
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An ultrafast molecular rotor based ternary complex in a nanocavity: a potential “turn on” fluorescence sensor for the hydrocarbon chain†
Sushant
Murudkar
a,
Aruna K.
Mora
a,
Prabhat K.
Singh
a,
Tusar
Bandyopadhyay
b and
Sukhendu
Nath
*a
aRadiation & Photochemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400 085, India. E-mail: snath@barc.gov.in; Fax: +91-22-5505151; Tel: +91-22-25593771
bTheoretical Chemistry Section, Bhabha Atomic Research Centre, Trombay, Mumbai 400 085, India
First published on 27th January 2015
Abstract
Formation of a ternary complex by an ultrafast molecular rotor (UMR) with a macrocyclic cavitand has been investigated for the sensitive detection of the alkyl chain of a surfactant. A benzothiazole based UMR, Thioflavin-T (ThT), has been used as a fluorescent probe. It is shown that ThT forms a very weak inclusion complex with γ-cyclodextrin (γ-CD) with an association constant of 8.8 M−1. However, the addition of a small amount of surfactant results in a significant increase in the emission intensity of ThT in γ-CD solution. From detailed steady-state and time-resolved fluorescence measurements and NMR studies, it has been inferred that the addition of the surfactant results in the formation of a ternary complex through the inclusion of its alkyl chain inside the γ-CD nanocavity. In such a ternary complex, the non-radiative torsional motion in ThT is largely prevented due to a large increase in the frictional force inside the nanocavity and results in a significant fluorescence enhancement. The formation of the binary and the ternary complexes in the present system has been further supported by the molecular docking and subsequent molecular dynamics simulation studies. The present result indicates that the inclusion complex with an UMR as a guest could be a potential candidate for the efficient detection of insoluble organic molecules, especially hydrocarbons.
1. Introduction
Owing to their extensive practical applications in all branches of science, supramolecular assemblies, though discovered decades earlier, are still a subject of active research.1 Studies on the supramolecular assemblies are mainly based on the binary inclusion complexes containing a guest and a host molecule.2,3 Such inclusion complexes are exclusively held together by non-bonded interactions like hydrophobic interactions,4 hydrogen bonding,5 dipole–dipole interactions,6 π–π stacking,7etc. Inclusion of a smaller guest molecule into the cavity of a macrocyclic host molecule does not only provide fundamental insight into the intermolecular forces that govern the complexation process, but also opens up a new field of practical applications ranging from analyte sensing to drug delivery, and synthesis of novel materials, etc.8 The use of macrocyclic host molecules for sensing applications has become very attractive due to their ability to form inclusion complexes with smaller guest molecules and modulate their physicochemical properties to a great extent.6,9Cyclodextrins (CDs), made up of D-glucopyranose units, are well known macrocyclic host molecules and form inclusion complexes with several classes of guest molecules.10,11 The exterior hydroxyl groups of CD make them soluble in water and their hydrophobic interior allows nonpolar organic molecules to reside in their nanocavity through hydrophobic interactions.10,12 The most remarkable property of CDs is their ability to modify the physicochemical properties of guest molecules through several types of interactions. Such modulation in guest's properties leads to the extensive use of CDs in analyte sensing application.3,13,14 Among three CDs, namely, α-, β-, and γ-cyclodextrins, the most popular for the analyte sensing activity is the β-CD due to its relatively rigid structure due to strong intramolecular hydrogen bonding between its secondary hydroxyl groups.10 However, with some exception,14,15 its analyte sensing activity is mainly limited by the formation of a binary inclusion complex due to the fact that the size of its nanocavity can accommodate mostly one guest molecule.
Apart from binary inclusion complexes, the formation of a ternary complex by CDs, where co-inclusion of a second guest molecule in the host molecular framework takes place, has also been reported.14,15 The inclusion of a third component in the nanocavity of CDs has been utilized to increase the solubility of hydrophobic drugs.16 Such ternary inclusion complex formation not only increases the bioavailability of the drug but also significantly reduces the amount of CDs required in the formulation.17 Because of such importance, ternary complexes have been investigated with a primary aim to understand the fundamental interactions that are responsible for their formation. However, such ternary complexes have an enormous potential application in the analyte sensing activity which is not explored adequately.
Being a very sensitive technique, fluorescence based methods have been extensively used to explore the possible applications of the ternary complex of CDs for the detection of different organic molecules. For example, some studies show that the fluorescence characteristics of aromatic hydrocarbons trapped in the nanocavity of CDs change in the presence of third additives, like alcohol,18 amines,19 surfactants,20etc. However, the change in the fluorescence intensity for such inclusion complexes is relatively small and is not suitable for the sensitive detection of analytes. Recently,21 a sensitive detection of Hg+2 has been reported based on the ternary complex where the emission intensity of the fluorophore decreases due to the addition of Hg+2, a “turn off” method. Such “turn off” methods have several intrinsic limitations, like low sensitivity, limited dynamic range, chances of false positive signal, etc.22,23 Hence its counterpart, the “turn on” methods, where the emission intensity of the probe increases due to the addition of the analyte, is more desirable.23,24
Ultrafast molecular rotors (UMR) are a class of fluorophores characterized by an ultrafast non-radiative barrierless torsional motion in their excited state. Due to such efficient non-radiative torsional motion, the emission yield of UMR is extremely low (<10−4) in low viscous solvents.25–27 The emission yield of an UMR is exclusively governed by the surrounding viscosity.28–30 Thus, the emission yield of such UMR increases by several folds in viscous solvents,28,29 confined environments,26,31–34etc., where the torsional motion in its excited state is restricted. Thioflavin T (ThT), a well-known benzothiazole based UMR (see Fig. 1 for molecular structure), has been used widely as an extrinsic fluorescence sensor for the detection of amyloid fibrils that are responsible for various neurodegenerative diseases.35 It has been shown that the emission yield of ThT and ThT based molecular rotors is very low in water and increases in high viscous solvent.28,29,36 It has been shown theoretically that the dihedral angle (ϕ) between the benzothiazole and the anilino moieties in its ground state is ∼39°.32,37 However, on photoexcitation, it undergoes very fast torsional motion around the central C–C single bond and ϕ becomes 90° (cf.Fig. 1).32 Femtosecond time-resolved fluorescence studies show that in water the time constant for the torsional motion in the excited state is ∼570 fs32 and the torsional time-constant increases with the increase in the solvent viscosity. Such torsional motion is largely restricted in amyloid fibrils and leads to very large fluorescence enhancement. Because of such characteristics, ThT has also been used to detect small structural changes due to premelting in the natural DNA27 and the weak hydrophobic interaction between ionic liquids and DNA,38 which otherwise could not be detected by common spectroscopic techniques including circular dichroism. Such an extremely high sensitivity of ThT fluorescence to the microviscosity indicates that its inclusion complex with CD could be a potential candidate for analyte sensing applications.
 | ||
| Fig. 1 Schematic presentation of the potential energy surface for ThT in its ground and first excited electronic states. The curved arrow in the molecular structure of ThT indicates the bond that undergoes torsional motion in its excited state. | ||
In the present work, we have investigated the formation of a ternary inclusion complex between ThT, γ-CD and surfactants through steady-state and time-resolved fluorescence experiments and by all atom molecular dynamics simulation. Our results show that such an inclusion complex with UMR could be a potential fluorescence sensor for the sensitive detection of the several organic molecules, especially hydrocarbons in the aqueous solution.
2. Materials and methods
Chloride salt of ThT, sodium dodecyl sulfate (SDS) and TritonX-100 (TX100) were purchased from Sigma. Cetyl pyridinium chloride (CPC) was obtained from E. Merck and used as received. ThT was recrystallized from double distilled water. Its purity was checked through absorption,39 emission,39 and proton NMR spectroscopy.40 γ-CD was purchased from TCI, India, and used without further purification. Double distilled water was used for all solution preparations. The concentration of ThT was determined by measuring the absorbance of its aqueous solution using extinction coefficient ε412 = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.41 The ground-state absorption spectra were recorded using a JASCO spectrophotometer (model V-650). All absorption spectra were recorded in a thermostated cell with 1 cm optical path. The steady-state emission spectra were recorded using a Hitachi spectrofluorimeter (model F-4500). For all steady-state emission measurements, the samples were excited with 412 nm light. Proton NMR spectra were recorded in deuterated water (D2O) using a Bruker Ascend TM-400 spectrometer operating at 400 MHz. The signal from residual HOD at δ = 4.79 was used as an internal lock.
000 M−1 cm−1.41 The ground-state absorption spectra were recorded using a JASCO spectrophotometer (model V-650). All absorption spectra were recorded in a thermostated cell with 1 cm optical path. The steady-state emission spectra were recorded using a Hitachi spectrofluorimeter (model F-4500). For all steady-state emission measurements, the samples were excited with 412 nm light. Proton NMR spectra were recorded in deuterated water (D2O) using a Bruker Ascend TM-400 spectrometer operating at 400 MHz. The signal from residual HOD at δ = 4.79 was used as an internal lock.
Pico-second time-resolved fluorescence measurements were carried out using a Horiba-Yovin (UK) spectrometer based on the time-correlated single-photon-counting (TCSPC) technique. Samples were excited by 406 nm, 1 MHz, 100 ps pulsed diode laser. Emission was detected at right angle to the excitation pulse using a photomultiplier tube based detection module (model TBX4; Horiba-Yovin). All TCSPC measurements were carried out with an emission polarizer set at the magic angle (54.7°) orientation with respect to the vertically polarized excitation pulse to eliminate the effect of rotational diffusion of the probe in the measured fluorescence decay traces.42 The instrument response function (IRF) was measured by collecting the scattered light from the suspended TiO2 particles in water. Fluorescence decays were fitted with a multi-exponential function using the standard convolute-and-compare nonlinear least square method.43
To predict the preferable binding orientation between guest and host molecules, we have performed blind molecular docking studies using AutoDock-4.2 program44 with the aid of the AutoDock Tools-1.5.4.45 All docking experiments were performed using the Lamarckian Genetic Algorithm (LGA) implemented in AutoDock-4 with an adaptive local search method.46 Default values of all docking parameters set by the program were used for the docking. However, the maximum number of energy evaluation was set to 2.5 × 106 and 100 runs were performed for each docking study. The grid size used for docking was selected in such a way that the entire inclusion complex can be encompassed in a cuboid having dimensions of 80 Å × 70 Å × 80 Å with a grid spacing of 0.375Å. All torsional angles in host molecules were set free for flexible docking. The resulting docking conformers were subsequently clustered with a root-mean-square-deviation (RMSD) tolerance of 2 Å and ranked according to their binding energy values.
The molecular structure of γ-CD was obtained from the CD-glycogen phosphorylase B complex (PDB ID-1P2G) after removing the protein moiety. Furthermore, the geometry of γ-CD was optimized by the PM3 semi-empirical method using Gaussian-03 package.47 Geometries of guest molecules (ThT and surfactant) were optimized using the DFT method implemented in Gaussian-03 package. B3LYP functional along with a 6-31++(d,p) basis set was used for their structural optimization.48 The contribution of bulk water was included through a conductor-like polarizable continuum model (CPCM).49
All the molecular dynamics (MD) simulations of the docked binary and ternary complexes were performed by the GROMACS 4.5.5 suit.50 The force fields for the γ-CD, ThT and SDS were all parametrized using Antechamber, a widely used parametrization scheme for novel molecules based on Amber force fields. The General Amber Force Field (GAFF) was used for the bonded and nonbonded terms.51 As an Amber-like force field, all the components of the complexes studied should be compatible with each other. The TIP3P water model was used to solvate the complexes.52
About 2000 water molecules were placed around each complex kept in a cuboid box, having a dimension which allows the solute outer surface to be at least 10 Å away from the box wall so as to ensure that no wall effect appeared in the simulated results. Required numbers of ions were added wherever needed to maintain electroneutrality. For binary inclusion complexes, a 10 ns-long trajectory was generated using the NVT simulating conditions. Long range electrostatics was handled using the particle mesh Ewald calculation method.53 The equations of motion were numerically integrated with a 2 fs time-step. Covalent bonds involving hydrogen atoms were constrained by the SHAKE algorithm.54 Of the 10 ns long equilibrium trajectories, the last 8 ns data were found to produce stable RMSD values, differing ∼3Å from the starting inclusion complexes. Orientations of ThT in the cavity of the CD can have a multitude of values. In order to quantify their relative orientations, we used time-dependent distribution of ϕ, defined as the angle between the molecular planes of CD and ThT (or rather their respective normals) and are shown in Fig. 2A. Each of the molecular planes was defined by three atoms (cf. Fig. S1 in ESI†). Eleven structures (indicated by arrows in Fig. 2B) of γ-CD–ThT binary complexes, having widely different ϕ values were extracted from the trajectory and were further subjected to blind docking for the inclusion of the surfactant molecule. The most stable docked ternary inclusion complex was then solvated in explicit TIP3P medium and MD simulations were carried out for sufficiently long time (50 ns) after a stable RMSD value has been attained during the first 3 ns.
 | ||
| Fig. 2 (A) Schematic presentation of angle ϕ between the normal of the molecular planes of CD and ThT. (B) Distribution of ϕ during 8 ns equilibrium MD run of the γ-CD–ThT binary complex. Arrows indicate the eleven orientations of ThT in the cavity of the CD which were further subjected to blind docking for the inclusion of the surfactant molecule. | ||
3. Results and discussion
3.1 Binary ThT–γ-CD system
 | ||
| Fig. 3 Ground state absorption spectra of ThT (10 μM) in aqueous solution at different concentrations of γ-CD. | ||
 | ||
| Fig. 4 The emission spectra of ThT (10 μM) aqueous solution in the presence of different concentrations of γ-CD: (1) 0, (2) 6, (3) 10, (4) 17, (5) 20 and (6) 25 mM. All emission spectra were corrected for the change in the absorbance due to the addition of γ-CD. Inset: emission spectra of ThT (10 μM) in the presence of 15 mM (a) β-CD and (b) γ-CD. | ||
The binding constant for the association of ThT with the γ-CD molecule is calculated using the following complexation reaction.
 | (1) |
:1 complexation model and is represented by the following equation.60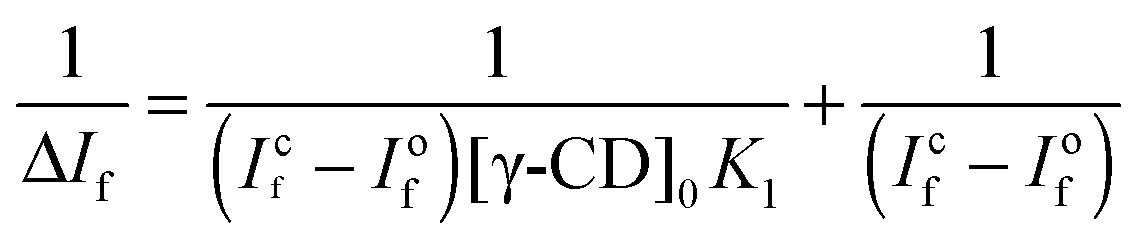 | (2) |
:1 inclusion complex. The binding constant estimated from the fitting is ∼8.8 M−1. Such a low value for the binding constant indicates a very weak association of ThT with the nanocavity of γ-CD due to the size mismatch of the guest and the host cavity. A similar low binding constant has been reported for the inclusion complex of pyrene in the CD nanocavity.61 Furthermore, the binding constant for the ThT–β-CD inclusion complex is reported to be ∼75 M−1.33 Thus, the present results further support the fact that the small cavity size of β-CD confines the ThT more strongly than that of the γ-CD.
 | ||
| Fig. 5 Double reciprocal plot (presented on a log–log scale) for the variation in the ThT emission intensity with the concentrations of γ-CD. The dashed line is the fitting of the experimental data by eqn (2). | ||
3.2 Ternary ThT–γ-CD–SDS system
 | ||
| Fig. 6 (A) Ground state absorption spectra of ThT (10 μM) and γ-CD (10 mM) in aqueous solution at different concentrations of SDS: solid lines – (1) 0, (2) 0.2, (3) 2 and (4) 8.3 mM SDS, dashed lines – (5) 13, (6) 18 (7) 25 mM SDS. (B) Normalized absorption spectra of ThT–γ-CD solution at different concentrations of SDS and the dashed curve shows the absorption spectra of ThT in 25 mM SDS solution. | ||
 | ||
| Fig. 7 Emission spectra of ThT–γ-CD aqueous solution in the presence of SDS: (A) 0–8 mM and (B) 9–25 mM. The dashed curve in the panel B shows the emission spectra of ThT in 25 mM SDS solution. Intensity of emission spectra was corrected for the change in the absorbance due to the addition of SDS. (C) Variation in the emission intensity of ThT–γ-CD solution at 490 nm with SDS concentrations. The dashed line is the guide to the eye. The triangle shows the emission intensity of ThT in 25 mM SDS solution. | ||
As mentioned earlier that the emission intensity of ThT primarily depends on the microviscosity around it, the present results clearly indicate that the microviscosity around ThT in the γ-CD cavity increases due to the addition of SDS (<8 mM). Such an increase in the emission intensity of ThT can be explained on the basis of the formation of a ternary complex between ThT–γ-CD and the SDS molecule. Due to its smaller size, ThT does not fit very tightly in a relatively large γ-CD nanocavity, and substantial space would be available for the SDS molecules to form a ternary complex. Formation of such a ternary complex is also favoured due to the fact that the alkyl chain of SDS has relatively higher affinity towards the nanocavity of CDs.63 Besides such hydrophobic interactions between the SDS alkyl chain and the γ-CD nanocavity, electrostatic interaction between the negatively charged SDS head group and the positively charged ThT also favours the formation of the ternary complex. Thus, SDS can act as a space regulator for the ThT–γ-CD inclusion complex by inserting its alkyl chain in the nanocavity and can modulate the volume of its empty space. Such space modulation in the nanocavity by SDS results in the formation of a much tighter inclusion complex and hence, ThT will experience large frictional force that will restrict its torsional motion in its excited state leading to significant increase in its emission intensity. Such changes in the emission intensity of different fluorophores in the nanocavity of CDs have been reported due to the formation of the ternary complex with different additives, like alcohol,18 amines,19 polymers,64etc. However, in all these cases reported earlier, the change in the emission intensity of the fluorescent probe was nominal. The enormous increase in the emission intensity of ThT (∼10 times) in the present study indicates that the ThT–γ-CD template could be a potential candidate for the efficient detection of several kinds of water insoluble organic molecules, especially hydrocarbons.
However, on further increase in the SDS concentration, the formation of the ternary complex will be completed and the excess SDS will form the micelles. It is evident from Fig. 7C that the emission yield of ThT in the SDS micelle is quite lower than that in the ternary complex. Thus, decrease in its emission intensity is expected due to the formation of SDS micelles at higher SDS concentration. Hence, a sharp decrease in the ThT emission beyond 8 mM SDS is the onset of the micelle formation. The present result indicates that the CMC of the SDS is substantially changed in the presence of γ-CD. Thus, the CMC of SDS in the presence of 10 mM γ-CD is estimated to be ∼13.1 mM. Such an increase in the CMC value in CD solution is known for several other surfactants.65 However, once the formation of micelles is completed at ∼15 mM of SDS, no further change in the emission intensity of ThT was observed. This result clearly indicates that in the presence of excess SDS, ThT is quantitatively transferred from the γ-CD nanocavity to the SDS micelles. Such relocation of ThT from the ternary complex to micelle is mainly attributed to the strong electrostatic attraction between the large negative charge density on the SDS micellar surface and positive charge on ThT.
The binding constant for the ternary complex was calculated by the method proposed by Hamai.61 Following equilibrium equations are considered for the calculation of the binding constant.
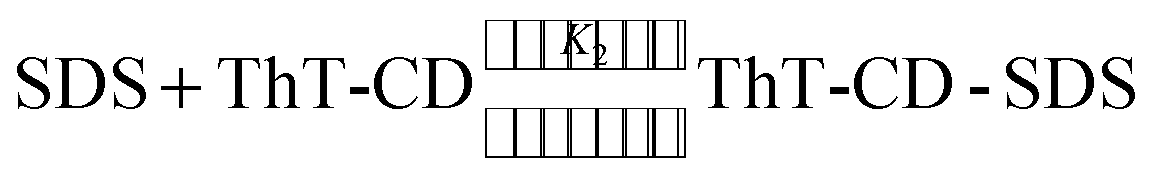 | (3) |
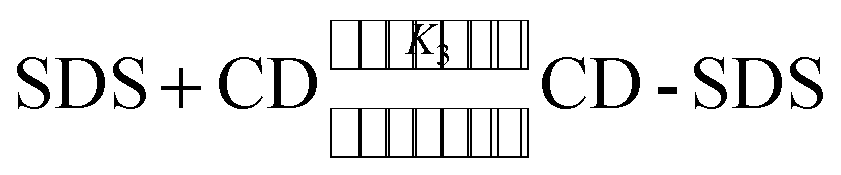 | (4) |
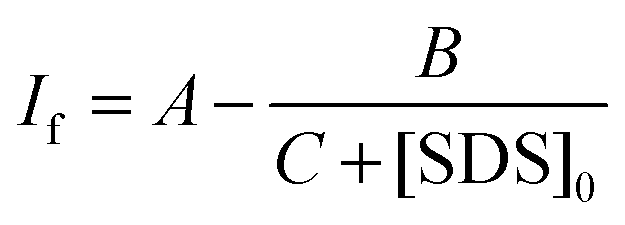 | (5) |
 | (6) |
The variation in the emission intensity of ThT–γ-CD solution with SDS concentration is shown in Fig. 8. The solid line is the fitting of the experimental data following eqn (5) and the fitting parameters are A = 1.356, B = 0.00396 and C = 0.0032. It is important to mention that for fitting purpose we have considered the experimental data up to the region where the increase in the emission intensity of ThT–γ-CD solution due to the addition of SDS is observed. As we have mentioned earlier that on further increase in the SDS concentration, the micellization of SDS takes place and leads to the displacement of ThT from the γ-CD nanocavity to SDS micelles. Considering the values of K1 = 8.8 M−1 and K3 = 940 M−1,63 the value of K2 was calculated from the value of C and found to be ∼40300 M−2.
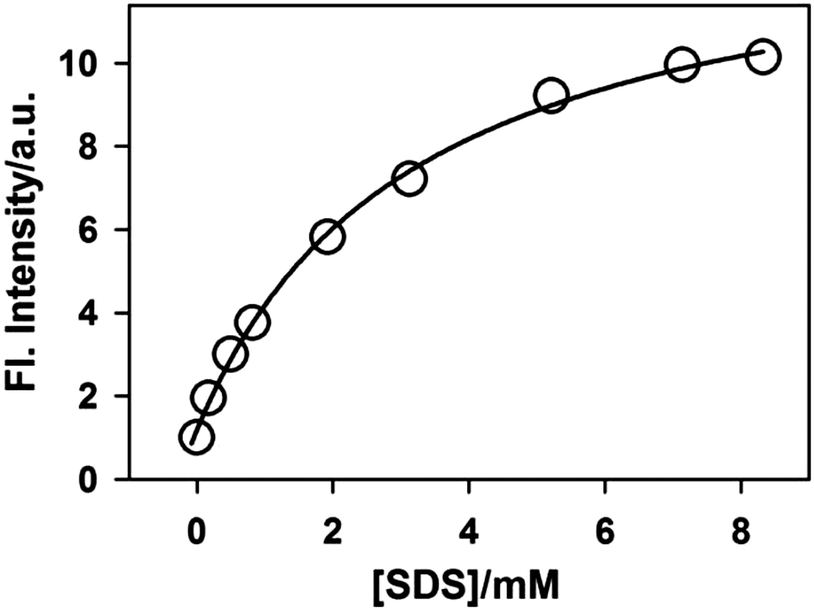 | ||
| Fig. 8 Variation in the emission intensity of ThT in 10 mM γ-CD solution with SDS concentrations. The solid line is the fitting to the experimental data following eqn (5). All emission intensity values were corrected for the change in the absorbance due to the addition of SDS. | ||
The more than one order of magnitude higher value for K2 than K3 clearly indicates that the propensity of SDS to bind with the γ-CD nanocavity increases enormously in the presence of ThT. Such increase in the binding ability can be explained on the basis of the following facts. The incorporation of one ThT molecule results in the displacement of a large number of water molecules from the γ-CD nanocavity leading to an increase in the hydrophobicity of the nanocavity to a large extent. As the interaction between the γ-CD nanocavity and the alkyl chain of SDS is hydrophobic in nature, the binding of SDS becomes stronger in the presence of ThT in the γ-CD nanocavity. Furthermore, due to the presence of opposite charges on SDS and ThT, the electrostatic attraction between them provides an additional driving force for the extraordinary binding of SDS to the ThT–γ-CD inclusion complex.
However, it is very important to know whether the formation of such a ternary complex is very specific for SDS or a general phenomenon for other organic molecules having a hydrophobic moiety. To check such possibility, we have further investigated the formation of a ternary complex by different classes of surfactants shown in Scheme 1. The changes in the emission intensity of the ThT–γ-CD complex due to the addition of different surfactant molecules are shown in Fig. 9. It is evident from the figure that irrespective of the charges of the head group and the nature of the hydrophobic tail, all surfactants lead to the formation of a ternary complex with the ThT–γ-CD complex, which results in an increase in the emission intensity of the solution. However, unlike SDS, for a cationic and neutral surfactant we have not observed any decrease in the emission intensity (cf.Fig. 7) at higher surfactant concentration. Such a difference in emission behavior is due to the fact that at a relatively high concentration when the surfactant forms a micelle, the positively charged ThT molecule in the ternary complex is quantitatively transferred to the negatively charged SDS micelles due to strong electrostatic attraction. On the other hand, the absence of such electrostatic interactions in the case of neutral micelles (TX100) and the presence of strong electrostatic repulsion in the case of positively charged micelles (CPC) prevent any such transfer of ThT from the ternary complex to the neutral and cationic micelles. Hence, for neutral and cationic surfactants, at relatively higher surfactant concentration the emission intensity attains a plateau. Thus, these results clearly indicate that the formation of the ternary complex by SDS with a ThT–γ-CD complex is not specific but rather a general phenomenon and can be accomplished by any organic species having a hydrophobic moiety, which can form an inclusion complex with the γ-CD nanocavity.
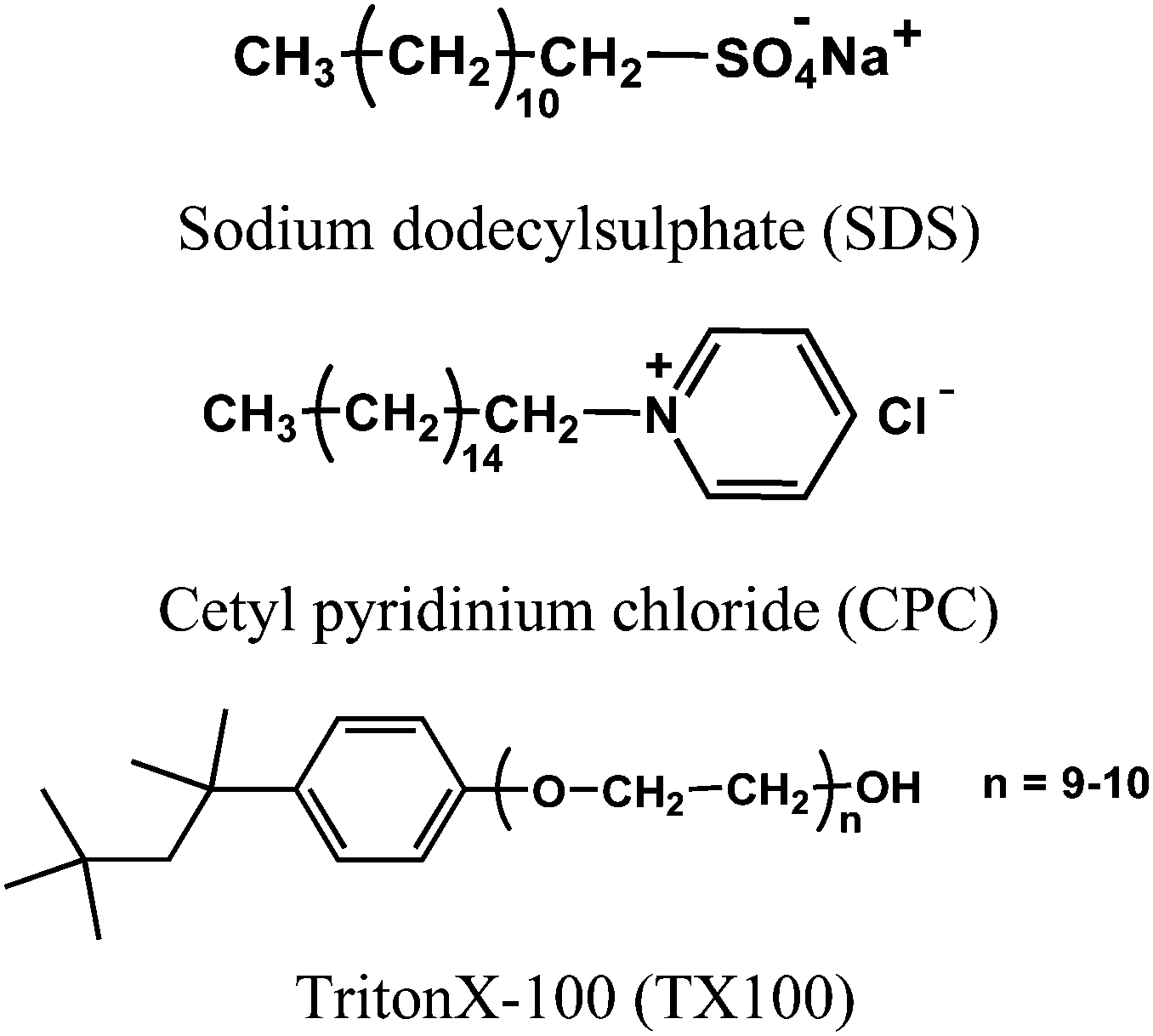 | ||
| Scheme 1 Molecular structure of surfactants used in the present study. | ||
 | ||
| Fig. 9 Variation in the emission intensity of 10 μM ThT in 10 mM γ-CD solution with the concentration of surfactants: (A) CPC and (B) TX-100. All emission intensities were corrected for the change in the absorbance due to the addition of surfactants. The dashed line is the guide to eye. | ||
The effect of SDS on the emission intensity of the ThT–β-CD system has also been investigated to check whether the binary complex has sufficient space to accommodate the alkyl chain of the surfactant. The variation of emission intensity of the ThT–β-CD system with SDS concentrations is shown in Fig. 10. It is seen that due to the initial addition of SDS the emission intensity of the ThT–β-CD complex decreases and at higher concentration of SDS the emission intensity is seen to increase to a large extend. The decrease in the emission intensity at lower SDS concentration indicates that the alkyl chain of the added surfactant replaces the ThT from the β-CD nanocavity resulting in the decrease in the emission intensity. Such replacement of ThT by the alkyl chain is also supported by the fact that the binding constant for the formation of the inclusion complex with the β-CD nanocavity is considerably larger for SDS (21000 M−1)66 as compared to that for ThT (75 M−1).34 This result clearly indicates that the size of the β-CD nanocavity is not large enough to accommodate the alkyl chain of the surfactant in the presence of ThT molecule. Thus, the formation of the ternary complex is not feasible due to the limited availability of the space in the β-CD nanocavity. The large increase in the emission intensity at higher SDS concentrations is due to the formation of the micelles and the re-localization of ThT from the bulk water to the micellar phase. It has been shown earlier that the emission intensity of ThT increases to a large extent due to its incorporation in the SDS micelles.34,67
 | ||
| Fig. 10 Variation in the emission intensity of ThT–β-CD solution with the concentration of SDS. The dashed line is the guide to the eye. | ||
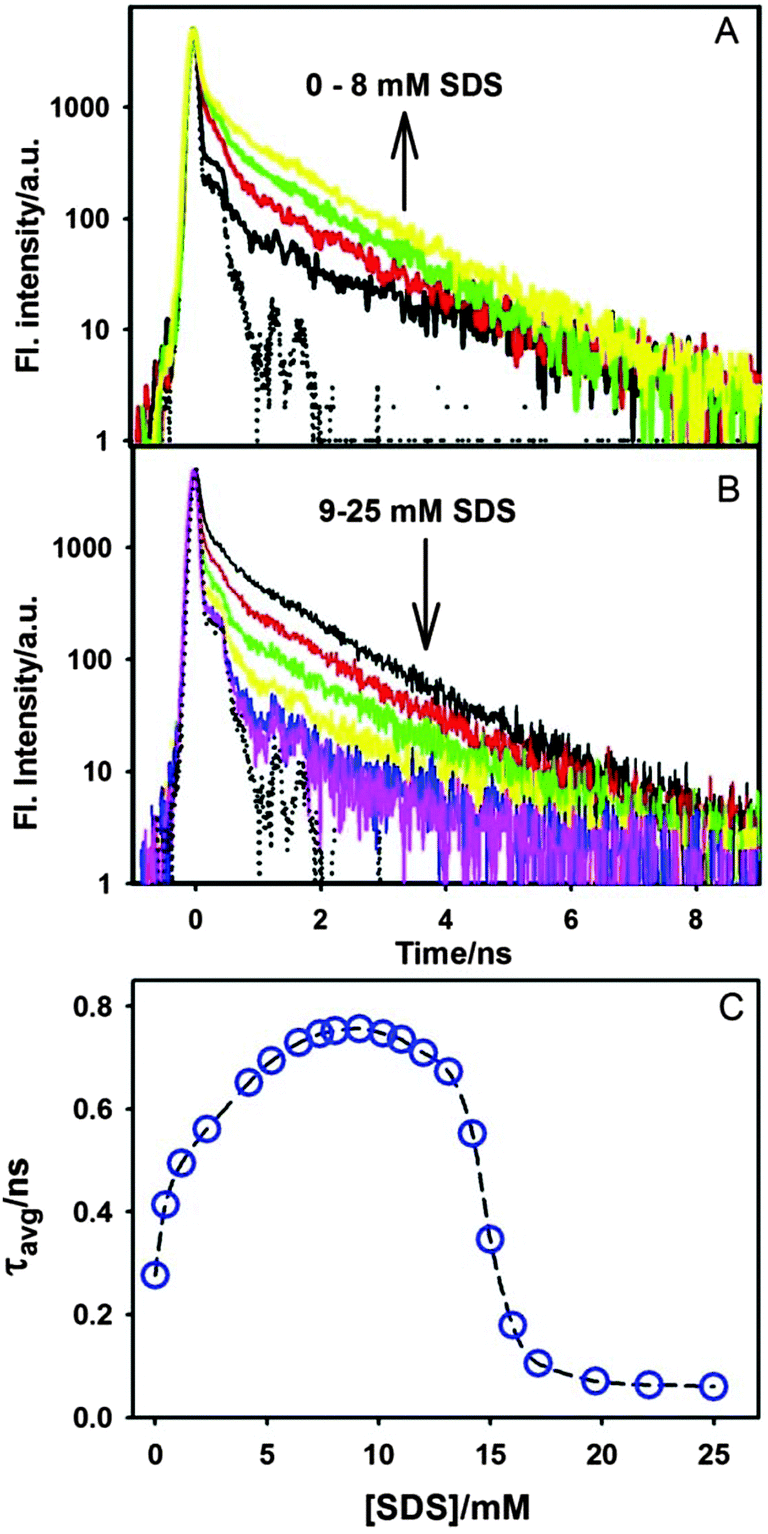 | ||
| Fig. 11 Transient fluorescence decays of the ThT–γ-CD complex at different concentrations of SDS (A) 0–8 mM SDS and (B) 9–25 mM SDS. The dotted curve in A and B panels shows the instrument response function. (C) Variation in the average lifetime (τavg) of ThT–γ-CD solution with the SDS concentrations. The dashed line is the guide to the eye. | ||
The variation in the average lifetime of the excited state ThT in γ-CD solution with the SDS concentration is shown in Fig. 11C. Such an increase in the excited state lifetime of ThT–γ-CD solution clearly indicates that the torsional motion of the ThT molecule in the γ-CD nanocavity is retarded due to the addition of SDS (up to 8mM). The present result further supports the fact that the addition of SDS results in the formation of the ternary complex which provides additional frictional force for the torsional motion in ThT resulting in the increase in its excited state lifetime in the γ-CD nanocavity. However, on further increase in the SDS concentration, as mentioned earlier, SDS forms a negatively charged micelle and consequently drags the positively charged ThT from the nanocavity. The excited state lifetime of ThT in the SDS micelle is found to be ∼≤60 ps, which is very close to our instrument limited value. Such faster decay in SDS indicates that the torsional motion of ThT in SDS micelles is relatively less restricted as compared to that in the ternary complex. This result can be explained by the fact that ThT binds on the surface of the micelles due to large negative charge density on the micellar surface. For such surface binding, the frictional force experienced by ThT in the micelles will be relatively less as compared to that in the nanocavity of γ-CD. Hence, excited state lifetime of ThT will be shorter in the micelles as compared to that in the nanocavity.
3.3 NMR studies
NMR spectra of SDS in the ternary complex were recorded and shown in Fig. 12. The NMR spectrum for SDS in water is also shown in Fig. 12 for comparison. The chemical shift values for different protons of free SDS (4 mM) in D2O and in ThT–γ-CD–SDS ternary complex are presented in Table 1. It is evident from Fig. 12 that the methyl proton (Ha) in SDS shows a triplet signal at 0.85 ppm and the protons of the intermediate nine methylene groups (Hb) show two overlapping signals at 1.27 ppm. The resonance signal at 1.67 and 4.05 was assigned to the Hc and Hd respectively.68 The proton in the hydrophobic tail group (a and b) experiences a relatively large low field shift whereas the proton close to the head group (c & d) shows a high field shift due to the formation of the ternary complex. The large positive Δδ values clearly indicate that the alkyl chain of SDS gets buried inside the nanocavity of γ-CD.68,69 The high field shift for c and d protons might be due to the decrease in the charge density of the sulphate head group due to the electrostatic interaction with positively charged ThT.69 Thus, NMR results clearly indicate the inclusion of the alkyl chain of the surfactant in the γ-CD nanocavity to form the ternary complex. | ||
| Fig. 12 Proton NMR spectra of SDS in water (A) and in ThT–γ-CD–SDS ternary complex (B). The concentration of SDS in both cases is 4 mM. | ||
| System | Ha | Hb | Hc | Hd |
|---|---|---|---|---|
| SDS | 0.85 | 1.27 | 1.67 | 4.05 |
| ThT–γ-CD–SDS | 1.00 | 1.42 | 1.61 | 3.96 |
| Δδ | 0.15 | 0.15 | −0.06 | −0.09 |
3.4 Molecular docking and MD simulation studies
To gain molecular level insight into the ternary complex in the water medium, molecular docking along with the MD simulation of the inclusion complexes has been carried out. Molecular docking of the ThT in the γ-CD nanocavity shows that a substantial part of the former gets incorporated into the nanocavity. Ten independent docking runs were performed for the ThT–γ-CD system. A 10 ns molecular dynamics run was carried out using the most stable docked structure of the binary complex and an average structure of the binary complex, extracted from the last 8 ns data, is shown in Fig. 13A. It is evident from the present results that in the most stable ThT–γ-CD complex system, the dimethylanilino moiety of ThT gets incorporated in the nanocavity of the γ-CD. Formation of such an inclusion complex is mainly responsible for the enhancement of ThT emission in the presence of γ-CD. | ||
| Fig. 13 Molecular structure of (A) ThT–γ-CD binary and (B) ThT–γ-CD–SDS ternary inclusion complex obtained from the MD simulations. | ||
Using eleven stable structures of the binary complex (one of them is shown in Fig. 13A), blind docking was performed using the ThT–γ-CD complex as the rigid host molecules and the SDS as the flexible ligand. The docking results show that the binding energy of SDS into the ThT–γ-CD complex ranges from 6.6 to 3.7 kcal mol−1. In such docking studies, invariably it is observed that the long hydrocarbon chain of the SDS molecule gets incorporated inside the γ-CD nanocavity containing a ThT and forms a sterically rigid ternary complex. Energetically the most stable docked structure was further utilized to perform the MD simulation upto 50 ns. The typical structure of the stable ternary complex is shown in Fig. 13B. It is evident from the figure that the hydrocarbon chain of the SDS molecule partially wraps around ThT molecule in the nanocavity. Due to such molecular arrangement, the torsional motion in ThT is expected to be further retarded in the ternary complex, which can result in the large emission enhancement in the presence of SDS. Thus, as we inferred from the absorption and fluorescence measurements, the docking and the MD simulation results further support our proposition on the formation of the ternary complex between ThT, γ-CD, and SDS.
From the last 40 ns of the 50 ns MD trajectories for the ternary complex, 4000 snapshots were taken at regular intervals for the binding free energy (ΔGbind) calculation using the solvated interaction energy (SIE) method.70 The SIE function is given as,
| ΔGbind(ρ, Din, γ, C) = α[EC + ΔGrbind + Evdw + γΔMSA] + C | (7) |
The SIE function fitted on a set of protein–ligand complexes gave absolute binding affinity predictions, the definitions and values of the best fitted parameters of which are: the AMBER van der Waals radii linear scaling coefficient, ρ = 1.1; the solute interior dielectric constant, Din = 2.25; the global proportionality coefficient related to the loss of configurational entropy upon binding α = 0.1048, the molecular surface area coefficient γ = 0.0129 kcal (mol Å2)−1 and a constant C = −2.89 kcal mol−1. In the SIE function for the binding free energy, EC and Evdw are the AMBER molecular mechanics force field based intermolecular Coulomb and van der Waals interaction energies in the bound state, respectively. ΔGrbind is the change in the reaction field energies between the bound and the free states and is calculated by solving the Poisson equation with the boundary element method program BRI BEM and using a molecular surface generated with a variable-radius solvent probe.71 The ΔMSA term is the change in the molecular surface area upon binding. Note that although none of the molecules involved in our ternary complex system are protein, the use of GAFF in our work ensures that the SIE fitted parameters (discovered through AMBER force field) for protein–ligand complexes can also be applied in the present system.
A time series plot of the calculated snapshot of the ΔGbind values was prepared to make sure that its average value could be extracted over a stable part of the MD trajectory with little drift in the mean value over time. The discovered average values of the SIE terms in the unit of kcal mol−1 were: 〈EC〉 = −34.85, 〈Evdw〉 = −30.39, ΔGrbind = 34.71, 〈γΔMSA〉 = −4.71, and 〈ΔGbind〉 = −6.57. Clearly, the present MD ensemble averaged binding free energy (−6.57 kcal mol−1) for the association of SDS into the ThT–γ-CD binary inclusion complex closely matched with our experimental value (−6.27 kcal mol−1) determined from steady-state fluorescence measurements.
4. Conclusions
The photophysical properties of the ThT–γ-CD inclusion complex have been investigated in the presence of different surfactants. ThT forms a very weak inclusion complex with γ-CD and leaves a large empty space in the nanocavity of γ-CD. The alkyl chain of the surfactant acts as a space regulator and occupy the empty space in the γ-CD nanocavity. Such space regulation by the surfactant alkyl chain increases the frictional forces for the torsional motion of ThT and leads to an enormous increase in its emission quantum yield. In the presence of ThT, the hydrophobicity of the γ-CD nanocavity increases leading to a stronger association with the hydrophobic alkyl chain of the surfactant. Such strong association of the surfactant leads to a tighter ternary inclusion complex and increases the excited state lifetime of ThT in the nanocavity. The formation of binary as well as the ternary complex was supported from the blind docking and the MD simulation studies. The large increase in the emission intensity of ThT due to the addition of surfactants indicates that the inclusion complex of UMR can be used efficiently for the detection of the several organic molecules especially hydrocarbons. Use of UMR based inclusion complexes with macrocyclic molecules for the detection of volatile organic compounds has been initiated in our laboratory and will be communicated in the future.Acknowledgements
The authors are thankful to Prof. A. Datta, Indian Institute of Technology, Bombay, for his help in the quantum chemical calculations. We are grateful to Dr S. Dey, Chemistry Division, for his help in the NMR measurements. We are also thankful to Dr H. Pal, Head, Molecular photochemistry Section, Dr D. K. Palit, Head, Radiation and Photochemistry Division, Bhabha Atomic Research centre for their constant encouragement and support during this work. SM thanks Board of Research in Nuclear Sciences (BRNS) for scholarship. The present work is funded by Department of Atomic Energy, India.Notes and references
- I. Vayá, V. Lhiaubet-Vallet, M. C. Jiménez and M. A. Miranda, Chem. Soc. Rev., 2014, 43, 4102–4122 RSC; N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084–4101 RSC; P. Kaur and K. Singh, RSC Adv., 2014, 4, 11980–11999 RSC; S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507–518 CrossRef CAS; X. Lin and M. W. Grinstaff, Isr. J. Chem., 2013, 53, 498–510 CrossRef; J. Wang, J. Newman Jr., S. L. H. Higgins, K. M. Brewer, B. S. J. Winkel and K. J. Brewer, Angew. Chem., Int. Ed., 2013, 52, 1262–1265 CrossRef PubMed.
- A. Harada, A. Hashidzume, H. Yamaguchi and Y. Takashima, Chem. Rev., 2009, 109, 5974–6023 CrossRef CAS PubMed; Y.-L. Zhao and J. F. Stoddart, Langmuir, 2009, 25, 8442–8446 CrossRef; M. Megyesi, L. Biczók and I. Jablonkai, J. Phys. Chem. C, 2008, 112, 3410–3416 Search PubMed; F. van de Manakker, M. van der Pot, T. Vermonden, C. F. van Nostrum and W. E. Hennink, Macromolecules, 2008, 41, 1766–1773 CrossRef; W. M. Nau, M. Florea and K. I. Assaf, Isr. J. Chem., 2011, 51, 559–577 CrossRef.
- M. Xu, S. Wu, F. Zeng and C. Yu, Langmuir, 2010, 26, 4529–4534 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons Ltd, Chichester, 2009 Search PubMed.
- F. Maffei, P. Betti, D. Genovese, M. Montalti, L. Prodi, R. D. Zorzi, S. Geremia and E. Dalcanale, Angew. Chem., Int. Ed., 2011, 50, 4654–4657 CrossRef CAS PubMed; J. Y. Lee, S. K. Kim, J. H. Jung and J. S. Kim, J. Org. Chem., 2005, 70, 1463–1466 CrossRef PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS PubMed; L. S. Kaanumalle and V. Ramamurthy, Chem. Commun., 2007, 1062–1064 RSC.
- A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; R. Martínez-Máñez and F. Sancenón, Chem. Rev., 2003, 103, 4419–4476 CrossRef PubMed; S. L. Wiskur, H. Ait-Haddou, J. J. Lavigne and E. V. Anslyn, Acc. Chem. Res., 2001, 34, 963–972 CrossRef PubMed; N. Bertrand, M. A. Gauthier, P. M. Céline Bouvet, A. Petitjean, J. C. Leroux and J. Leblond, J. Controlled Release, 2011, 155, 200–210 CrossRef PubMed; R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2012 CrossRef PubMed.
- L. Fabbrizzi and A. Poggi, Chem. Soc. Rev., 1995, 24, 197–202 RSC; R. Pinalli, M. Suman and E. Dalcanale, Eur. J. Org. Chem., 2004, 451–462 CrossRef CAS; E. V. Anslyn, J. Org. Chem., 2007, 72, 687–699 CrossRef PubMed; R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef PubMed; B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef.
- J. Szejtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS PubMed.
- E. M. M. D. Valle, Process Biochem., 2004, 39, 1033–1046 CrossRef; G. Wenz, Angew. Chem., Int. Ed., 1994, 33, 803–822 CrossRef.
- K. Takahashi, Chem. Rev., 1998, 98, 2013–2034 CrossRef CAS PubMed.
- S. Li and W. C. Purdy, Chem. Rev., 1992, 92, 1457–1470 CrossRef CAS; T. Ogoshi and A. Harada, Sensors, 2008, 8, 4961–4982 CrossRef PubMed; R. Yang, K. Li, K. Wang, F. Zhao, N. Li and F. Liu, Anal. Chem., 2003, 75, 612–621 CrossRef.
- A. Ponnua and E. V. Anslyna, Supramol. Chem., 2010, 22, 65–71 CrossRef.
- P. Mura, F. Maestrelli and M. Cirri, Int. J. Pharm., 2003, 260, 293–302 CrossRef CAS; M. Milewski, W. Augustyniak and A. Maciejewski, J. Phys. Chem. A, 1998, 102, 7427–7434 CrossRef; S. Sonzini, S. T. J. Ryan and O. A. Scherman, Chem. Commun., 2013, 49, 8779–8781 RSC; H.-J. Kim, J. Heo, W. S. Jeon, E. Lee, J. Kim, S. Sakamoto, K. Yamaguchi and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 1526–1529 CrossRef.
- J. C. de Miranda, T. E. A. Martins, F. Veiga and H. G. Ferraz, Braz. J. Pharm. Sci., 2011, 47, 665–681 CrossRef CAS PubMed; J. S. Smith, R. J. Macrae and M. J. Snowden, Eur. J. Pharm. Sci., 2005, 60, 73–80 Search PubMed; T. Loftsson and D. Duchene, Int. J. Pharm., 2007, 329, 1–11 CrossRef PubMed.
- T. Loftsson and H. Fridriksdottir, Int. J. Pharm., 1998, 163, 115–121 CrossRef CAS; P. Mura, M. T. Faucci and G. P. Bettinetti, Eur. J. Pharm. Sci., 2001, 13, 187–194 CrossRef.
- G. Nelson, G. Patonay and I. M. Warner, J. Inclusion Phenom., 1988, 6, 277–289 CrossRef CAS; A. M. de la Pena, T. T. Ndou, J. B. Zung, K. L. Greene, D. H. Live and I. M. Warner, J. Am. Chem. Soc., 1991, 113, 1572–1577 CrossRef.
- K. Kano, I. Takenoshita and T. Ogawa, J. Phys. Chem., 1982, 86, 1833–1838 CrossRef CAS; S. Hamai, J. Phys. Chem., 1988, 92, 6140–6144 CrossRef.
- J. Andreaus, J. Draxler, R. Marr and A. Hermetter, J. Colloid Interface Sci., 1997, 193, 8–16 CrossRef CAS; G. Astray, A. Cid, L. García-Río, C. Lodeiro, J. C. Mejuto, O. Moldes and J. Morales, Prog. React. Kinet. Mech., 2010, 35, 105–129 CrossRef.
- K. Kanagaraj, K. Bavanidevi, T. J. Chow and K. Pitchumani, RSC Adv., 2014, 4, 11714–11722 RSC.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838–9839 CrossRef CAS PubMed.
- J. Du, M. Liu, X. Lou, T. Zhao, Z. Wang, Y. Xue, J. Zhao and Y. Xu, Anal. Chem., 2012, 84, 8060–8066 CrossRef CAS PubMed.
- H. Wang, Y. Wang, J. Jin and R. Yang, Anal. Chem., 2008, 80, 9021–9028 CrossRef CAS PubMed; D. Huang, C. Niu, X. Wang, X. Lv and G. Zeng, Anal. Chem., 2013, 85, 1164–1170 CrossRef PubMed; Z. Wang, J. H. Lee and Y. Lu, Chem. Commun., 2008, 6005–6007 RSC.
- N. Amdursky, R. Gepshtein, Y. Erez and D. Huppert, J. Phys. Chem. A, 2011, 115, 2540–2548 CrossRef CAS PubMed; P. Changenet, H. Zhang, M. J. van der Meer, M. Glasbeek, P. Plaza and M. M. Martin, J. Phys. Chem. A, 1998, 102, 6716–6721 CrossRef; M. A. Haidekker, M. Nipper, A. Mustafic, D. Lichlyter, M. Dakanali and E. A. Theodorakis, in Advanced Fluorescence Reporters in Chemistry and Biology I, Fundamentals and Molecular Design, ed. A. P. Demchenko, Springer-Verlag, Berlin, 2010, vol. 8, pp. 267–308 Search PubMed; M. A. Haidekker and E. A. Theodorakis, Org. Biomol. Chem., 2007, 5, 1669–1678 Search PubMed; M. A. Haidekker and E. A. Theodorakis, J. Biol. Eng., 2010, 4, 11 CrossRef PubMed; C. Singh, B. Modak, J. A. Mondal and D. K. Palit, J. Phys. Chem. A, 2011, 115, 8183–8196 CrossRef PubMed; N. Amdursky, Y. Erez and D. Huppert, Acc. Chem. Res., 2012, 45, 1548–1557 CrossRef PubMed.
- T. Hutter, N. Amdursky, R. Gepshtein, S. R. Elliott and D. Huppert, Langmuir, 2011, 27, 7587–7594 CrossRef CAS PubMed.
- S. Murudkar, A. K. Mora, P. K. Singh and S. Nath, Chem. Commun., 2012, 48, 5301–5303 RSC.
- P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, J. Phys. Chem. B, 2010, 114, 5920–5927 CrossRef CAS PubMed.
- V. I. Stsiapura, A. A. Maskevich, V. A. Kuzmitsky, V. N. Uversky, I. M. Kuznetsova and K. K. Turoverov, J. Phys. Chem. B, 2008, 112, 15893–15902 CrossRef CAS PubMed.
- R. O. Loutfy and B. A. Arnold, J. Phys. Chem., 1982, 86, 4205–4211 CrossRef CAS; B. D. Allen, A. C. Benniston, A. Harriman, S. A. Rostron and C. Yu, Phys. Chem. Chem. Phys., 2005, 7, 3035–3040 RSC.
- P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, J. Phys. Chem. B, 2009, 113, 8532–8538 CrossRef CAS PubMed; P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, Chem. Commun., 2011, 47, 6912–6914 RSC; P. K. Singh and S. Nath, J. Photochem. Photobiol., A, 2012, 248, 42–49 CrossRef PubMed.
- P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, J. Phys. Chem. B, 2010, 114, 2541–2546 CrossRef CAS PubMed.
- P. K. Singh, M. Kumbhakar, H. Pal and S. Nath, Phys. Chem. Chem. Phys., 2011, 13, 8008–8014 RSC.
- P. K. Singh and S. Nath, J. Phys. Chem. B, 2013, 117, 10370–10375 CrossRef CAS PubMed.
- H. LeVine, III, Protein Sci., 1993, 2, 404–410 CrossRef CAS PubMed; T. Ban, K. Yamaguchi and Y. Goto, Acc. Chem. Res., 2006, 39, 663–670 CrossRef PubMed; F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef PubMed; J. E. Gestwicki, G. R. Crabtree and I. A. Graef, Science, 2004, 306, 865–869 CrossRef PubMed; S. Gilead and E. Gazit, Angew. Chem., Int. Ed., 2004, 43, 4041–4044 CrossRef PubMed.
- A. Srivastava, P. K. Singh, M. Kumbhakar, T. Mukherjee, S. Chattopadyay, H. Pal and S. Nath, Chem. – Eur. J., 2010, 16, 9257–9263 CrossRef CAS PubMed.
- W. Dzwolak and M. Pecul, FEBS Lett., 2005, 579, 6601–6603 CrossRef CAS PubMed; V. Fodera, M. Groenning, V. Vetri, F. Librizzi, S. Spagnolo, C. Cornett, L. Olsen, M. van de Weert and M. Leone, J. Phys. Chem. B, 2008, 112, 15174–15181 CrossRef PubMed.
- P. K. Singh, J. Sujana, A. K. Mora and S. Nath, J. Photochem. Photobiol., A, 2012, 246, 16–22 CrossRef CAS PubMed.
- J. C.-C. Hsu, E. H.-L. Chen, R. C. Snoeberger, F. Y. Luh, T.-S. Lim, C.-P. Hsu and R. P.-Y. Chen, J. Phys. Chem. B, 2013, 117, 3459–3468 CrossRef CAS PubMed.
- K. J. Robbins, G. Liu, G. Lin and N. D. Lazo, J. Phys. Chem. Lett., 2011, 2, 735–740 CrossRef CAS.
- M. Groenning, L. Olsen, M. van de Weert, J. M. Flink, S. Frokjaer and F. S. Jørgensen, J. Struct. Biol., 2007, 158, 358–369 CrossRef CAS PubMed.
- J. R. Lackowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- D. V. O'Connor and D. Phillips, Time correlated single photon counting, Academic, New York, 1984 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef CAS PubMed; R. Huey, G. M. Morris, A. J. Olson and D. S. Goodsell, J. Comput. Chem., 2007, 28, 1145–1152 CrossRef PubMed.
- M. F. Sanner, J. Mol. Graphics Modell., 1999, 17, 57–61 CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785–789 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollamn and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- A. A. Maskevich, V. I. Stsiapura, V. A. Kuzmitsky, I. M. Kuznetsova, O. I. Povarova, V. N. Uversky and K. K. Turoverov, J. Proteome Res., 2007, 6, 1392–1401 CrossRef CAS PubMed.
- P. Friedhoff, A. Schneider, E.-M. Mandelkow and E. Mandelkow, Biochemistry, 1998, 37, 10223–10230 CrossRef CAS PubMed; E. S. Voropai, M. P. Samtsov, K. N. Kaplevskii, A. A. Maskevich, V. I. Stepuro, O. I. Povarova, I. M. Kuznetsova, K. K. Turoverov, A. L. Fink and V. N. Uverskiid, J. Appl. Spectrosc., 2003, 70, 868–874 CrossRef.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- M. R. H. Krebs, E. H. C. Bromley and A. M. Donald, J. Struct. Biol., 2005, 149, 30–37 CrossRef CAS PubMed.
- M. L. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef.
- S. Nigam and G. Durocher, J. Phys. Chem., 1996, 100, 7135–7142 CrossRef CAS; B. Jana, S. Senapati, D. Ghosh, D. Bose and N. Chattopadhyay, J. Phys. Chem. B, 2012, 116, 639–645 CrossRef PubMed.
- S. Hamai, J. Phys. Chem., 1989, 93, 2074–2078 CrossRef CAS.
- K. Kalyanasundaram, Photochemistry in microheterogeneous systems, Academic Press Inc., Orlando, 1987 Search PubMed.
- V. T. Liveri, G. Cavallaro, G. Giammona, G. Pitarresi, G. Puglisi and C. Ventura, Thermochim. Acta, 1992, 199, 125–132 CrossRef.
- P. Mura, M. T. Faucci and G. P. Bettinetti, Eur. J. Pharm. Sci., 2001, 13, 187–194 CrossRef CAS; R. L. Carrier, L. A. Miller and I. Ahmed, J. Controlled Release, 2007, 123, 78–99 CrossRef PubMed.
- E. A. M. Al-Sherbini, Colloids Surf., A, 2009, 352, 1–4 CrossRef CAS PubMed; L. Jiang, Y. Yan and J. Huang, Adv. Colloid Interface Sci., 2011, 169, 13–25 CrossRef PubMed.
- W. M. Z. Wan Yunus, J. Taylor, D. M. Bloor, D. G. Hall and E. Wyn-Jones, J. Phys. Chem., 1992, 96, 8979–8992 CrossRef CAS.
- S. Kumar, A. K. Singh, G. Krishnamoorthy and R. Swaminathan, J. Fluoresc., 2008, 18, 1199–1205 CrossRef CAS PubMed.
- B. Z. Idiyatullin, K. S. Potarikina, Y. F. Zuev, O. S. Zueva and O. G. Us'yarov, Colloid J., 2013, 75, 532–537 CrossRef CAS.
- D. Zhao, Y. Chen and Y. Liu, Chin. Chem. Lett., 2014 DOI:10.1016/j.cclet.2014.11.028.
- Q. Cui, T. Sulea, J. D. Schrag, C. Munger, M.-N. Hung, M. Naïm, M. Cygler and E. O. Purisima, J. Mol. Biol., 2008, 379, 787–802 CrossRef CAS PubMed; M. Naïm, S. Bhat, K. N. Rankin, S. Dennis, S. F. Chowdhury, I. Siddiqi, P. Drabik, T. Sulea, C. I. Bayly, A. Jakalian II and E. O. Purisima, J. Chem. Inf. Model., 2007, 47, 122–133 CrossRef PubMed.
- E. O. Purisima, J. Comput. Chem., 1998, 19, 1494–1504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04636f |
| This journal is © the Owner Societies 2015 |