with copper, followed by a thin coating (<1 nm) of graphene can yield nanostructures with a stable SERS efficiency of hot spots for cell labelling and imaging.9 Such nanostructures can enable facile conjugation with hydrophilic/hydrophobic molecules, and can prevent contamination, due to protection from the outer graphene layer. Moreover, graphene coating on a AgCu alloy surface sustains the optical properties in the presence of aggressive substances such as hydrogen peroxide, hydrogen sulfide or nitric acid. In another example, Au–Fe alloy nanoparticles can be used as contrast agents for the SERS imaging of cell toxicity and for in vivo studies of tumour permeability and retention effects.10 The Au–Fe nanoalloys can be easily conjugated with thiolated molecules without cytoxicity and exhibit a long retention time in tumour cells (which is a great advantage over mono-Au or mono-Fe nanostructures).
Plasmonic hot spots with a great variety of geometries have been produced ever since the first prediction of higher values of the electromagnetic field increasing at the sharp edges of nanostructures.11 For example, silver nanoparticles with a triangular, rectangular or oval shape exhibit strong E-field enhancement contours, with enhancement factors reaching up to 103 (see Fig. 1).12 In contrast, the electromagnetic field increase is one order of magnitude less efficient in spherical silver nanoparticles (see Fig. 2). We calculated the electromagnetic field distribution around two types of silver nanoparticles with a spherical geometry, namely (i) a silica-core (30 nm) with a thin silver layer (5 nm), and (ii) a silver nanoparticle coated with a thin silica layer (5 nm). There was a slight difference of electromagnetic field increase at two excitation wavelengths (532 nm and 785 nm) (see Fig. 2A–D), but the values of the electromagnetic field increase did not exceed 102 (see Fig. 2E). However, well-defined gold nanobridged particles (Au–NNP) with a uniform and hollow gap (∼1 nm) in a single nanosphere can produce higher enhancement factors, but over a narrow distribution from 1 × 108 to 5 × 109 (see Fig. 3).13 One advantage is that a quantitative analysis of the number and position of Raman reporters (i.e. Raman active dyes) can be performed by this approach.
 | ||
| Fig. 1 Colorful E-field enhancement contours of silver nanoparticle monomers with different shapes: (a) and (b) a triangular prism polarized along the two different primary symmetry axes; (c) and (d) rod and spheroid polarized along their long axes. The arrows show the maximum of the E-field. Reprinted with permission from ref. E. Hao, G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366. | ||
 | ||
| Fig. 2 (A–D) Electromagnetic field distribution around two types of plasmonic nanoparticles with a spherical geometry: (i) silica-core (30 nm) and a thin silver layer (5 nm) and (ii) silver-core (30 nm) and a thin silica layer (5 nm) at two excitation wavelength (532 nm and 785 nm). The incident electromagnetic plane wave is directed along the z-axis with the polarization along the y-axis, and the x-axis is perpendicular to the zy-plane. (E) Calculated distribution of the SERS enhancement factor (EF) in the wavelength region from 400 nm to 1100 nm. | ||
 | ||
| Fig. 3 (A) Gold nanobridged nanogap particles (Au–NNPs) that are DNA-anchored, and (B) and (C) calculated electromagnetic field increase distributions at the nanobridges within the Au–NNP (indicated by white arrows) and silica-insulated core–shell structure. Reprinted with permission from ref. D.-K. Lim, K.-S. Jeon, J.-H. Hwang, H. Kim, S. Kwon, Y. D. Suh, J.-M. Nam, Nat. Nanotechnol. 2011, 6, 452–460. | ||
SERS entirely depends on the strongest plasmon resonances on the surface, either with a relatively uniform enhancement or with large variations.14 The latter, i.e. plasmonic hot spots, enable single molecule detection. However, strong enhancement is not the only requirement for the SERS detection of individual molecules, and the surface area needs to be considered as well. As SERS is a surface spectroscopy tool, a larger surface area with a periodic array of plasmonic hot spots can increase the detection of a greater potential number of individual molecules. Direct contact of the analyte with plasmonic hot spots in the first surface layers is another SERS requirement. Below, we examine the main questions about single molecule detection in SERS and assess possible approaches to answer these questions.
1.2. Single molecule problem for plasmonic hot spots
Single molecule detection by SERS occurs at plasmonic hot spots with electromagnetic field increases higher than 107–108.15,16 Various plasmonic nanostructures other than spherical18,19 or triangular,17 from a rod-like geometry18 to plasmonic nanostars (∼1010),19 have been designed in order to produce reliable hot spots for single molecule detection. However, single molecule detection by SERS is full of controversy and problems. Fluctuations in the signals, as well as a lack of reproducibility and understanding of the origins of the single molecule phenomenon under SERS conditions highlight the need for intense research to fix these problems. SERS, as a powerful diagnostic tool, can enable chemical imaging at the molecular level in real time. This is especially interesting if one desires to carry out the direct visualization of molecular processes in living cells.Understanding how the local electromagnetic field environment contributes to the SERS studies of single molecules has promoted intense scientific interest. It is now well accepted that single molecules under SERS originate at hot spots in colloidal aggregates from adjacent nanoparticle pairs6 or from highly branched individual nanostructures. Analysis of the regions using maps of the polarization surface charge density shows that some hot spots are formed due to the collective and phase coherent excitation of localized surface plasmon resonances.20 Other hot spots derive from the interference of plasmonic excitations due to scattering from interparticle junctions and surfaces.20 The former can provide the most spatially delocalized regions of high electromagnetic field strength, whereas the latter can generate intense local fields at a certain excitation energy. Some regions of high electromagnetic field strength can be spatially delocalized, due to the phase coherent excitation of local surface plasmon resonance. Other regions may be formed due to the collective and coherent excitation of conduction electrons. Both types of resonance can support a variety of optical processes in nearby plasmonic hot spots. In this way, a greater understanding of the electron dynamics at the plasmon resonance can be helpful for the interaction with a single molecule under SERS conditions.21
For single molecule detection, plasmonic hot spots can be engineered by fulfilling three criteria: (a) the required extent of characterization, (ii) availability and/or (iii) ease of fabrication. Hot spots can be formed in silver nanocube or nanorod dimers or nanoparticle–nanowire couples by nanofabrication techniques or self-assembly methods.22,23 However, the number of plasmonic nanostructures is relatively small, and a single value of the enhancement factor is difficult to assess for each cluster geometry. Even the most successful examples face challenges from uncontrolled aggregation and localization. This leads to a change of SERS intensity and the appearance of new spectral peaks, which mostly are caused by the rearrangement of colloids over time.
In a biological environment, the SERS detection of single molecules requires the sustainability of plasmonic hot spots and functionalization for better selectivity and multiplexing. The former properties can be designed from great variety of nanostructure geometries (e.g. spheres, triangles, rods, holes, wires, rice, eggs, etc.) by developing colloidal self-assembly, lithography and other methods. The latter needs creativity in the interplay of the chemical functional groups to meet specific tasks (e.g. targeted binding, selective localization, multiple sensing, etc.).
1.3. Nanostructuring of plasmonic hot spots for single molecule SERS detection
In metallo-dielectric nanostructures, the plasmonic hot spots are highly sensitive to the geometry, e.g. nanoshells, nanoeggs and nanocups.24 Their optical properties can be predicted by numerical methods of plasmon hybridization solutions (see Fig. 4).25 Although the intrinsic plasmon modes at the hot spots of these geometries can be larger than in concentric nanostructures, the enhancement values can be comparable to the individual dimer junctions.26 Plasmonic hot spots of an oval geometry can offer a larger region of maximum field enhancement, in contrast to a narrowly confined junction.27 | ||
| Fig. 4 (A) An energy-level diagram describing the plasmon hybridization in metal nanoshells resulting from the interaction between the sphere and cavity plasmons. The two nanoshell plasmons are in an antisymmetrically coupled (antibonding) plasmon mode and a symmetrically coupled (bonding) plasmon mode. (B) Diagrams depicting the concentric nanoshell geometry described by the concentric radius of the core (a1), inner shell (b1), spacer layer (a2), and outer shell (b2). Reprinted with permission from ref. E. Prodan, C. Radloff, N. J. Halas, P. Nordlander, Science, 2003, 302, 419–422. | ||
Several approaches have recently been developed to advance the strength and sustainability of plasmonic hot spots. One strategy is to use alumina28 or a silica coating layer over plasmonic nanostructures of different geometries. This silica coating has been developed into a new successful diagnostic tool ‘SHINERS’ (shell-isolated nanoparticle-enhanced Raman spectroscopy).29 The ultrathin silica coating can conform to different contours of substrates, thereby effectively contributing to the separation from the direct contact with a probed material.30 Moreover, SERS signals can be measured at hot spots that are spread over non-metallic surfaces (e.g. semi-conducting Si or organic substances). For neutral pH values (between 5 and 8), alumina is mostly supposed not to dissolve. However, at lower pH values it releases Al3+ cations and at higher pH values Al(OH)4− anions.31 Silica provides silicates (H4SiO4) in solution for the whole pH range, until an increase in pH > 9, when it then provides H3SiO4− or H2SiO42−.
Another strategy introduces the laser illumination of plasmonic nanostructures for the dynamic localization of hot spots,32 or self-assembly33 with capillarity constructed reversibility.34 A number of possible ways have been demonstrated to predict an analyte positioning at hot spots within uniform plasmonic nanocrystals35 or nanorods.36
Another successful example demonstrates the well-ordered plasmonic hot spots in the nanostructure of periodic arrays of Ag nanorods on prepatterned polycarbonate sheets. (see Fig. 5A).37 This method allows one to define nucleation sites for the subsequent guided growth of plasmonic hot spots at the junctions between adjacent nanorods (see Fig. 5B). Moreover, this approach offers a proof-of-principle fabrication method for highly ordered hot spots with elongated geometry by using the polymeric template in order to control the nucleation process.
 | ||
Fig. 5 (A) SEM images of Ag nanorod arrays on polymer films prepared under (a) 0° deposition, (b) 85° deposition (top view), (c) 85° deposition (cross-section), (d) 85°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85° deposition, (e) 0°:85° deposition, and (f) 0°:85°:−85° deposition. The scale bar in each image represents 1 μm. (B) Electromagnetic field increase distribution of (a) 85°, (b) 85°:−85°, (c) 0°:85°, and (d) 0°:85°:−85° arrays. All the internal fields are set to zero for clarity. Reprinted with permission from ref. M. Keating, S. Song, G. Wei, D. Graham, Y. Chen, F. Placido, J. Phys. Chem. C, 2014, 118, 4878–4884. :85° deposition, (e) 0°:85° deposition, and (f) 0°:85°:−85° deposition. The scale bar in each image represents 1 μm. (B) Electromagnetic field increase distribution of (a) 85°, (b) 85°:−85°, (c) 0°:85°, and (d) 0°:85°:−85° arrays. All the internal fields are set to zero for clarity. Reprinted with permission from ref. M. Keating, S. Song, G. Wei, D. Graham, Y. Chen, F. Placido, J. Phys. Chem. C, 2014, 118, 4878–4884. | ||
Hot spots can be immobilized in plasmonic nanostructures on the solid substrates (e.g. glass) due to functionalization by silanes, amines, thiols, carboxylates, epoxides or aldehydes. The conjugation reaction can be performed directly or through spacers by using bifunctional cross-linkers. Biotags can be modified by silanes for extended chemical functionality, by a variety of cross-linkers and conjugations schemes38 or can be prepared with –SH, –COOH, –NH2 functional groups on the surface for controlled positioning.
Different chain lengths of alkanethiols, which form SAMs (self-assembled monolayers), can be used to control the distance between an attached analyte and a surface with plasmonic hot spots, thereby enabling ‘on’ or ‘off’ SERS active states.39 As another advantage, SAM can be used on colloidal Ag surfaces in order to eliminate the Ag2O layer (2 nm), due to the displacement upon the covalent attachment of molecules onto the surface.40 Moreover, SAM can be advantageous for the consistent localization of analytes at plasmonic hot spots, excluding when there is contamination by air.41,42 The quantitative evaluation of SERS enhancement can be carried out by SAM on glass with a predicted packing density of highly ordered hexagonally closed-packed gold nanoparticles and incorporated Raman reporter molecules (e.g. p-mercaptoaniline, pMA).43 In such a configuration, plasmonic hot spots can be preserved, yielding sufficient enhancement factors for single molecule detection.
Colloidal self-assembly can be used as another efficient strategy in order to sustain the structure of hot spots with dimensions as low as one nanometer.44 The self-assembly methods range from the electrostatic interactions between colloids to a more complex functionalization by polymers (e.g. DNA hybridization). The latter offers the reversibility of plasmonic hot spots for specific sequence recognition (e.g. adenosine). Colloids larger than 20 nm can provide sufficient surface plasmon resonance to support single molecule detection.45 One such successful example introduces hot spots in ‘3D Au nanoparticles’ consisting of a silica shell-isolated Au nanoparticle core with catalytically active small Au satellites on top of the silica shell (1.5 nm). However, the choice of detected single molecules can be restricted to those bearing a sulfur-specific binding group. Alternatively, various types of single molecules can be detected by using gold–silver nanodumbbells, which provide a good platform for an extended bioconjugation with biomolecules (e.g. proteins) and a multiplexing capability.40 The latter is especially useful for in vitro and in vivo bio-labelling, with improved quantification.
1.4. Polymeric approach of plasmonic hot spots for single molecule SERS detection
Embedding plasmonic hot spots into a thin polymeric film can protect its structure against a low pH solution or against organic solvents (an advantage over silica coating).46 Moreover, SERS spectra can be collected with a high degree of reproducibility, good signal-to-noise ratio and qualitative class discrimination.47 Stimuli responsive polymers can facilitate the plasmonic hot spot assembly through the closer contact to analytes in a reversible48 and multifunctional manner.49 The single molecule detection of an analyte at different resonant states50–52 can be advanced through the attraction by charged ligands (e.g. CTAB or citrate) to plasmonic hot spots by varying its dimensions53 or the ionic strength of the solution. Difficulties in the quantification and prediction of an analyte assessment at the plasmonic hot spots can be overcome by linking, polymer coating or molecular permeation,54 thus enabling multiplexed labelling.Plasmonic hot spots formed in the matrix of polyvinyl pyrrolidone with embedded silver nanoparticles can maintain their SERS enhancement over several cycles during water treatment.55 Thin polymeric films of plasmonic hot spots can be employed as a long-range ordering porous structure with a controlled nanoscale roughness.56 The SERS detection of amino acids L-phenylalanine, L-glutamin and L-histidine can be advanced by plasmonic cellulose as a natural sponge in water or ethanol.57 Flexible membranes with plasmonic hot spots inside a natural rubber58 and cellulose-based dipsticks59 can extend the trace for different chemicals in real time.
Single molecules can be detected at plasmonic hot spots impregnated within flexible films (e.g. filter paper, polymer fibers, elastomers, plastics or carbon-based substances).60 However, the uniform distribution of hot spots on or within the substrate and their stability need to be further developed for real applications. One of the successful examples shows how flexible silica sheets with discrete and monodisperse hot spots in the junctions of gold nanorods can maintain plasmonic geometry and integrity for single molecule detection.61 As a great advantage for real applications, such a single molecule SERS platform can facilitate the detection of a known marker for illicit drugs or can be employed as nanoscale barcodes.
We recently demonstrated how plasmonic hot spots can be switched ‘on’ or ‘off’ selectively by controlling the polymeric chain length of polyethylene glycol (see Fig. 6).62 Preformed silver nanoparticles can assemble into a necklace structure within the matrix of polyethylene glycol bearing a longer chain length (PEG, 8000 Da) yielding hot spots at the interparticle junctions (see Fig. 6A and C). In contrast, a polymeric matrix with a shorter PEG chain length (1000 Da) is ineffective against particle attraction, and hot spots cannot be formed (see Fig. 6B and D). The SERS enhancement factor at such hot spots is several orders of magnitude higher than from random gold or silver aggregates, and is sufficient for single molecule detection.
 | ||
| Fig. 6 Schematic illustration of switch on (A) and off (B) in the “hot spot” formation of silver nanoparticles in the matrix of polyethylene glycol (PEG) with longer (8000 Da) and shorter chain lengths (1000 Da). (C) and (D) TEM images of Ag nanoparticles in the PEG matrix of 8000 Da and 1000 Da after heating at 70 °C. | ||
The design of plasmonic hot spots for single molecule SERS detection is impossible without an understanding of the electromagnetic coupling at the edges between two adjacent nanostructures. In the next section, we examine the factors responsible for the electromagnetic coupling mechanisms and enhancement at the hot spots of plasmonic nanostructures.
2. Electromagnetic coupling at plasmonic hot spots
The electromagnetic coupling between plasmonic nanoparticles of different shapes is highly complex, due to the asymmetric geometries and the configuration of the substrate with different dielectric compositions.63 For example, the strength of plasmonic hot spots can be controlled by the interaction of silver nanospheres with the underlying substrate through the shape, interparticle junction and refractive index. As the distance d between the plasmonic nanostructures decreases, the enhancement of the local electric field at the plasmonic hot spots grows considerably faster than 1/d for the hybridized plasmon modes.64–66 An oscillation of the charge densities of opposite signs occurs across the interparticle junction, followed by a frequency shift of those particular hybridized modes to the red with decreasing d. The evolution of the plasmon modes as a function of the interparticle separation can be examined by the electron energy loss spectroscopy.67 The enhancement factors for single molecule detection can be found at the hotspots of plasmonic nanostructures excited at the frequency of the mode with a linear symmetric combination of the dipolar resonances.68 Single molecule detection can be collected by SERS at such hot spots,69 either in an aggregated state70 or by the self-assembly of plasmonic dimers linked by analytes.71 To note, the hybridized nature of the plasmon modes becomes more obvious when examined at extended electrodes: the spectral positions and relative intensities in the SERS spectrum can be controlled by the hot spot geometry through the polarizability tensor of the analyte/hot spot pair.It is recognized that the electromagnetic field coupling between two spheres can yield enhancement factors of ∼105 in the framework of a quasistatic approximation. These enhancement factors have been used in the interpretation of single molecules SERS detection during the oxygen transport of protein haemoglobin (Hb).72 In this study, the minimal aggregation number for effective single molecule SERS detection is the dimer, which consists of a pair of Ag NPs bridged by a Hb molecule. Evidence for the single molecule detection limit has been provided by the striking spectral fluctuations and clear differences in the SERS peak positions and relative intensities. Higher SERS intensity can arise at hot spots of a dimer with the axis oriented parallel to the incident electromagnetic field. The SERS enhancement decays from ∼1010 to 106 at the distance from a hot spot, in agreement with recent calculations.73 The origin of the temporal fluctuations in the single molecule SERS is unknown. There is a debate ongoing about whether the fluctuation effects are the signs of spontaneous variations in the protein conformation and adsorption state or due to photochemical effects near Ag surfaces with possible photonic forces induced by the electric field gradients. However, regardless, it is clear that these effects can lead to conformational changes and possible disintegration of the protein, which should be taken into account in future studies of single molecule SERS.
The electromagnetic coupling can be also introduced by the filling of isolated nanoholes in a metal film with preformed plasmonic nanoparticles, i.e. between a ‘positive’ (nanoparticle) and a ‘negative’ (nanohole) metal nanoparticle.74 Selective chemical modification in the holes can form a stable ‘hole–particle’ pair, strongly depending on the polarization of the incident excitation wave.
2.1. Extinction of plasmonic hot spots in single molecule SERS
The plasmon resonance spectra of plasmonic aggregates are usually complicated because of the overlap between the dipolar and multipolar plasmon modes.75 The dipolar plasmon should satisfy the following criteria: sufficient polarization dependence of the plasmon resonance maximum following a cosine squared law, and also the SERS maxima and plasmon resonance maxima should have the same polarization dependence relative to each other. If so, the coupling of the dipolar plasmon resonance with SERS can be identified.76 To recall, plasmon resonance changes its maximum wavelength according to the surrounding media, and the SERS spectra can also be changed by increasing the refractive index of the same medium. By replacing water with oil (i.e. with a refractive index closer to that of glass), the plasmon resonance of the plasmonic aggregates can be red shifted, followed by shifts of the Raman peaks in the SERS spectra.However, the SERS excitation profile (i.e. the SERS intensity plotted as a function of the wavelength of the excitation laser) does not, in general, track the extinction spectrum of the SERS substrate.77 This is a direct consequence of the fact that the SERS spectrum can be dominated by the resonances at hot spots, whereas the extinction spectrum of the SERS substrate can include contributions from the entire plasmonic nanostructure. For example, the SERS signal of molecules adsorbed on a closely packed plasmonic dimer can be dominated by the near fields in the interstice, while the extinction spectrum of the dimer involves absorption and scattering by the whole dimer. Several SERS excitation profiles can arise at multiple hot spots, each of them relevant to a residing specific molecule with a SERS spectrum. The SPR band can be shifted by the composition and geometry of the plasmonic nanostructure (dimensions and periodicity of hot spots). For example, a spherical silica core and a thin Au shell with various dimensions of a core–shell structure can move the surface plasmon band from the visible into the near-IR region.78
2.2. Plasmon hybridization theory of plasmonic hot spots
In the framework of the plasmonic theory, the foundation for the rational design of plasmonic nanostructures has been predicted by Fano resonance from interactions between plasmon modes. Several pioneering theoretical contributions describe how plasmons interact on adjacent nanostructures and hybridize to form collective modes (see Fig. 4).79 Original studies show that the plasmons of complex metallic nanostructures can be expressed as linear combinations of plasmons associated with their elementary parts. This is analogous to atomic orbital interaction and hybridization in molecular orbital theory. The model considers the electron gas in a nanoparticle as an incompressible fluid, and the plasmons of a complex nanoparticle can be viewed as a system of interacting plasmons supported on its elementary surfaces.80 This approach can be applied to plasmonic systems with a spherical geometry, such as single and concentric nanoshells, with the calculated plasmon energies being in agreement with the Mie scattering and a Drude dielectric function for the metallic shells. Plasmons can be mapped into a two level system consisting of a bare cavity and a solid sphere plasmon. Their interactions depend on the thickness of the shell, yielding a tunable symmetric and antisymmetric plasmon.The plasmon hybridization approach can be generalized to dimers and also to more complex plasmonic systems. Its extension can include the damping of the plasmons and the finite compressibility of the electron gas. In plasmonic dimers, this approach provides a detailed understanding of the energies and the extinction cross-sections of the plasmons in a nanosphere dimer.81 The dimer plasmons can be viewed as bonding–antibonding linear combinations of individual nanosphere plasmons. As the dimer separation is decreased, individual sphere plasmons with different angular momentum hybridize, resulting in dimer plasmons with finite dipole moments. Hot spots for single molecule detection can be formed by the admixture of dark multipolar plasmons. Plasmon modes in such complex plasmonic structures can be theoretically analyzed by group theory.82 The experimental verification in the self-assembled plasmonic nanoparticle clusters,83 nanorings84 and nanorods85 with localized surface plasmon resonance can exceed those for ‘standard’ plasmonic nanostructures. The coupling between plasmonic and excitonic systems resulting in hybrid mixed plasmonic and excitonic states, i.e. plexcitons, can be described by a quantum mechanical method based on the Zubarev's Green functions.86
Coupling of the local plasmons can also increase the photonic density of states at the emission frequency by a factor of g(ω′),2 where ω′ is the frequency of scattering. Two nanostructures can be coupled via the optical near field, followed by the formation of plasmon resonances of the individual nanostructures as a composite system with its own spectrum of plasmon resonances. This complex spectrum can be considered as a linear combination of the subsystem modes (see Fig. 4).79,81 Each nanostructure with a single dominant mode can be introduced as a combination of a system with ‘bonding’ and ‘antibonding’ hybrid modes. However, not every plasmon mode (of an individual or combined system) can be strongly coupled to a far-field radiation. Modes without a net dipole moment cannot be involved in the coupling, but can have profound consequences if hybridized with bright modes.87
Once we understand how the coupling of plasmons can occur at the interparticle junctions between two adjacent nanostructures, we examine the possible enhancement mechanisms that can be involved at SERS conditions of hot spots.
3. Enhancement mechanisms for single molecule detection by SERS
In general, three mechanisms can be involved in SERS increases: (i) electromagnetic, i.e. local electromagnetic field increase at resonance with the surface plasmons; (ii) chemical, i.e. charge transfer at the metal/molecule interface and (iii) molecular resonance, i.e. the resonance of molecules at excited states due to the interaction with the surface plasmons (SERRS).3.1. Electromagnetic mechanism
In conventional SERS, the enhancement factor is about 106, meaning that |E|2 would have to be 103, if assuming the full EF has an electromagnetic origin. In real applications, the typical average |E|2 values are of a few hundred, suggesting that most but not the entire enhancement is electromagnetic. In single molecule SERS, the properties which determine the peak |E|2 values are less known. Peak |E|2 values are relatively modest at the isolated spheres (∼102), being higher at the spheroids and nanoprisms (>103), as well as large at nanostars or other highly branched geometries (>107–108), due to the red-shifted plasmon resonance and sharp points with lightening rod effects.It is believed that the main contribution to the overall enhancement in SERS is electromagnetic in origin, due to the nature of the dielectric–metal interface. The enhancement factors can extend from 104 and higher, depending on the heterogeneity and dimensions of the plasmonic hot spots.88,89 Typical SERS enhancement values for single molecule detection lie in the order of 107 to 1010 at the hot spots of silver or gold nanostructures. One such classical examples is shown in Fig. 7.90 In the framework of the |E|4-approximation, the spatial distribution of the enhancement factors at the hot spots between two spherical nanoparticles can be obtained with the exact generalized Mie theory.75 The SERS enhancement decays drastically with the increased distance from the peak value at the shortest junction (see Fig. 7A). This value can vary by an order of magnitude over distances comparable to a few molecular dimensions (∼2–4 nm). However, the peak values of the SERS enhancement can be three orders of magnitude higher at hot spots for a gold tip above a planar gold surface (see Fig. 7B) and can be controlled by the excitation wavelength (see Fig. 7C). If the resonance in the second type (with the tip) is shifted into the red compared to the plasmonic colloids, a SERS enhancement of ∼1011 can be developed. The enhancement decreases with the distance from the hot spot up to a certain value, where it then starts increasing again (see Fig. 7D). This type of behaviour can be found in the plasmonic dimers using a log-scale.
 | ||
| Fig. 7 (A) and (B) Electromagnetic field increase with the calculated SERS enhancement factor (EF) distribution in the nanogap (2 nm) between two gold nanospheres (30 nm) with polarization along the vertical axis of the dimer and gold tip on the top of a gold nanosphere surface at two excitation wavelengths, 559 nm and 633 nm. (C) and (D) SERS EFs distribution versus excitation wavelength in the region from 400 nm to 700 nm and the distance from the hot spot (in nm). Reprinted with permission from ref. Etchegoin, E. C. Le Ru, Phys. Chem. Chem. Phys., 2008, 10, 6079–6089. | ||
Fundamentally, it is important to design a plasmonic substrate for a particular plasmonic resonance (excitation wavelength) in order to benefit from high SERS EFs and to carry out single molecule studies. As in most Raman experiments or instruments, the excitation laser is fixed (i.e. 532 nm, 633 nm or 785 nm) and so the properties of SERS active substrates have to be optimized for a particular excitation. For this reason, we calculated the electromagnetic field distribution around plasmonic dimers composed of two types of silver nanoparticles with a spherical geometry (see Fig. 8). These were preformed silver nanoparticles (30 nm) with a thin silica layer (5 nm) (see Fig. 8A and B) and silica nanoparticles (30 nm) with a thin silver layer (5 nm) (see Fig. 8C–E). In the first type of silver dimer, the highest SERS EFs could reach a value of ∼105 at 532 nm excitation wavelength (see Fig. 8A), and one order of magnitude less at 785 nm of the incident electromagnetic wave (see Fig. 8B). However, the second plasmonic configuration was more preferable, as the SERS EFs could reach about 1011 at 532 nm (see Fig. 8C) and 109 at 785 nm excitation wavelengths (see Fig. 8D). We also calculated the decay profiles of the peak values of the SERS enhancement, which decreased faster at 532 nm due to the longer separation between the SiO2–Ag NPs (see Fig. 8E).
 | ||
| Fig. 8 Calculated electromagnetic field distribution in the dimers consisting of two types of nanoparticles: (A)–(D) silver nanoparticles (30 nm) with a thin silica layer (5 nm) and silica nanoparticles (30 nm) with a thin silver layer (5 nm) at two excitation wavelength (532 nm and 785 nm). (E) Calculated SERS EFs versus distance between silica–silver nanoparticles at two excitation wavelengths. The abbreviation Sm11 indicates single molecule model between two identical silica spheres. | ||
At present, the SERS theory is incomplete, as it is largely based more on electrostatics than electrodynamics, with the SERS enhancement factors proportional to |E|4 in approximation. A more complete theory requires quantum mechanics and a dynamic approach.21 A quantum mechanical calculation (quantum plasmonics) showed that the field enhancement in a coupled nanoparticle dimer is strongly affected by the non-linear effects at the junctions, thereby reducing the field enhancement, as computed by linear theories.11
One of the first steps towards the SERS theory can be introduced by a detailed time-dependent density functional theory (TDDFT) investigation of the absorption and Raman spectra of a pyrazine molecule located at complex or at the junction between two silver clusters.91,92 From the analysis of the Raman scattering cross-sections, the overall SERS enhancement includes the following contributions: static chemical (∼10), charge-transfer (∼103) and electromagnetic (∼105).91,93
3.2. Chemical enhancement contribution
In SERS, the existence of the chemical enhancement is not fully resolved from the electromagnetic part for the following reasons. First, many different chemical effects can affect the SERS intensity (e.g. molecule adsorption or orientation), which are not strictly considered as SERS enhancement factors. Second, the chemical enhancement is difficult to measure experimentally. Third, the chemical enhancement depends on any modification of the Raman polarizability tensor upon adsorption of the molecule onto the metal surface (e.g. a change in the nature and identity of the adsorbate). One of the significant arguments for a chemical enhancement contribution can be derived from the existence of the spill out of the electrons, instead of a step function, for the electron density in the classical calculation.85,94 The clear signatures of a chemical enhancement can be explained by a recently proposed scheme based on the chemical interface damping the local surface plasmon resonances.95Resonant intramolecular and metal-molecule charge transfer (CT) mechanisms can constitute the chemical enhancement.21,96 In the CT mechanism, coupling of an analyte at the hot spots occurs through the transient charge transfer, yielding the formation of a transient molecular anion (cation) or a neutral biradical.21 This is due to the temporary residence of a hot electron (hole) followed by a return to the metal with a ‘memory’ of the molecule's vibrations.21 In the framework of the density functional theory (DFT), within a generalized gradient approximation, a reactive ligand might prefer to bind to uncoordinated clusters rather than to a single crystal face. Such chemical effects can be extended to all transition metals beyond silver or gold. However, complex formation alone cannot account for the six-order of magnitude enhancement factor unless the formation of the metal ligand bond is due to a CT mechanism with a greater molecular cross-section. At present, we need to understand the quantitative estimations of the formation of a metal–analyte chemical bond as a first layer effect, as well as the role of the hot electrons (holes) in a plasmonic decay, which might be accelerated by the chemisorption process.
3.3. Molecular resonance in SERRS
In another mechanism, the SERS enhancement factor can be more than six orders of magnitude higher if the excitation laser frequency matches the energy of an electronic transition of an analyte (i.e. resonance SERS, SERRS).96 SERRS is selective to particular excitation modes of analyte with a strong and long progression of overtones with the depolarization ratios of anomalous or inversed polarization. A main drawback of this is that the number of the chromophores can be limited by the choice of excitation frequency. Traces of fluorescent impurities can also prevent the acquisition of a SERRS spectrum. SERRS experiments can allow the measurement of the overall resonant Raman (RR) cross-section of dye molecules (e.g. rhodamine 6G) in order to understand the SERRS mechanism.97 The RR value of rhodamine 6G (at 514 nm excitation) can be of the order of 10−22 cm2 or two orders of magnitude less, i.e. 10−24 cm2, if directly measured by a method based on polarization difference.96 This is important for understanding SM-SERS for the characterization of biological systems by SERS, since high enhancement factors (>108) can be achieved with molecules bearing a relatively low scattering cross-section. This is in agreement with the statement that a large Raman cross-section does not mean a large SERS enhancement, according to the definition of the SERS intensity by using the following expression Is= ΓsαsNsG|E0|2, where Γs is the instrumental factor, αs is the molecular cross-section, Ns is the number of molecules contributing to the scattered signal, G is the electromagnetic contribution to the enhancement (plasmonic), and E0 is the amplitude of the incident electric field.21The next question that arises is how to predict and calculate what SERS enhancement is sufficient for single molecule detection. How can the scattered field at hot spots be computed and visualized in nanostructures of different complexity? Is the prediction of a SERS spectrum of individual molecules possible for comparison with experimentally collected data? Below, we discuss the existing theoretical methods that can be useful to answer these questions.
4. Theoretical methods for single molecule SERS enhancement
In most studies plasmonic hot spots can be adsorbed onto the underlying dielectric or metallic substrate, whose optical properties can be modelled by extending standard Mie theory.98 For the SERS enhancement factors, the biquadratic enhancement is often further approximated as simply the quadratic field enhancement at the excitation laser frequency, |E(ω)|4. However, it is important to keep in mind that the plasmon must have sufficient bandwidth to cover both the excitation and the Raman-shifted emission frequencies. In other words, a narrow plasmon resonance can limit the enhancement of the Raman shifted photons whose frequencies do not overlap the SPR spectral bandwidth.4.1. Methods for near-field nano-optics
There are numerous theoretical techniques for the evaluation of the optical properties of plasmonic hot spots.99,100 In the framework of the theoretical schemes and computational techniques dedicated to near-field nano-optics studies, the coupled dipole approximation (CDA, but also called the dipole–dipole approximation, DDA) can provide numerical values for the local electric field distribution in the plasmonic nanostructures. As an advantage, the complete electromagnetic field, either scattered or localized by the plasmonic nanostructures, can be computed. After an arbitrary discretization of the particles into elementary sources, the actual electromagnetic field outside can be represented as the coherent sum of the fields emitted by each elementary source.101,102Another method is based on the direct space integral equation (DSIEM), which belongs to the family of volume integration methods for the ab initio investigation of optical near-field distributions. One advantage of the DSIEM is that only a specification of the frequency dependent dielectric constant and the geometry parameters of the designed nanostructures are required. Alternatively, a well-defined surface can be described by a perturbative diffraction theory, providing an interesting framework for the weak fluctuation amplitudes of the surface. This can be represented as the first step towards nano-optics experiments and does not need extensive computational work.
There is also an important family of methodologies based on the concept of multipolar expansion for spatially localized nanoparticles of subwavelength dimensions in a complex environment.103 In this case, the optical response of the plasmonic nanoparticle is represented by a set of multipolar polarizabilities generally centered around a selected point or located inside the particle.
4.2. Methods for the electromagnetic coupling
In a description of the electromagnetic coupling in the framework of the electrostatic eigenmode, the asymptotic plasmon chain depends on the interparticle coupling strength and the dielectric constant of the surrounding (background) medium. However, a quantitative description of the plasmon resonances within the plasmonic chains of hot spots cannot be reproduced by either the boundary element (BEM or MoM, method of moments) or EEM methods. To remind us, BEM is a numerical computational method of solving linear partial differential equations that have been formulated as integral equations (i.e. in boundary integral form). However, BEM is significantly less efficient than volume-discretization methods (finite element method FEM, finite difference method FDM or finite volume method FVM). Boundary element formulations typically give rise to fully populated matrices. This means that the storage requirements and computational time will tend to grow according to the square of the problem size. By contrast, finite element matrices are typically banded (elements are only locally connected) and the storage requirements for the system matrices typically grow only linearly with the problem size. BEM is applicable to problems for which Green's functions can be calculated, typically in the linear homogeneous media, placing considerable restrictions on the range and generality of problems suitable for boundary elements.Volume-based methods are introduced by the following methods: (i) Finite Difference Time Domain (FDTD) for scattering computations of non-spherical and inhomogeneous particles;104,105 (ii) Transmission Line Matrix (TLM)111 for modelling complex and inhomogeneous scatterers with magnetic, dielectric and absorption parameters; (iii) Volume Integral Equation (VIE),106 which is based on two approaches: (a) the method of moments (MoM)107 for actual field formulation and (b) a coupled dipole method (CDM)108 with the concept of excited fields or discrete dipole approximation (DDA),109 which treats a shaped particle as a three-dimensional assembly of dipoles with a complex polarizability on a cubic grid; and finally, (iv) there is a set of the Finite Element Methods (FEM), with electromagnetic formulation in the frequency domain for handling complex scattering shapes.110 To note, each theory has its own range of applicability, which depends on the particle shape, its composition and refractive index, and its size relative to the wavelength of the incident wave. For calculation, one should keep in mind the respective method in terms of computer resources and memory, execution time and parameters for the accuracy of the final computational results. For example, a surface-based method will need less computer capacity than a volume-based method for the same scattering problem.
Over the last thirty years, the experimental work on small metallic aggregates in transparent dielectric materials has included stimulated investigations of the scattered light from the layered micro- and nanometer metallic particles deposited on a surface and the modification induced by adsorbed molecules.111 The understanding of the near-field optical signals along regular arrays or metal particle chains at the dielectric surface has been substantially improved by applying new concepts of optical near-field coupling between plasmonic nanoparticles.
4.3. Methods for a qualitative evaluation of the SERS
For a qualitative analysis it is often challenging to obtain a normal Raman spectrum of the analyte or to quantify the number of molecules adsorbed on a surface, yet these numbers are necessary for an accurate evaluation of the SERS enhancement. Many times, an overwhelming fluorescence signal or a weak Raman cross-section can prohibit obtaining a normal Raman spectrum of an analyte. For example, rhodamine 6G exhibits an extremely high fluorescence quantum yield (at 532 nm), and hence any possibility to observe a normal resonance Raman spectrum vanishes due to the high fluorescence background. In SERS, the fluorescence is quenched by electron transfer in a close proximity to the plasmonic surface. The enhancement factors for the fluorescent probe molecules can be estimated by calculation of the resonance Raman cross-section with the stimulated Raman methods97 or by extrapolating the off-resonance Raman cross-section.112 The scattering cross-section of weak Raman scatterers can be estimated by theoretical calculations, but the values may appear to be in a quantitative disagreement with the experimental results.Calculation of the SERS enhancement factor from the number of molecules contributing to the SERS signal is another challenge due to the uncertainties in the surface roughness, surface area, and/or packing densities. However, in this way, despite the lower limit for the SERS enhancement, the maximum possible number of contributing molecules can be estimated. For a known order of probe molecules on a surface, the number of adsorbed molecules can be calculated from the highest possible literature values given for the packing density. However, one should keep in mind that in SERS only a very small number of molecules contribute to the largest part of the signal.3,113 This means that at plasmonic hot spots, a single molecule can produce the same SERS intensity as hundreds or thousands of randomly adsorbed molecules (∼102–103 smaller than the maximum value). Therefore, the number of molecules contributing to the SERS signal cannot be determined from its absolute intensity, due to the extreme statistical distributions.
Alternatively, the single molecule SERS enhancement can be derived by using a bi-analyte procedure.114 The main idea here is to measure the SERS signal from a mixture of two molecular Raman-active species with distinguishable SERS spectra (although preferably with a comparable cross-section) for a given laser wavelength. If the concentration is such that there are many molecules at hot spots, the SERS signal will be a mixture of two analytes. The observation of a SERS signal of purely one type of analyte is clear evidence that it comes from a very small number of molecules. By carrying out several such experiments at decreasing concentrations, eventually only pure (non-mixed) signals will arise, i.e. single molecule SERS signals. The non-mixed signals may be attributed to either single molecules or a few molecules of the same type. The advantage of this method is that the ultra-low analyte concentrations, which are difficult to estimate correctly and reliably, will be replaced by the largest concentration (by a factor of 100), where most SERS signals originate from single molecules. In addition to its simplicity, this method can provide a direct proof of single molecule SERS, and with better and more reliable statistics of SM-SERS events.
4.4. Methods for the spectral and spatial imaging of single molecule SERS
Another method proposes a direct imaging of plasmonic hot spots evolution and development during molecular interaction. One such experiment using this method demonstrated how hot spot imaging could be performed using a photoactive polymer (e.g. commercial SU-8) with two-photon absorption and polymerization at 800 nm excitation. Here, the enhanced field intensities at the tips of the triangular nanoantennas promote this non-linear polymerization with a higher spatial confinement.115 The growth of this polymer can be observed at the tips of the nanostructure and not in the junction. Another photoresponsive polymer can be used to topographically image the hot spot distribution on lithographically-patterned silver nanorods, as well as on triangular nanoantenna structures.116,117 Silver nanorods can be coated with a photoresponsive polymer system, consisting of the photoswitchable azo-dye molecule dispersed red 1 (DR1) grafted as a side chain to PMMA, i.e. poly[(methyl methacrylate)-co-(disperse red 1 methacrylate)]. The thickness of this thin film nanostructure can vary from 50 nm to 100 nm. The mass transport can be monitored, due to illumination-induced trans-cis photoisomerization of the azobenzene moiety yielding a topographic change in the polymer in response to the field intensity. Each molecule can act as a nanoprobe of the optical near-field of the nanostructures at the sub-diffraction limit scale of the polymer film.Recent progress has been made in the development of the spectral and spatial super-resolution imaging of plasmonic hot spots.118 Although this method does not describe the SERS enhancement factors, it revealed that the location of the SERS emission depends both on the plasmonic properties of the SERS substrate, as well as on the location of the emitting analyte on the surface. The authors hypothesize the existence of two fundamentally different concepts, namely the electromagnetic theory, which is associated with the plasmon resonance, and SERS, which is defined by the molecular emitter, similar to the plasmon modes of the nanostructure. They suggest that the SERS hot spot (which we call a ‘plasmonic hot spot’ throughout this paper) is a convolution between the plasmon-enhanced electromagnetic fields and the position of the coupled molecular emitter on the surface. Therefore, the centroid position of the SERS emission will change as the position of the molecule changes, thus impacting how its emission is re-radiated into the far field by the plasmonic nanostructure.119
To move on, let us examine the empirical strategies that exist for capturing the characteristic vibrational signatures of single molecules. And next, let us take a look at the present real application of SERS as a diagnostic tool to detect small molecules (e.g. ions, glucose, lipids) from inside living cells (e.g. plants, yeast, HeLa, fibroblast, etc.) in vivo or in vitro.
5. Strategies for single molecule SERS detection
At present, the SERS detection of biological molecules is carried out by two strategies: (i) direct (label free) detection and (ii) indirect (Raman dye or Raman reporter molecules as labels) detection. Although the first method allows the reliable acquisition of intrinsic SERS spectra of target biomolecules, its sensitivity is lower than the second method. However, the reproducibility of the second method depends on the non-specific binding of the ligands and hence positive results can be falsified.In both approaches, the SERS analysis of biologically important molecules is based on the general motifs of the vibrational pattern.120 One commonly used example is the characteristic aromatic amino acid bands present at ∼950 cm−1 (C–COO− stretch) and ∼1400 cm−1 for the COO− symmetric stretch. Another commonly used spectral feature is the broad amide (CO–NH) I and III bands (1600–1700 cm−1) and 1200–1350 cm−1 shift, used in intrinsic peptide and protein analysis to describe both primary and secondary structure characteristics.121 Local pH variations at the molecular level can be monitored by using the indirect method.122,123 For example, plasmonic hot spots can be formed in hollow Au nanoshells, and mercaptobenzoic acid can be used as a Raman reporter label to correlate the spectral changes of the Raman bands (e.g. 1430 cm−1 and 1590 cm−1) with the local pH variations of the solution.95,123
A series of alternative strategies has been recently developed to advance single molecule detection by SERS. One of these strategies introduces a method based on the deposition of an active agent (e.g. a capture ligand or antibody) on the entire surface of plasmonic nanostructure, in order to facilitate the capturing of a desired analyte. Another approach employs the intercalation of a plasmonic nanostructure into an alkanethiol self-assembled monolayer (SAM) or lipid bilayer. The SERS EF can then be calculated using a ruler, as a single-stranded DNA with adenine bases bears a terminating fluorescein group (Raman reporter molecule). Furthermore, the positioning of the fluorescein terminus can be facilitated by varying the number of bases. The molecular packing density and nanoparticle coverage can be estimated by the ratio of SERS signals from the fluorescein functional group and the adenine, and can also be proven by the Mie theory.
5.1. Detection of ions
The rapid and ultrasensitive detection or recognition of inorganic atomic ions is a challenge for many conventional techniques, and require extensive sample preparation, which may even be destructive. In contrast, SERS can be used as a non-destructive, rapid and ultrasensitive technique for the indirect detection or recognition of atomic species. One of the strategies is to use a capping agent (e.g. antibodies or nucleic acid aptamers) with a relatively high SERS cross-section at plasmonic hot spots.124 Among the halogens, only chloride, bromide and iodide can be monitored by SERS. A chloride is of special interest due to its presence in the cytosol of cells.125The detection of iodide (I−) or thiocyanate (SCN−) is of particular interest in the human body and health science. For example, a deficiency or excess of iodide can be a cause of many diseases, such as goiter, hypothyroidism, and hyperthyroidism (Grave's disease).126 In human body fluids (e.g. serum, saliva and urine), a small amount of SCN− can be found, which is produced by the digestion of some vegetables or thiocyanate-containing foods (e.g. milk and cheese), but a higher level of SCN− can arise upon the inhalation of a tobacco smoke. The presence of SCN− in body fluids (saliva) can thus help to discriminate non-smokers and smokers.127,128 The selective SERS detection of these ions (concentration ∼0.01 × 10−6 M) can be performed indirectly at hot spots of a starch-reduced gold nanoparticles. This method can be used in a human serum system, due to its simplicity, biocompatibility (starch) and the possibility of for large-scale production.
Different inorganic oxoanions in natural water, or other species such as cyanide or sulfocyanide, can also be successfully detected by SERS.129 SERS signals can be enhanced in an electrostatic interaction by the surface charge or can be induced by the chemical affinity of the cyanide group. As an advantage, SERS can recognize the chemical element and also provide information about its chemical form, oxidation state or complexation, which are critical for assessing toxicity. The indirect SERS detection of protons, copper, zinc, mercury and cadmium can be carried out through the binding of an organic ligand at the plasmonic surface.
5.2. Detection of glucose
The development of new sensitive methods for glucose detection is vital for the diagnosis of diabetes. In a healthy organism, the normal concentration is 70–99 × 10−3 g per 10−10 L, while values above 100 × 10−3 g per 10−10 L indicate a pre-diabetic state and higher than 126 × 10−3 g per 10−10 L diagnose diabetes. A real-time detection of physiological concentrations of glucose (<450 × 10−3 g per 10−10 L) can be carried out in a complex biological medium (bovine plasma) by SERS at a roughened silver surface with the SAM of decanethiol.130 The SAM can serve as a partition layer for closer contact of an analyte to the silver surface. A quantitative measurement of glucose can be carried out by different methods based on a SERRS-ELISA technique. To remind us, ELISA (enzyme-linked immunosorbent assay) is a method for the indirect measurement of proteins developed in immunology. The latter can be also used for the recognition of either an antigen or an antibody in serum. The combination of ELISA with SERRS can overcome the problem of a high fluorescence background from biological media and an inability to detect interactions over distances greater than 10 nm. The quantitative detection analysis of glucose over a large clinically relevant concentration range can be also carried out by a chemometric method using the partial least squares (PLS) method.5.3. Detection of lipids
In biology, lipids are important structural elements of cell membranes with the functions of energy storage (as fats in adipose tissue) and signalling. SERS of lipids is a newly emerging area, although some investigations have been carried out with normal Raman spectroscopy.131 The SERS sensing of lipids is focused on the investigations of phospholipid bilayers and their properties during their interactions with various analytes. For example, lipids such as sphingosine-1-phosphate and platelet activating factor (as cellular signaling molecules) can be detected in complex mixtures by applying partition layer schemes.The molecular information from the intracellular compartments can be used to find out information about the molecular structure, interaction and compositional changes of biomolecules in real time. However, most of the Raman signals from a single cell are weak, due to the relatively low concentration of analytes and low excitation laser intensity (excluding degeneration of a cell). A collection of a Raman spectrum from a living cell can take hundreds of seconds, requiring long-data acquisition to build up a ‘Raman map’ in real time.132 Therefore, much effort has been concentrated on the development of biocompatible plasmonic nanostructures in order to extend SERS application to biological processes in real time.133
6. Intracellular SERS detection in live cells
Early SERS studies were performed with silver nanoparticles to monitor the intracellular distribution of drugs in the whole cell and to examine the antitumour drug–nucleic acid complexes.134,135 Later, label-free SERS detection was extended to bacteria. At present, the SERS approaches in biological measurements of live cells can be introduced by a great variety of functional substrates and SERS nanotags. Various strategies have been developed to overcome the experimental difficulties raised by contamination of the substrates, the limited dynamic range of analyte detection due to the saturation of a SERS signal and the strong dependence on the binding affinity of an analyte at hot spots (no attachment – no SERS signal at any concentration of solution).One approach introduces an indirect SERS detection strategy for tissue imaging by using a prostate specific antigen (PSA) as a Raman reporter target protein.136 Advanced spectral multiplexing and an increased signal strength can be produced by using silica-coated SAM on binary gold–silver nanoshells. Alternatively, plasmonic hot spots can be formed in aggregates of silver nanoparticles inside a polymeric structure with benzotriazole and 8-hydroxyquinoline groups as Raman reporters for a multiplexed analysis. In this indirect SERS method, the inner polymeric core connects the plasmonic hot spots, whereas the outer polymeric shell favours the attachment of oligonucleotide probes for the detection of specific DNA targets. In another approach, a Raman reporter could be encapsulated in a silica layer over plasmonic nanostructure for a selective SERS detection of proteins. A silica layer provided a protective barrier between a plasmonic core and surrounding medium. As a great advantage, multiple tags can be employed for multiplex targeting without SERS spectral overlap. One of these is a reactive isothiocyanate group, which can be used as a ‘molecular anchor’ for embedding organic dyes, or four multi-functional molecules on silver nanotags.26 In most of these approaches, the SERS EF is above 108 and is sufficient for single molecule detection.
Biocompatible Raman nanotags bearing indocyanine green on gold or silver nanostructures can be predictively positioned inside the cell in order to deliver information on the local molecular structure.137 The intracellular detection of pH can be monitored by a 4-mercaptobenzoic acid as another Raman nanotag, thereby reliably avoiding the fluctuation of the SERS signal intensity.138 However, this approach requires careful studies of the cellular uptake and intracellular distribution and many efforts should be directed into studying the stability of plasmonic hot spots under physiological conditions.139 The intracellular distribution of pH values can be quantified by using a SERS image from Raman nanotags, which are sensitive to the change of chemical conditions.140–143 Among these, the latter quinone moieties, as another type of Raman nanotags on plasmonic nanostructures, can enable the intracellular sensing of the redox potential. The intracellular detection of DNA, RNA, proteins or phospholipids in real time can be advanced by a glass nanopipette decorated with plasmonic hot spots.144 This direct method avoids the problem of uncontrolled colloidal aggregation inside the cell, but the orientation of the analytes at hot spots should be taken into account.
To note, SERS signals can be improved by at least two orders of magnitude by using the spatial offset Raman scattering (SORS) technique. In this method, a scattered light is collected from regions offset from the point of excitation, in order to improve the resolution depth in the sample analysis. The combination of SERS and SORS has given birth to a new bioanalytical tool: SESORS.145
6.1. Intracellular SERS study of bacteria
One of the first SERS studies was performed with silver nanoparticles adsorbed on the cell wall of bacteria.146 The SERS enhancement from Ag NPs on bacteria in water was about 9 times higher than in PBS solution, but then increased by 30-fold in a mixture of Ag NPs and a bacterial suspension. A SERS spectral discrimination of three strains of E. coli and one strain of S. epidermidis could be performed by this approach. Moreover, reduced assay time, simple handling and low reactant volumes were possible due to the higher sensitivity and selectivity of this technique.147–150A layer-by-layer approach has been successfully extended by SERS studies for single bacterial cell identification.151 The binding affinity of analytes at hot spots for SERS can be controlled by the negative charge of the bacterial surface, which is sensitive to pH value, ionic strength, and the concentration of surfactants in the suspending medium.152 Biochemical information can be also collected from pathogenic bacteria by in situ SERS measurements.153 SERS barcodes can be constructed from seven different strains of the marine pathogen Vibrio parahaemolyticus in their natural state and can be used as a successful model system. This approach is promising for the genetic identification of the strain-level differences in SERS spectra. Moreover, it can be advanced through the incorporation of genomic data, thus further extending the successful application of SERS to environmental monitoring and biomedical diagnostics.
The intracellular SERS spectrum from Escherichia coli can distinguish multiplexing spectral modes with a relatively high sensitivity.154 A SERS study of single cells can discriminate pathogenic or non-pathogenic bacteria in situ without cultivation and for screening out infection and contamination. SERS spectra from single cells or their ensemble show different peaks from extracellular and intracellular locations and can thus be attributed to the constituents on and inside the cell of E. coli.
6.2. SERS examination of plants, yeast, HeLa and other cells
In plant cells, analysis of the self-repair mechanisms of lacerated hypocotyls of Phaseolus vulgaris var. saxa can be performed by SERS from Au nanoparticles reduced on the plant tissue surface.143 Specifically, Au cyanides or a build-up of lignin and suberin can be identified in the wound healing process. SERS from nicotinamide adenine dinucleotide (NAD) and other adenine-containing materials can be monitored in plant tissue seeds, green tea leaves and in red cabbage.155 Other SERS peaks can arise from flavins, chlorophyll or lipids without antioxidants.In yeast cells, SERS studies can be carried out using a layer-by-layer approach for the encapsulation of living fungi cells with plasmonic nanostructures.156 Yeast cells are interesting because their walls act as the first line of defense against a fungal infection in pathology and chemotherapy.157 The surface of yeast cells and their membrane dynamics have thus been recently studied by SERS with a single molecule sensitivity.158
One of the critical limitations of intracellular SERS studies arises from the complex matrix of molecules within the cell which produce the SERS signals.159 This leads to a complicated spectrum, which is difficult to interpret. Another problem lies in the destabilization of plasmonic hot spots by the intracellular environment and is critical for real medical applications. Several intracellular SERS studies of live HeLa cells demonstrate the improved stability of hot spots by using silver and iron oxide nanoparticles160 or carbon nanotubes.161 Another successful example demonstrated the SERS monitoring of a neurotransmitter release,162 cancer diagnostics,163 and drug detection in cells.164
In recent years, SERS has been applied to the analysis of various biological functions and phenomena such as apoptosis, cell division and embryonic stem cell differentiation. In the analysis of the apoptotic cell, plasmonic nanoparticles could be introduced into human osteosarcoma cells.165 The SERS spectra could be detected from the nucleus of the HSC-3 cell, and cell division could be monitored. The spectral features of SERS revealed the phase-dependent differences during the cell cycle from G1, S to G2/M.166 In a related study, SERS analysis of the mouse embryonic stem cells could distinguish each differentiation stage.167 This is important for multiplexing, because many SERS peaks usually overlap due to the biochemical contributions, which is a common problem in complex biological environments of live cells.
Moreover, higher proliferation rates of cells and the dynamics of cellular activity can be examined by SERS in real time.168 It is important to note that SERS spectra can be strongly influenced by the surface change of the plasmonic nanostructure inside a live cell. Therefore, the surface functionalization of the nanostructures is critical in order to control the destination of the SERS active particle in a cell and to reduce the internal biomolecular complexity. One of the approaches is to embed plasmonic nanostructures into the cellular nucleus by using a nuclear localization signal (NLS) peptide.169
Overall, SERS enables the detection of single biomolecules from cells, but the SERS spectra have a time and position dependence, due to the dynamic interactions between plasmonic hot spots and biomolecules. The intracellular challenge lies in the dynamic nature of the cellular compartments, accompanied by various factors, e.g. flexibility, mobility, accessibility, polarity and exposed surface at the plasmonic/analyte interface.
6.3. Intracellular SERS in vivo
The successful development of SERS as a diagnostic tool inside live cells170 expedites in vivo studies.148,171 In this new investigation avenue, SERS relies on the indirect detection of analytes by using Raman nanotags. The latter can exhibit more than 200 times brighter SERS intensity with sharp molecular signatures in real time.148 Different biomarkers can be targeted simultaneously in vivo, in order to provide faster and more accurate multiplexed analysis (a great progress for in vivo diagnostics).The first in vivo application of SERS was used for the quantitative measurement of glucose in a rat.172 In this study, a silver film over a nanosphere (FON-type) surface with a two-component SAM layer could improve the detection of a glucose Raman signal by positioning within the first few nanometers of the SERS active surface. The selectivity of the SERS surface could be achieved by the designed hydrophilic/hydrophobic parts with decanethiol/mercaptohexanol (DT/MH) SAM, which is analogous to the ethylene glycol-terminated SAMs. In this way, the shorter hydroxyl-terminated chains act as hydrophilic pockets for closer contact with glucose, excluding the proteins. The stability of the plasmonic hot spots could then be derived by the FON geometry and single molecule analytes were collected. The in vivo studies could be carried out directly from the FONs, which were implanted along the midline of the rat's back. A reversible binding of glucose to the SERS active surface could then be spectroscopically monitored in real time. This approach can be used as a successful treatment and in the care of diabetics.
Other in vivo studies by SERS have been recently demonstrated by the targeting of tumours with Raman nanotags in live mice.148 Plasmonic hot spots in the matrix of polyethylene glycol can be encapsulated with a crystal violet as a Raman nanotag for the conjugation with tumour-targeting ligands (e.g. ScFv antibody). SERS intensity higher than two orders of magnitude in comparison to the brightness of quantum dots can be detected at a relatively large penetration depth (∼1–2 cm). The latter is of great advantage for tissue studies, as multiple scattering and absorption on the surface and throughout the surface layers can weaken the signal intensity, even causing its complete loss. As another advantage, tumour imaging and treatment based on the photothermal effect can be examined by this approach.
We dare to make a statement that, at present, SERS has reached a stage of its development where it is able to perform the early steps in the chemical imaging diagnostics of living cells. Although this newly emerged field is in an embryonic state, it will undoubtedly experience successful development, followed by making a great impact in medicine and the human prediction and diagnostics of diseases at the molecular level in real time. Let us examine the initial steps towards this success existing now.
7. SERS chemical imaging of live cells
Raman-assisted SERS and SERS imaging of live cells is a new growth field involving intensive study. One of the successful studies demonstrated how a SERS non-invasive imaging of a living plant (tobacco leaf of Nicotiana benthamiana) can be used to monitor the distribution of Raman tags inside the leaf in vivo.173 The time-resolved transportation of the SERS active nanoparticle through the live cell could be traced in real time.174 The dynamics of biomolecules in living cells could be detected spatially and temporally in parallel by using a slit scanning Raman microscope.We recently developed a simple, but efficient approach to perform chemical imaging from enhanced Raman signatures of live cellular compartments in real time (see Fig. 9).175 In our approach, plasmonic hot spots can sustain the SERS activity in the matrix of polyacrylic acid in the form of a thin film inside a live cell (e.g. NIH/3T3 fibroblast). The intracellular organelles can be distinguished by using a hypercluster analysis (HCA) from the strongest Raman bands (see Fig. 9A). To note, the HCA can provide label-free methods for the visualization of intracellular components and processes based on strong Raman/SERS signals. The regions of the nucleus and cytoplasm can be recognized, due to the contrast differences (brighter regions are due to the stronger SERS activity). The numbered spots with a violet or red colour indicate closely-packed SERS hot spots appearing at the membrane interface or at the outer surface of the cell (see Fig. 10b). Strong SERS spectra exhibit multiple peaks over the entire spectral region, with different shape profiles from the nucleus and periphery membrane of the nucleus or the cytoplasm. Single molecule detection can be proved by the detection of an adenine.113
 | ||
| Fig. 9 (A) Raman spectroscopic imaging of a live NIH/3T3 fibroblast with an embedded SiO2@Ag–PAA particle in a coloured spectral map (scale bar is 4 μm). The map reflects the differences found in the Raman data and the SERS active Ag–PAA nanofilms. This map is a linear combination of the averaged single spectra and characteristic for the cell compartments (green cytoplasm and blue-nucleus). The intense green and blue colours of the spots separated by a darker background contrast indicate the presence of SERS effective SiO2@Ag–PAA particles inside the cellular medium. Spots with violet and red colors show SiO2@Ag–PAA particles that are located at the membrane interface or at its surface (highlighted with numbers). A confocal Raman image is generated by the integration of the intensity of the strongest bands in the three spectral ranges: (i) <1000 cm−1, (ii) 1000–2000 cm−1 and (iii) 2500–3500 cm−1 after a local baseline subtraction using the hypercluster analysis (HCA) as one of the efficient label-free methods for the visualization of intracellular components and processes. (B) Selected SERS spectra that are collected from the nucleus (2 in A) and periphery membranes of the nucleus (5 in A) or cytoplasm (6 in A). The laser excitation wavelength was 532 nm and the grating was 600 g mm−1 (BLZ = 500 nm) and at a spectral resolution of 3 cm−1. Reprinted with permission from ref. D. Radziuk, R. Schuetz, A. Masic, H. Moehwald, Phys. Chem. Chem. Phys., 2014, 16, 24621–24634. | ||
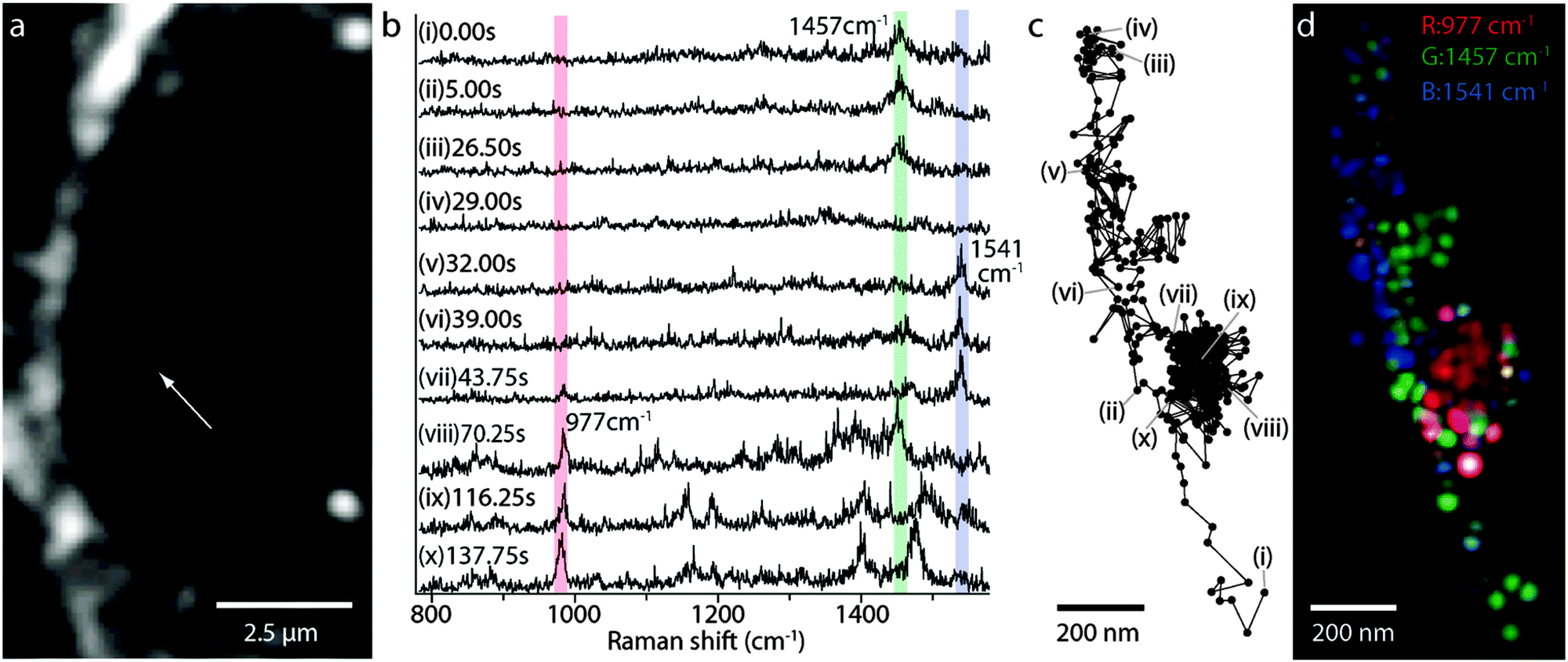 | ||
| Fig. 10 Dynamic SERS imaging of cellular transport pathways with endocytosed gold nanoparticles. (a) An image of a J774A.1 macrophage cell in the dark-field microscope mode. The white arrow indicates a gold nanoparticle as a small white spot. (b) SERS spectra from the nanoparticle in panel a. Three Raman peaks are overlaid with bars in red, green and blue. (c) Trajectory of the nanoparticle, marked by a white arrow in panel a, obtained in the dark-field mode images. (d) An RGB color map of the molecular distribution displayed on the nanoparticle trajectory. The green spots show the Raman intensity distribution of 1457 cm−1, blue spots of 1541 cm−1, and red spots of 977 cm−1. The green and blue colors are highlighted during the linear paths, whereas the red color appears during the confined zone random walk. The spatial resolution is determined as ∼65 nm, resulting from a particle diameter of ∼50 nm and a measurement accuracy of ∼15 nm. Reprinted with permission from ref. J. Ando, K. Fujita, N. I. Smith and S. Kawata, Nano Lett., 2011, 11, 5344–5348. | ||
The dynamics of the cellular SERS trace imaging in a live cell can also be mapped in real time (see Fig. 10a).176 The molecular transport of organelle and the accumulation of lysosomes can be monitored. Multiplex spectral and trajectory SERS spectra (see Fig. 10b) can enable the imaging of membrane protein diffusion, nuclear entry and rearrangement of the cellular cytoskeleton. A moving particle (see Fig. 10c) can be traced by a SERS image (see Fig. 10d), which can then be reconstructed from the vibrational bands of CH2 and CH3 of lipids and proteins (e.g. amide II). The time interval to detect the Raman signal can take as long as 250 ms, which is sufficient to follow a typical kinesin movement.
An entire Raman/SERS image of a single cell can be scanned in only 10 s9 and the Raman signals can be collected throughout the cytoplasm following an accurate localization of the embedded plasmonic nanostructures. As the sustained SERS activity is of a very good quality in a biological environment, this approach can be potentially used to monitor endocytosis inside a cell in real time.
Three-dimensional SERS spectral maps can be collected from a variety of biomolecules (e.g. glutathione, β-carotene, chlorophyll, hydroxyquinoline, NAD, reductase enzyme and other proteins) involved in the intracellular processes in green algae or fungi.177 A SERS image can be produced in several minutes, followed by a quick principle component analysis (PCA) or cluster analysis. This approach can be useful to study physico-chemical processes at the plasmonic/biological membrane and in the local cell environment in real time, in order to benefit from a best fit of the hot spots to the needs of a specific intracellular targeting.
Conclusions and outlook
Sustainable plasmonic hot spots can be formed through a successful combination of the geometry and metal dielectric function in complex nanostructures beyond silver or gold. A thin coating of alumina, silica or graphene enables the stability of the SERS efficiency by preventing the plasmonic surface from contamination and an undesired contact with the aggressive substances. The protective surface layer can facilitate conjugation with hydrophilic/hydrophobic molecules, thereby reducing cytoxicity, followed by long retention time inside the cells.For single molecule detection, individual plasmonic nanospheres provide an insufficient enhancement of the electromagnetic field, in contrast to the other geometries, such as nanorods, nanorice or nanostars, among other highly branched nanostructures. Plasmonic hot spots arising at the electromagnetic coupling between two adjacent nanostructures enable stronger SERS enhancement within the distance <10 nm. If such hot spots appear as periodic arrays in an elongated geometry over a larger micrometer surface, the SERS detection of the potential number of single molecules can be significantly increased. Understanding the non-linear coupling of plasmon resonances at hot spots can yield plasmon mapping and stable SERS efficiency of complex hybrid nanostructures and lead to reliable single molecule studies.
For the prediction of the SERS excitation profile, we need to keep in mind that the plasmon resonance depends on the refractive index of the surrounding medium and can be shifted, followed by shifts of the Raman peaks in the SERS spectra. However, this is not the case with the extinction spectrum: the SERS excitation does not track the extinction spectrum of the SERS substrate. This is a direct consequence of the SERS spectrum being dominated by the plasmon resonances at hot spots, whereas the extinction spectrum can include contributions from the entire plasmonic nanostructure.
Still we need to understand the origins of the signal fluctuations in the single molecule detection under SERS conditions. By studying the interaction between the excitation field and a localized surface plasmon resonance, the interference of plasmonic excitations in the scattering from interparticle junctions and surfaces can help to reveal the resonance supporting high enhancement under SERS conditions. A helpful step in figuring out the stable SERS conditions for single molecule detection could be explained by the electron dynamics in the optical properties in nearby plasmonic hot spots. The problem of the calculation of the SERS enhancement factor for the number of molecules contributing to the SERS signal is still present due to the uncertainties in the surface roughness, area and packing molecular density. However, new approaches based on the ratio of SERS signals from functional groups at hot spots and detected analytes can yield a reliable estimation of the molecular packing density, as proven by theoretical methods or from previous studies.
There is slight understanding on how to facilitate analyte positioning exactly at plasmonic hot spots in order to benefit from SERS detection. Recent empirical attempts have employed some exotic methods such as the laser illumination of plasmonic nanostructures and their dynamic self-assembly or even the principle of reversibility using the capillary effects. However, one of the most promising approaches can be the guided growth of plasmonic hot spots within a polymeric matrix bearing a great variety of functional groups for a specific targeting of single analytes. The nature and chain length of a polymer can enable the switching ‘on’ or ‘off’ of plasmonic hot spots, thereby selectively separating the SERS enhancement mechanisms (e.g. electromagnetic, chemical/charge transfer or molecular resonance). This could be a proof-of-principle for highly ordered plasmonic hot spots with an elongated geometry and for surpassing the conjugation for a controlled positioning of single analytes at a desired space. Raman nanotags or Raman reporter molecules can be encapsulated or attached at the plasmonic surface at different stages within the nucleation process. The method of self-assembly of the monolayers can still be advantageous as it enables the consistency of the surface monolayers and excludes harmful contamination and oxidation processes.
In a biological milieu, the interplay of the chemical functional groups and the Raman active reporters at the plasmonic hot spots is necessary in order to sustain the SERS enhancement for the selective and multiplex detection of single molecules. Great progress has been made in the sensitivity of the SERS detection of ions and glucose, thus enabling rapid indirect detection and recognition of the early stages of many diseases. SERS of lipids is a newly emerging field, despite a few investigations being carried out with normal Raman spectroscopy. This is important as the properties of lipids during their interactions with various analytes (e.g. cellular signaling molecules) are almost unknown.
The bright future of single molecule SERS offers the chemical imaging at the molecular level in real time. The speed of SERS imaging has been significantly increased and can take only several seconds to scan a single cell. This is particularly exciting if one imagines a direct visualization of molecular processes in living cells. Moreover, the development of single molecule SERS can facilitate the detection of a known marker for drugs and can be employed as nanoscale barcodes. One of the most exciting future outcomes can be that by the chemical visualization, we can learn about the distribution of drugs and their interaction with the molecules of nucleic acid or other complexes inside the cell by single molecule SERS in real time. Other fantastic outcomes of single molecule SERS important for environmental monitoring and biomedical diagnostics can be expected in the discrimination of pathogenic or non-pathogenic bacteria in situ, excluding cultivation, infection and contamination. Additional examples can include the examination of the apoptosis, cell division and differentiation at the molecular level in real time. Moreover, SERS can allow the tracing of individual particles inside the living cell and learn so much about the molecular transport of organelles, the accumulation of lysosomes, membrane protein diffusion and nuclear entry and the rearrangement of cellular cytoskeleton and many others.
Overall, it is evident that single molecule SERS has a great potential to become an established analytical tool in biomedical diagnostics. For single molecule SERS development, a lot more advances can be expected in the area of colloidal lithography and self-assembly, with impacts on the reversibility and dynamic properties of hot spots for specific needs. An extensive library of Raman nanotags and bioconjugation strategies will be produced in order to address the problem of analyte positioning. More efforts should be directed to the understanding of the fundamental aspects of the SERS enhancement mechanisms with a strong focus on single molecule spectroscopy.
Acknowledgements
We thank the Max Planck Society and the Deutsche Forschungsgemeinschaft for the financial support.References
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- E. C. Le Ru and P. G. Etchegoin, MRS Bull., 2013, 38, 631–640 CrossRef.
- M. L. Weber, J. P. Litz, D. J. Masiello and K. A. Willets, ACS Nano, 2012, 6, 1839–1848 CrossRef CAS PubMed.
- K. A. Willets and S. M. Stranahan, Proc. SPIE, 2012, 8228, 82280P CrossRef PubMed.
- S. Maier, Plasmonics: Fundamentals and Applications, Springer, New York, 2007 Search PubMed.
- L. Novotny and B. Hecht, Principles of Nano-optics, Cambridge University Press, 2012 Search PubMed.
- G. V. Naik, V. M. Shalaev and A. Boltasseva, Adv. Mater., 2013, 25, 3264–3294 CrossRef CAS PubMed.
- Z. L. Song, Z. Chen, X. Bian, L. Y. Zhou, D. Ding, H. Liang, Y. X. Zou, S. Wang, L. Chen, C. Yang, X. B. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 13558–13561 CrossRef CAS PubMed.
- V. Amendola, S. Scaramuzza, L. Litti, M. Meneghetti, G. Zuccolotto, A. Rosato, E. Nicolato, P. Marzola, G. Fracasso, C. Anselmi, M. Pinto and M. Colombatti, Small, 2014, 10, 2476–2486 CrossRef CAS PubMed.
- D. C. Marinica, A. K. Kazansky, P. Nordlander, J. Aizpurua and A. G. Borisov, Nano Lett., 2012, 12, 1333–1339 CrossRef CAS PubMed.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, J. H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J. M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- E. C. Le Ru and P. G. Etchegoin, Principles of Surface Enhanced Raman Spectroscopy (and related Plasmonic Effects), Elsevier, Amsterdam, 2009 Search PubMed.
- S. Emory and S. M. Nie, Anal. Chem., 1997, 69, 2631–2635 CrossRef CAS.
- J. P. Camden, J. A. Dieinger, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2008, 130, 12616–12617 CrossRef CAS PubMed.
- S. Campione, S. M. Adams, R. Ragan and F. Capolino, Opt. Express, 2013, 21, 7957–7973 CrossRef PubMed.
- W. Wei, K. Chen and G. Ge, Adv. Mater., 2013, 25, 3863–3868 CrossRef CAS PubMed.
- L. Rodriguez-Lorenzo, R. A. Alvarez-Puebla, I. Pastoriza-Santos, S. Mazzucco, O. Stephan, M. Kociak, L. M. Liz-Marzan and F. J. Garcia de Abajo, J. Am. Chem. Soc., 2009, 131, 4616–4618 CrossRef CAS PubMed.
- J. P. Litz, J. P. Camden and D. J. Masiello, J. Phys. Chem. Lett., 2011, 2, 1695–1700 CrossRef CAS.
- M. Moskovits, Phys. Chem. Chem. Phys., 2013, 15, 5301–5311 RSC.
- H. Wie and H. Xu, Nanoscale, 2013, 5, 10794–10805 RSC.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2012, 12, 165–171 CrossRef PubMed.
- M. W. Knight and N. J. Halas, New J. Phys., 2008, 10, 105006 CrossRef.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- J. B. Lassiter, M. W. Knight, N. A. Mirin and N. J. Halas, Nano Lett., 2009, 9, 4326–4332 CrossRef CAS PubMed.
- H. Wang, D. W. Brandl, F. Le, P. Nordlander and N. J. Halas, Nano Lett., 2006, 6, 827–832 CrossRef CAS PubMed.
- Z. Hu, Z. Liu, L. Li, B. Quan, Y. Li, J. Li and C. Gu, Small, 2014, 10, 3933–3942 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Y. Ding, Z. L. Yang, S. B. Li, X. S. Zhou, F. R. Fan, W. Zhang, Z. Y. Zhou, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- J. F. Li, X. D. Tian, S. B. Li, J. R. Anema, Z. L. Yang, Y. Ding, Y. F. Wu, Y. M. Zeng, Q. Z. Chen, B. Ren, Z. L. Wang and Z. Q. Tian, Nat. Protoc., 2013, 8, 52–65 CrossRef CAS PubMed.
- Metal Ion Adsorption on Silica, Alumina, and related Surfaces, in Encyclopedia of Surface and Colloid Science, ed. P. Somasundaran, Taylor & Francis group, 2nd edn, 2006, vol. 5, p. 3618 Search PubMed.
- C. T. Ertsgaard, R. M. McKoskey, I. S. Rich and N. C. Lindquist, ACS Nano, 2014, 8, 10941–10946 CrossRef CAS PubMed.
- P. P. Patra, R. Chikkaraddy, R. P. N. Tripathi, A. Dasgupta and G. V. P. Kumar, Nat. Commun., 2014, 5, 4357–4365 CAS.
- H. L. Liu, Y. D. Sun, Z. Jin, L. B. Yang and J. H. Liu, Chem. Sci., 2013, 4, 3490–3496 RSC.
- A. Klinkova, H. Therein-Aubin, A. Ahmed, D. Nykypanchuk, R. M. Choueiri, B. Gagnon, A. Muntyanu, O. Gang, G. C. Walker and E. Kumacheva, Nano Lett., 2014, 14, 6314–6321 CrossRef CAS PubMed.
- H. Huang, J. H. Wang, W. Jin, P. Li, M. Chen, H. H. Xie, X. F. Yu, H. Wang, Z. Dai, X. Xiao and P. K. Chu, Small, 2014, 10, 4012–4019 CrossRef CAS PubMed.
- M. Keating, S. Song, G. Wei, D. Graham, Y. Chen and F. Placido, J. Phys. Chem. C, 2014, 118, 4878–4884 CAS.
- T. Schiestel, H. Brunner and G. E. M. Tovar, J. Nanosci. Nanotechnol., 2004, 4, 504–511 CrossRef CAS PubMed.
- C. S. Levin, B. G. Janesko, R. Bardhan, G. E. Scuseria, J. D. Hartgerink and N. J. Halas, Nano Lett., 2006, 6, 2617–2621 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, H. M. Kim, J. M. Nam and Y. D. Suh, Nat. Mater., 2010, 9, 60–67 CrossRef CAS PubMed.
- N. L. Netzer, C. Qui, Y. Zhang, C. Lin, L. Zhang, H. Fong and C. Jiang, Chem. Commun., 2011, 47, 9606–9608 RSC.
- A. Gutes, R. Maboudian and C. Carraro, Langmuir, 2012, 28, 17846–17850 CrossRef CAS PubMed.
- H. X. Xu, J. Aizpurua, M. Kall and P. Apell, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2000, 62, 4318–4324 CrossRef CAS.
- S. L. Kleinman, R. R. Frontiera, A.-I. Henry, J. A. Dieringer and R. P. Van Duyne, Phys. Chem. Chem. Phys., 2013, 15, 21–36 RSC.
- W. Xie, B. Walkenfort and S. Schlücker, J. Am. Chem. Soc., 2013, 135, 1657–1660 CrossRef CAS PubMed.
- W. F. Dong, G. Sukhorukov and H. Möhwald, Phys. Chem. Chem. Phys., 2003, 5, 3003–3012 RSC.
- O. E. Rivera-Betancourt, E. S. Sheppard, D. C. Krause and R. A. Dluhy, Analyst, 2014, 139, 4287–4295 RSC.
- R. A. Alvarez-Puebla, R. Contreras-Caceres, I. Pastoriza-Santos, J. Perez-Juste and L. M. Liz-Marzan, Angew. Chem., Int. Ed., 2009, 48, 138–143 CrossRef CAS PubMed.
- N. Pazos-Perez, C. S. Wagner, J. M. Romo-Herrera, L. M. Liz-Marzan, F. J. Garcia de Abajo, A. Wittemann, A. Fery and R. A. Alvarez-Puebla, Angew. Chem., Int. Ed., 2012, 51, 12688–12693 CrossRef CAS PubMed.
- S. L. Kleinman, E. Ringe, N. Valley, K. L. Wustholz, E. Phillips, K. A. Scheidt, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2011, 133, 4115–4122 CrossRef CAS PubMed.
- J. A. Dieringer, R. B. Lettan, K. A. Scheidt and R. P. Van Duyne, J. Am. Chem. Soc., 2007, 129, 16249–16256 CrossRef CAS PubMed.
- S. E. J. Bell and M. R. McCourt, Phys. Chem. Chem. Phys., 2009, 11, 7455–7462 RSC.
- K. L. Wustholz, A. I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. Van Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS PubMed.
- G. B. Braun, S. J. Lee, T. Laurence, N. Fera, L. Fabris, G. C. Bazan, M. Moskovits and N. O. Reich, J. Phys. Chem. C, 2009, 113, 13622–13629 CAS.
- W. Lin, Appl. Phys. A: Mater. Sci. Process., 2011, 102, 121–125 CrossRef CAS.
- L. Lu, I. Randjelovic, R. Capek, N. Gaponik, J. Yang, H. Zhang and A. Eychmuller, Chem. Mater., 2005, 17, 5731–5736 CrossRef CAS.
- P. A. A. P. Marques, H. I. S. Nogueira, R. J. B. Pinto, C. P. Neto and T. Trindade, J. Raman Spectrosc., 2008, 39, 439–443 CrossRef CAS.
- F. C. Cabrera, P. H. B. Aoki, R. F. Aroca, C. J. L. Constantino, D. S. dos Santos and A. E. Job, J. Raman Spectrosc., 2012, 43, 474–477 CrossRef CAS.
- W. W. Yu and I. M. White, Analyst, 2013, 138, 1020–1025 RSC.
- L. Polavarapu and L. M. Liz-Marzan, Phys. Chem. Chem. Phys., 2013, 15, 5288–5300 RSC.
- K. D. Osberg, M. Rycenga, G. R. Bourret, K. A. Brown and C. A. Mirkin, Adv. Mater., 2012, 24, 6065–6070 CrossRef CAS PubMed.
- D. Radziuk and H. Moehwald, Nanoscale, 2014, 6, 6115–6126 RSC.
- S. P. Zhang and H. X. Xu, ACS Nano, 2012, 6, 8128–8135 CrossRef CAS PubMed.
- P. K. Jain, W. Huang and M. A. El-Sayed, Nano Lett., 2007, 7, 2080–2088 CrossRef CAS.
- A. L. Demming, F. Festy and D. Richards, J. Chem. Phys., 2005, 122, 184716 CrossRef CAS PubMed.
- B. Pettinger, K. F. Domke, D. Zhang, G. Picardi and R. Schuster, Surf. Sci., 2009, 603, 1335–1341 CrossRef CAS PubMed.
- K. J. Savage, M. M. Hawkeye, R. Esteban, A. G. Borisov, J. Aizpurua and J. J. Baumberg, Nature, 2012, 491, 574–577 CrossRef CAS PubMed.
- J. Jiang, K. Bosnick, M. Maillard and L. Brus, J. Phys. Chem. B, 2003, 107, 9964–9972 CrossRef CAS.
- E. J. Blackie, E. C. L. Ru and P. G. Etchegoin, J. Am. Chem. Soc., 2009, 131, 14466–14472 CrossRef CAS PubMed.
- Z. Wang, S. Pan, T. D. Krauss, H. Du and L. J. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8638–8643 CrossRef CAS PubMed.
- T. Dadosh, J. Sperling, G. W. Bryant, R. Breslow, T. Shegai, M. Dyshel, G. Haran and I. Bar-Joseph, ACS Nano, 2009, 3, 1988–1994 CrossRef CAS PubMed.
- H. Xu, E. J. Bjerneld, M. Käll and L. Börjesson, Phys. Rev. Lett., 1999, 83, 4357–4360 CrossRef CAS.
- V. Amendola and M. Meneghetti, Adv. Funct. Mater., 2012, 22, 353–360 CrossRef CAS.
- H. Wei, U. Håkanson, Z. L. Yang, F. Hook and H. X. Xu, Small, 2008, 4, 1296–1300 CrossRef CAS PubMed.
- T. Itoh, V. Biju, M. Ishikawa, Y. Kikkawa, K. Hashimoto, A. Ikehata and Y. Ozaki, J. Chem. Phys., 2006, 124, 134708 CrossRef PubMed.
- K. Yoshida, T. Itoh, H. Tamaru, V. Biju, M. Ishikawa and Y. Ozaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 115406 CrossRef.
- M. D. Doherty, A. Murphy, J. McPhillips, R. J. Pollard and P. Dawson, J. Phys. Chem. C, 2010, 114, 19913–19919 CAS.
- H. Wang, D. W. Brandl, P. Nordlander and N. J. Halas, Acc. Chem. Res., 2007, 40, 53–62 CrossRef CAS PubMed.
- E. Prodan, C. Radloff, N. J. Halas and P. Nordlander, Science, 2003, 302, 419–422 CrossRef CAS PubMed.
- E. Prodan and P. Nordlander, J. Chem. Phys., 2004, 120, 5444–5454 CrossRef CAS PubMed.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- D. W. Brandl, N. A. Mirin and P. Nordlander, J. Phys. Chem. B, 2006, 110, 12302–12310 CrossRef CAS PubMed.
- J. A. Fan, C. Wu, K. Bao, R. Bardhan, N. J. Halas, V. N. Manoharan, P. Nordlander, G. Shvets and F. Capasso, Science, 2010, 328, 1135–1138 CrossRef CAS PubMed.
- F. Hao, Y. Sonnefraud, P. V. Dorpe, S. A. Maier, N. J. Halas and P. Nordlander, Nano Lett., 2008, 8, 3983–3988 CrossRef CAS PubMed.
- J. Zuloaga, E. Prodan and P. Nordlander, ACS Nano, 2010, 4, 5269–5276 CrossRef CAS PubMed.
- A. Manjavacas, F. J. Garcia de Abajo and P. Nordlander, Nano Lett., 2011, 11, 2318–2323 CrossRef CAS PubMed.
- J. B. Lassiter, H. Sobhani, J. A. Fan, J. Kundu, F. Capasso, P. Nordlander and N. J. Halas, Nano Lett., 2010, 10, 3184–3189 CrossRef CAS PubMed.
- Y. Fang, N.-H. Seong and D. D. Dlott, Science, 2008, 321, 388–391 CrossRef CAS PubMed.
- A. Laurence, G. B. Braun, N. O. Reich and M. Moskovits, Nano Lett., 2012, 12, 2912–2917 CrossRef PubMed.
- P. G. Etchegoin and E. C. Le Ru, Phys. Chem. Chem. Phys., 2008, 10, 6079–6089 RSC.
- L. Zhao, L. Jensen and G. C. Schatz, J. Am. Chem. Soc., 2006, 128, 2911–2919 CrossRef CAS PubMed.
- L. L. Zhao, L. Jensen and G. C. Schatz, Nano Lett., 2006, 6, 1229–1234 CrossRef CAS PubMed.
- R. Esteban, A. Borisov, P. Nordlander and J. Aizpurua, Nat. Commun., 2012, 3, 1–9 Search PubMed.
- F. Hubenthal, Appl. Phys. B: Lasers Opt., 2014, 117, 1–5 CrossRef CAS.
- A. T. Zayak, Y. S. Hu, H. Choo, J. Bokor, S. Cabrini, P. J. Schuck and J. B. Neaton, Phys. Rev. Lett., 2011, 106, 083003 CrossRef CAS.
- E. C. Le Ru, L. C. Schroete and P. G. Etchegoin, Anal. Chem., 2012, 84, 5074–5079 CrossRef CAS PubMed.
- S. Shim, C. M. Stuart and R. A. Mathies, ChemPhysChem, 2008, 9, 697–699 CrossRef CAS PubMed.
- K. C. Vernon, A. M. Funston, C. Novo, D. E. Gomez, P. Mulvaney and T. J. Davis, Nano Lett., 2010, 10, 2080–2086 CrossRef CAS PubMed.
- T. Wriedt, Part. Part. Syst. Charact., 1998, 15, 67–74 CrossRef CAS.
- C. Girard, Rep. Prog. Phys., 2005, 68, 1883–1933 CrossRef.
- C. Girard, A. Dereux and J. C. Weeber, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 1081–1085 CrossRef CAS.
- W. H. Weber and G. W. Ford, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125429 CrossRef.
- G. W. Ford and W. H. Weber, Phys. Rep., 1984, 113, 195–287 CrossRef CAS.
- A. Taflove, Computational electrodynamics: the finite-difference time-domain method, Artech House, Boston, 1995 Search PubMed.
- K. S. Yee, Numerical solution of initial boundary value problems involving Maxwell's equations in isotropic media. IEEE Trans. Antennas Propag. AP-14, 1966, pp. 302–307.
- A. Lakhatakia and G. W. Mulholland, J. Res. Natl. Inst. Stand. Technol., 1993, 98, 699–716 CrossRef.
- M. F. Caterdra, E. Gago and L. Nuno, IEEE Trans. Antennas Propag., 1989, 37, 528–537 CrossRef.
- J. I. Hage and J. M. Greenberg, J. Astrophys., 1990, 361, 251–259 CrossRef CAS.
- B. T. Draine, J. Astrophys., 1988, 333, 848–872 CrossRef CAS PubMed.
- J. L. Volakis, A. Chatterjee and L. C. Kempel, J. Opt. Soc. Am. A, 1994, 11, 1422–1433 CrossRef.
- C. Girard, Rep. Prog. Phys., 2005, 68, 1883–1933 CrossRef.
- S. A. Meyer, E. C. Le Ru and P. G. Etchegoin, J. Phys. Chem. A, 2010, 114, 5515–5519 CrossRef CAS PubMed.
- S. K. Kim, T. H. Joo, S. W. Suh and M. S. Kim, J. Raman Spectrosc., 1986, 17, 381–386 CrossRef CAS.
- E. C. Le Ru, M. Meyer and P. G. Etchegoin, J. Phys. Chem. B, 2006, 110, 1944–1948 CrossRef CAS PubMed.
- A. Sundaramurthy, P. J. Schuck, N. R. Conley, D. P. Fromm, G. S. Kino and W. E. Moerner, Nano Lett., 2006, 6, 355–360 CrossRef CAS PubMed.
- M. L. Juan, J. Plain, R. Bachelot, A. Vial, P. Royer, S. K. Gray, J. M. Montgomery and G. P. Wiederrecht, J. Phys. Chem. A, 2009, 113, 4647–4651 CrossRef CAS PubMed.
- C. Hubert, R. Bachelot, J. Plain, S. Kostcheev, G. Lerondel, M. Juan, P. Royer, S. Zou, G. C. Schatz, G. P. Wiederrecht and S. K. Gray, J. Phys. Chem. C, 2008, 112, 4111–4116 CAS.
- K. A. Willets, Chem. Soc. Rev., 2014, 43, 3854–3864 RSC.
- K. A. Willets and S. M. Stranahan, Nano Lett., 2010, 10, 3777–3784 CrossRef PubMed.
- X. Han, B. Zhao and Y. Ozaki, Anal. Bioanal. Chem., 2009, 394, 1719–1727 CrossRef CAS PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, New York, 2001 Search PubMed.
- A. M. Scwartzberg, T. Y. Oshiro, J. Z. Zhang, T. Huser and C. E. Talley, Anal. Chem., 2006, 78, 4732–4736 CrossRef PubMed.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS PubMed.
- D. Tsoutsi, J. M. Montenegro, F. Dommershausen, U. Koert, L. M. Liz-Marzan, W. J. Parak and R. A. Alvarez-Puebla, ACS Nano, 2011, 5, 7539–7546 CrossRef CAS PubMed.
- H. F. Lodish, Mol. Cell. Biol., Scientific American Books, New York, 1999 Search PubMed.
- M. P. Cecchini, V. A. Turek, J. Paget, A. A. Kornyshev and J. B. Edel, Nat. Mater., 2012, 12, 165–171 CrossRef PubMed.
- L. M. Galanti, Clin. Chem., 1997, 43, 184–185 CAS.
- J. S. Torano and H. J. M van Kan, Analyst, 2003, 128, 838–843 RSC.
- D. J. Aberasturi, J.-M. Montenegro, I. R. Larramendi, T. Rojo, T. A. Klar, R. Alvarez-Puebla, L. M. Liz-Marzan and W. J. Parak, Chem. Mater., 2012, 24, 738–745 CrossRef.
- W. Xie and S. Schlucker, Phys. Chem. Chem. Phys., 2013, 15, 5329–5344 RSC.
- C. Fox, R. Uibel and J. Harris, J. Phys. Chem. B, 2007, 111, 11428–11436 CrossRef CAS PubMed.
- K. Kneipp, A. S. Haka, H. Kneipp, K. Badizadegan, N. Yoshizawa, C. Boone, K. E. Shafer-Peltiler, J. T. Motz and R. R. Dasari, Appl. Spectrosc., 2002, 56, 150–154 CrossRef CAS.
- Y. Kitahama, T. Itoh, P. Pienpinijtham, S. Ekgasit, X. X. Han and Y. Ozaki, in Functional Nanoparticles for Bioanalysis, Nanomedicine, and Bioelectronic Devices, Volume 2, ed. M. Hepel and C. J. Zhong, 2012, ch. 9, vol. 1113, pp. 181–234 Search PubMed.
- I. Nabiev, H. Morjani and M. Manfait, Eur. Biophys. J., 1991, 19, 311–316 CrossRef CAS.
- H. Morjani, J. F. Riou, I. Nabiev, F. Lavelle and M. Manfait, Cancer Res., 1993, 53, 4784–4790 CAS.
- B. Kustner, M. Gellner, M. Schutz, F. Schoppler, A. Marx, P. Strobel, P. Adam, C. Schmuck and S. Schlucker, Angew. Chem., Int. Ed., 2009, 48, 1950–1953 CrossRef PubMed.
- J. Kneipp, H. Kneipp, W. L. Rice and K. Kneipp, Anal. Chem., 2005, 77, 2381–2385 CrossRef CAS PubMed.
- C. E. Talley, L. Jusinski, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 7064–7068 CrossRef CAS PubMed.
- I. A. Larmour, E. A. Argueta, K. Faulds and D. Graham, J. Phys. Chem. C, 2012, 116, 2677–2682 CAS.
- C. E. Talley, L. Jusinski, C. W. Hollars, S. M. Lane and T. Huser, Anal. Chem., 2004, 76, 7064–7068 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Witting and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, B. Witting and K. Kneipp, J. Phys. Chem. C, 2010, 114, 7421–7426 CAS.
- C. A. R. Auchinvole, P. Richardson, C. McGuinnes, V. Mallikarjun, K. Donaldson, H. McNab and C. J. Campbell, ACS Nano, 2012, 6, 888–896 CrossRef CAS PubMed.
- S. Busch, K. Schmitt, C. Erhardt and T. Speck, J. Raman Spectrosc., 2010, 41, 490–497 CrossRef CAS.
- N. Stone, K. Faulds, D. Graham and P. Matousek, Anal. Chem., 2010, 82, 3969–3973 CrossRef CAS PubMed.
- H. Zhou, D. Yang, N. P. Ivleva, N. E. Mircescu, R. Niessner and C. Haisch, Anal. Chem., 2014, 86, 1525–1533 CrossRef CAS PubMed.
- A. Walter, A. Marz, W. Schumacher, P. Rosch and J. Popp, Lab Chip, 2011, 11, 1013–1021 RSC.
- X. M. Qian, X. H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. M. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- M. Kahraman, A. I. Zamaleeva, R. F. Fakhrullin and M. Culha, Anal. Bioanal. Chem., 2009, 395, 2559–2567 CrossRef CAS PubMed.
- L. Zeiri, B. V. Bronk, Y. Shabtai, J. Eichler and S. Efrima, Appl. Spectrosc., 2004, 58, 33–40 CrossRef CAS PubMed.
- M. Kahraman, A. I. Zamaleeva, R. F. Fakhrullin and M. Culha, Anal. Bioanal. Chem., 2009, 395, 2559–2567 CrossRef CAS PubMed.
- J. Li and L. A. McLandsborough, Int. J. Food Microbiol., 1999, 53, 185–193 CrossRef CAS.
- J. Xu, J. W. Turner, M. Idso, S. V. Biryukov, L. Rognstad, H. Gong, V. L. Trainer, M. L. Wells, M. S. Strom and Q. Yu, Anal. Chem., 2013, 85, 2630–2637 CrossRef CAS PubMed.
- L. Zeiri, B. V. Bronk, Y. Shabtai, J. Eichler and S. Efrima, Appl. Spectrosc., 2004, 58, 33–40 CrossRef CAS PubMed.
- L. Zeiri, J. Raman Spectrosc., 2007, 38, 950–955 CrossRef CAS.
- R. F. Fakhrullin, A. I. Zamaleeva, M. V. Morozov, D. I. Tazetdinova, F. K. Alimova, A. K. Hilmutdinov, R. I. Zhdanov, M. Kahraman and M. Culha, Langmuir, 2009, 25, 4628–4634 CrossRef CAS PubMed.
- M. Osumi, Micron, 1998, 29, 207–233 CrossRef CAS.
- A. Sujith, T. Itoh, H. Abe, A. A. Anas, K. Yoshida, V. Biju and M. Ishikawa, Appl. Phys. Lett., 2008, 92, 103901 CrossRef PubMed.
- K. Willets, Anal. Bioanal. Chem., 2009, 394, 85–94 CrossRef CAS PubMed.
- P. Candeloro, L. Tirinato, N. Malara, A. Fregola, E. Casals, V. Puntes, G. Perozziello, F. Gentile, M. L. Coluccio, G. Das, C. Liberale, F. De Angelis and E. Di Fabrizio, Analyst, 2011, 136, 4402–4408 RSC.
- P. Knief, C. Clarke, E. Herzog, M. Davoren, F. M. Lyng, A. D. Meade and H. J. Byrne, Analyst, 2009, 134, 1182–1191 RSC.
- R. J. Dijkstra, W. J. M. Scheenen, N. Dam, E. W. Roubos and J. J. ter Meulen, J. Neurosci. Methods, 2007, 159, 43–50 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, Nano Lett., 2007, 7, 1591–1597 CrossRef CAS PubMed.
- I. R. Nabiev, H. Morjani and M. Manfait, Eur. Biophys. J., 1991, 19, 311–316 CrossRef CAS.
- H. W. Tang, X. B. Yang, J. Kirkhamand and D. A. Smith, Appl. Spectrosc., 2008, 62, 1060–1069 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, Nano Lett., 2012, 12, 5369–5375 CrossRef CAS PubMed.
- R. R. Sathuluri, H. Yoshikawa, E. Shimizu, M. Saito and E. Tamiya, PLoS One, 2011, 6, e22802 Search PubMed.
- J. Ando, K. Fujita, N. I. Smith and S. Kawata, Nano Lett., 2011, 11, 5344–5348 CrossRef CAS PubMed.
- W. Xie, L. Wang, Y. Zhang, L. Su, A. Shen, J. Tan and J. Hu, Bioconjugate Chem., 2009, 20, 768–773 CrossRef CAS PubMed.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- C. L. Zavaleta, B. R. Smith, I. Walton, W. Doering, G. Davis, B. Shojaei, M. J. Natan and S. S. Gambhir, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13511–13516 CrossRef CAS PubMed.
- D. A. Stuart, J. M. Yuen, N. Shah, O. Lyandres, C. R. Yonzon, M. R. Glucksberg, J. T. Walsh and R. P. Van Duyne, Anal. Chem., 2006, 78, 7211–7215 CrossRef CAS PubMed.
- A. Shen, J. Guo, W. Xie, M. Sun, R. Richards and J. Hu, J. Raman Spectrosc., 2011, 42, 879–884 CrossRef CAS.
- K. Fujita, S. Ishitobi, K. Hamada, N. I. Smith, A. Taguchi, Y. Inouye and S. Kawata, J. Biomed. Opt., 2009, 14, 024038 CrossRef PubMed.
- D. Radziuk, R. Schuetz, A. Masic and H. Moehwald, Phys. Chem. Chem. Phys., 2014, 16, 24621–24634 RSC.
- J. Ando, K. Fujita, N. I. Smith and S. Kawata, Nano Lett., 2011, 11, 5344–5348 CrossRef CAS PubMed.
- R. H. Lahr and P. J. Vikesland, ACS Sustainable Chem. Eng., 2014, 2, 1599–1608 CrossRef CAS.
| This journal is © the Owner Societies 2015 |