
Noteworthy performance of La1−xCaxMnO3 perovskites in generating H2 and CO by the thermochemical splitting of H2O and CO2†
Sunita
Dey
a,
B. S.
Naidu
 a,
A.
Govindaraj
ab and
C. N. R.
Rao
*ab
a,
A.
Govindaraj
ab and
C. N. R.
Rao
*ab
aChemistry and Physics of Materials Unit, New Chemistry Unit, Sheikh Saqr Laboratory, International Centre for Materials Science (ICMS) and CSIR Centre of Excellence in Chemistry, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bangalore-560064, India. E-mail: cnrrao@jncasr.ac.in
bSolid state and Structural Chemistry Unit, Indian Institute of Science, Bangalore-560012, India
First published on 5th November 2014
Abstract
Perovskite oxides of the composition La1−xCaxMnO3 (LCM) have been investigated for the thermochemical splitting of H2O and CO2 to produce H2 and CO, respectively. The study was carried out in comparison with La1−xSrxMnO3, CeO2 and other oxides. The LCM system exhibits superior characteristics in high-temperature evolution of oxygen, and in reducing CO2 to CO and H2O to H2. The best results were obtained with La0.5Ca0.5MnO3 whose performance is noteworthy compared to that of other oxides including ceria. The orthorhombic structure of LCM seems to be a crucial factor.
Increasing interest in sustainable energy sources and climate change has encouraged research in the production of H2 from H2O. Similarly, there is much interest in the reduction of CO2 to CO by simple means. One of the significant methods being explored for the generation of H2 and CO is the thermochemical splitting of H2O and CO2, respectively, by using oxide materials. The basic reactions of thermochemical H2O and CO2 splitting cycles employing metal oxides MOn are the following:
| Endothermal step: MOn → MOn−δ + δ/2O2 | (1) |
| Exothermal step: MOn−δ + δCO2 → MOn + δCO |
| MOn−δ + δH2O → MOn + δH2 | (2) |
For this purpose, ZnO, ferrites, CeO2 and related oxides have been employed.1–10 Recently, oxide perovskites such as La1−xSrxMnO3 and La1−xSrxFeO3 were employed for thermochemical fuel production,11–13 and results have been encouraging. Thus, Sr and Mn doped LaAlO3 splits H2O and CO2 better than CeO2.14 Because of the contemporary relevance of this subject, we considered that it is important to investigate other perovskite oxides for the thermochemical splitting of H2O and/or CO2 with the expectation that they may produce superior results. The materials employed by us for this purpose belong to the La1−xCaxMnO3 (LCM) family which possesses Mn3+ and Mn4+ ions and has paramagnetic insulator properties at high temperatures unlike paramagnetic metals having La1−xSrxMnO3 (LSM) compositions. Furthermore, LCMs are orthorhombic while LSMs are rhombohedral. We carried out measurements on LCMs in comparison with LSMs and ceria. Besides, we also tested the activity of La1−xMnO3 and La1−xSrxFeO3. We have studied the reduction of CO2 by thermogravimetric analysis and the reduction of H2O by employing an experimental set-up fabricated locally.
La1−xCaxMnO3 perovskites with x = 0.35, 0.5, 0.65 (designated here as LCM 35, LCM 50 and LCM 65) possess an orthorhombic structure (refer to ESI,† Fig. S1) with the lattice parameters decreasing slightly with the increasing Ca content. This is because, the content of the smaller Mn4+ (ionic radius 0.54 Å) increases over that of Mn3+ (ionic radius 0.65 Å) upon the substitution of trivalent La with bivalent Ca (refer to ESI,† Fig. S2).15 We calculated the atomic % of La, Ca and Mn present in LCMs (refer to ESI,† Table S1 and Fig. S3) by EDS analysis. The composition of LCMs was established from the background corrected La3d (∼836 eV), Ca2p (∼286 eV) and Mn2p (∼642 eV) XP survey scan spectra (refer to ESI,† Fig. S4, eqn (3)). The calculated ratios among La, Ca and Mn in LCM 35 and LCM 50 are 0.66/0.37/1 and 0.5/0.5/1, respectively (refer to ESI,† Table S2).
Thermogravimetric analysis of the three LCM compositions shows that upon heating above 1000 °C, O2 production increases with the Ca2+ or Mn4+ content. The mass loss due to reduction starts around 1000 °C and reaches a plateau after 1400 °C. The O2 released from LCM 35, LCM 50 and LCM 65 is 109, 316 and 653 μmol g−1, respectively (Fig. 1a). It is to be noted that the amount of O2 evolved by LCM 50 is greater than that evolved by La0.5Sr0.5MnO3 (LSM 50), the value in the case of the latter being 201 μmol g−1. Ceria evolves only 63 μmol g−1 of O2 (Fig. 1b). Thus, the amount of O2 evolved by LCM 50 is 1.6 times that of LSM 50 and five times that of ceria. We studied the splitting of CO2 by the LCM compounds. A reversible mass gain is observed in all the compounds during the CO2 injection at 1100 °C (Fig. 1a).
 | ||
| Fig. 1 Representative TGA of thermochemical CO2 splitting of (a) La1−xCaxMnO3 (x = 0.35, 0.5, 0.65); (b) La0.5Ca0.5MnO3 (LCM 50) and La0.5Sr0.5MnO3 (LSM 50) in comparison with CeO2. Reduction and oxidation temperatures are 1400 °C and 1100 °C, respectively. | ||
A detailed study was carried out by changing the calcium content systematically. CO produced by LCM 35, LCM 50 and LCM 65 is 175, 525 and 810 μmol g−1, respectively, over the same time scale. The CO production activity of LCM 50 is also about 1.6 times and 5 times more than that of LSM 50 and ceria, respectively (Table 1).
| O2 released (μmol g−1) | CO produced (μmol g−1) | |
|---|---|---|
| La0.65Sr0.35MnO3 | 100 | 137.5 |
| La0.5Sr0.5MnO3 | 201.3 | 325 |
| La0.65Ca0.35MnO3 | 109.4 | 175 |
| La0.5Ca0.5MnO3 | 315.6 | 525 |
| La0.35Ca0.65MnO3 | 653.1 | 350 |
| Ceria | 62.5 | 112 |
The kinetics of CO production appears to become sluggish at a high Ca content. The reoxidation peak of LCM 65 can be deconvoluted in two parts depending on its shape. The initial straight increase in the mass (394 μmol g−1) corresponds to the reaction-controlled regime followed by the slower constant gain (450 μmol g−1) corresponding to the diffusion-controlled regime (Fig. 1a). The fuel production activity of LCM 50 was tested during three successive cycles (Fig. 2) in the temperature range of 1400–1000 °C. This study has revealed reversibility in both O2 evolution and CO production, though the kinetics of reoxidation appears to become slow on repeated cycling. We have carried out the reduction of the LCM 50 compound at 1350 °C followed by reoxidation at 1000 °C (Fig. 2 and refer to ESI,† Table S3; cycle 3). We found that the amount of O2 evolved (237 μmol g−1) and CO produced (481 μmol g−1) over the temperature range of 1350–1000 °C is more than that evolved by the activity of Sr, Mn doped LaAlO3, although the experimental set-up in the two cases is not identical.14 As-synthesized LCM 50 is composed of few micron size particles as observed from FESEM images. No significant change in the morphology of LCM 50 was observed after TGA analysis (refer to ESI,† Fig. S5).
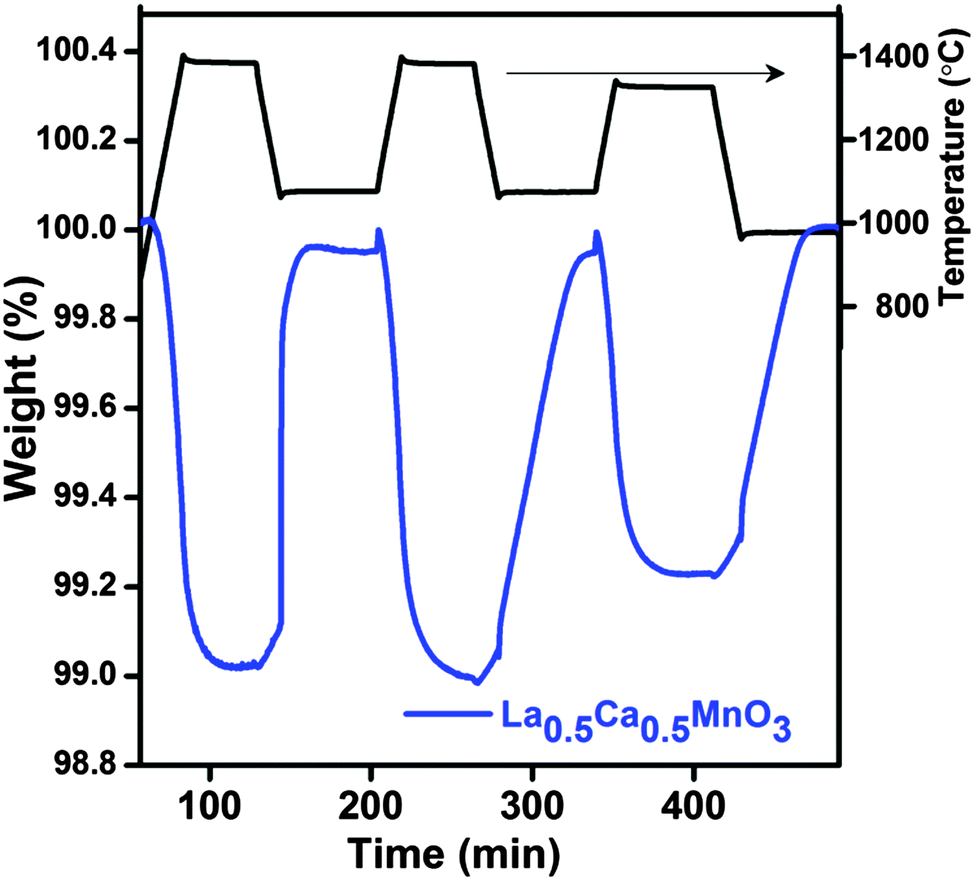 | ||
| Fig. 2 TGA of three consecutive thermochemical CO2 splitting cycles of La0.5Ca0.5MnO3 (LCM 50) performed in the temperature range of 1400–1000 °C. | ||
In order to understand the superior performance of LCM 50, we prepared a perovskite of the composition Sm0.5Sr0.5MnO3 (SmSM 50) and carried out TG analysis. SmSM 50 is also an orthorhombic perovskite (refer to ESI,† Fig. S6) with the tolerance factor (t = 0.982) comparable with that of orthorhombic LCM 50 (t = 0.985).16 Interestingly, both LCM 50 and SmSM 50 produce comparable amounts of O2 and CO during TG analysis in the temperature range of 1400–1100 °C, unlike LSM 50 (Fig. 3). This indicates that the orthorhombic perovskites show superior activity than the rhombohedral perovskites. This could be due to the distortion in octahedral MnO6 in orthorhombic perovskites compared to the rhombohedral one.15,16
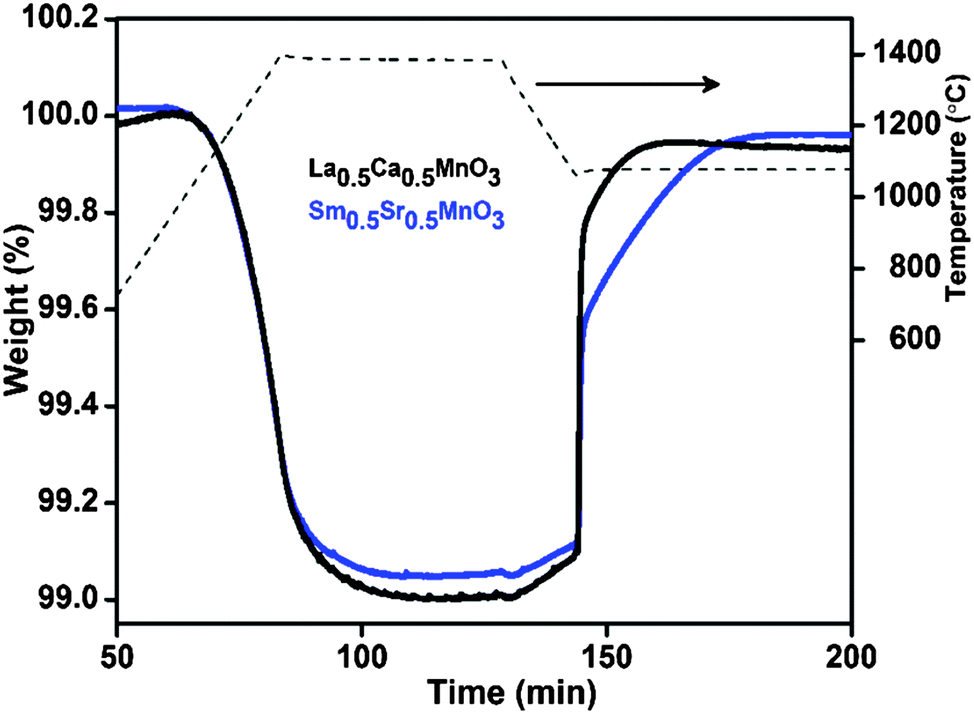 | ||
| Fig. 3 Representative TGA of thermochemical CO2 splitting of Sm0.5Sr0.5MnO3 (SmSM 50) in comparison with that of La0.5Ca0.5MnO3 (LCM 50). Reduction and oxidation temperatures are 1400 °C and 1100 °C, respectively. | ||
We investigated the splitting of water by employing the experimental set-up shown in Fig. 4 (refer to ESI,† details are provided in the reactivity tests part). The H2O splitting activity of LCM 50 was tested in comparison with that of LSM 50 with the help of the set-up presented in Fig. 4. The sample was placed inside the tubular furnace and heated up to 1400 °C under a constant N2 flow.
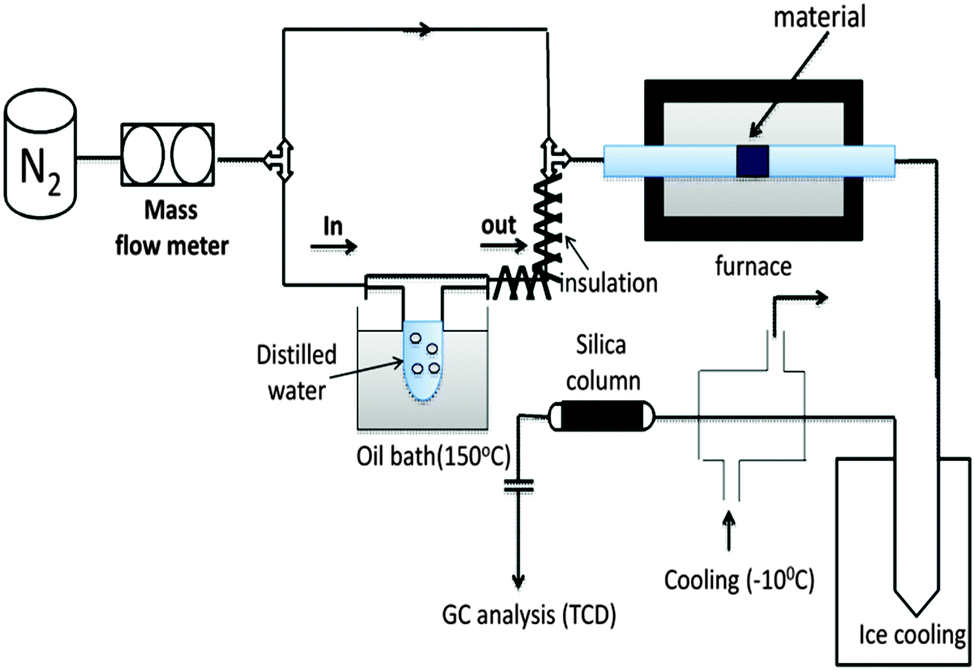 | ||
| Fig. 4 Experimental set-up for thermochemical water splitting. | ||
As shown in Fig. 5a, O2 evolution starts at around 1000 °C, and approaches the plateau region at 1400 °C. Both LCM 50 and LSM 50 show the complete O2 production within 30 min after reaching 1400 °C. The amount of O2 produced in the case of LCM 50 and LSM 50 is 272 and 193 μmol g−1, respectively, close to the values obtained from TGA measurements discussed earlier. The H2O splitting activity was tested by introducing steam in a continuous N2 flow. The splitting temperature of H2O is 1000 °C. Fig. 5b shows the rate of H2 production. Production of H2 starts immediately after the entry of H2O in the gas stream. The amount of H2 produced in a span of 100 min is 407 and 308 μmol g−1 for LCM 50 and LSM 50, respectively. Notably, H2 production is not complete even after 100 min and exhibits an extended tail over a long duration. The slow kinetics of H2 evolution can arise due to factors such as a decrease in chemical diffusivity, changes in the surface reaction constant, steam concentrations as well as the intrinsic thermodynamic driving forces.13
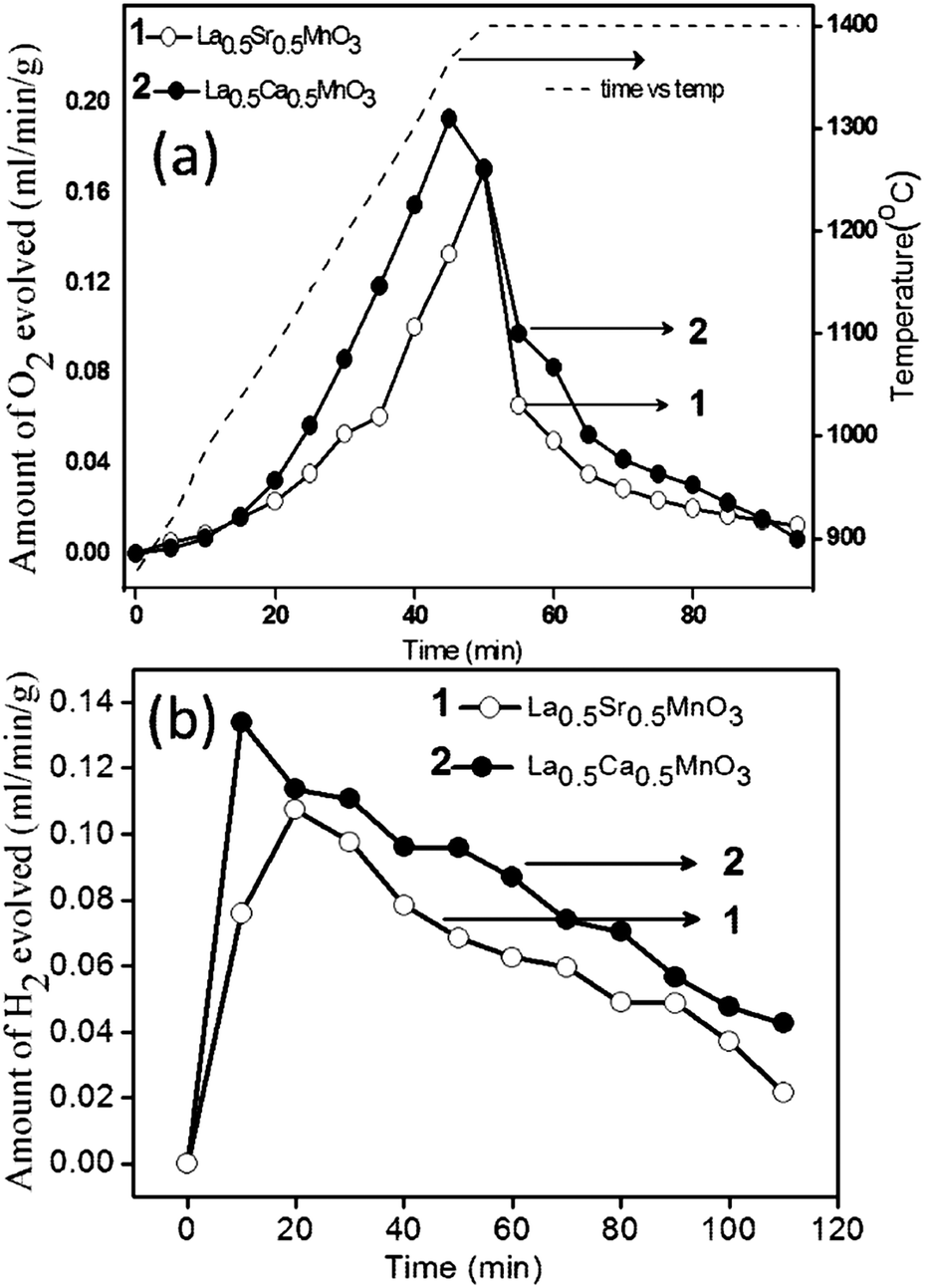 | ||
| Fig. 5 (a) Oxygen and (b) hydrogen evolution profiles of La0.5A0.5MnO3 (A = Sr, Ca). Reduction and oxidation temperatures are 1400 °C and 1000 °C, respectively. | ||
We tested the CO2 splitting activity of La1−xMnO3 (x = 0.1, 0.2) as it contains a high concentration of Mn4+ (Fig. 6a).17 But phase transformation from the rhombohedral to the orthorhombic structure takes place under the high temperature reaction conditions (refer to ESI,† Fig. S7). Therefore, La1−xMnO3 cannot be used for this purpose. We also investigated the activity of La1−xSrxFeO3 (x = 0.3) inspired by the literature data available for its H2 production activity in membrane reactors.12,18 The O2 evolution of La0.7Sr0.3FeO3 starts at a low temperature, with the total amount of O2 produced being 360 μmol g−1 at 1300 °C (Fig. 6b). Unfortunately, the CO production yield is low (20% of reduction yield).
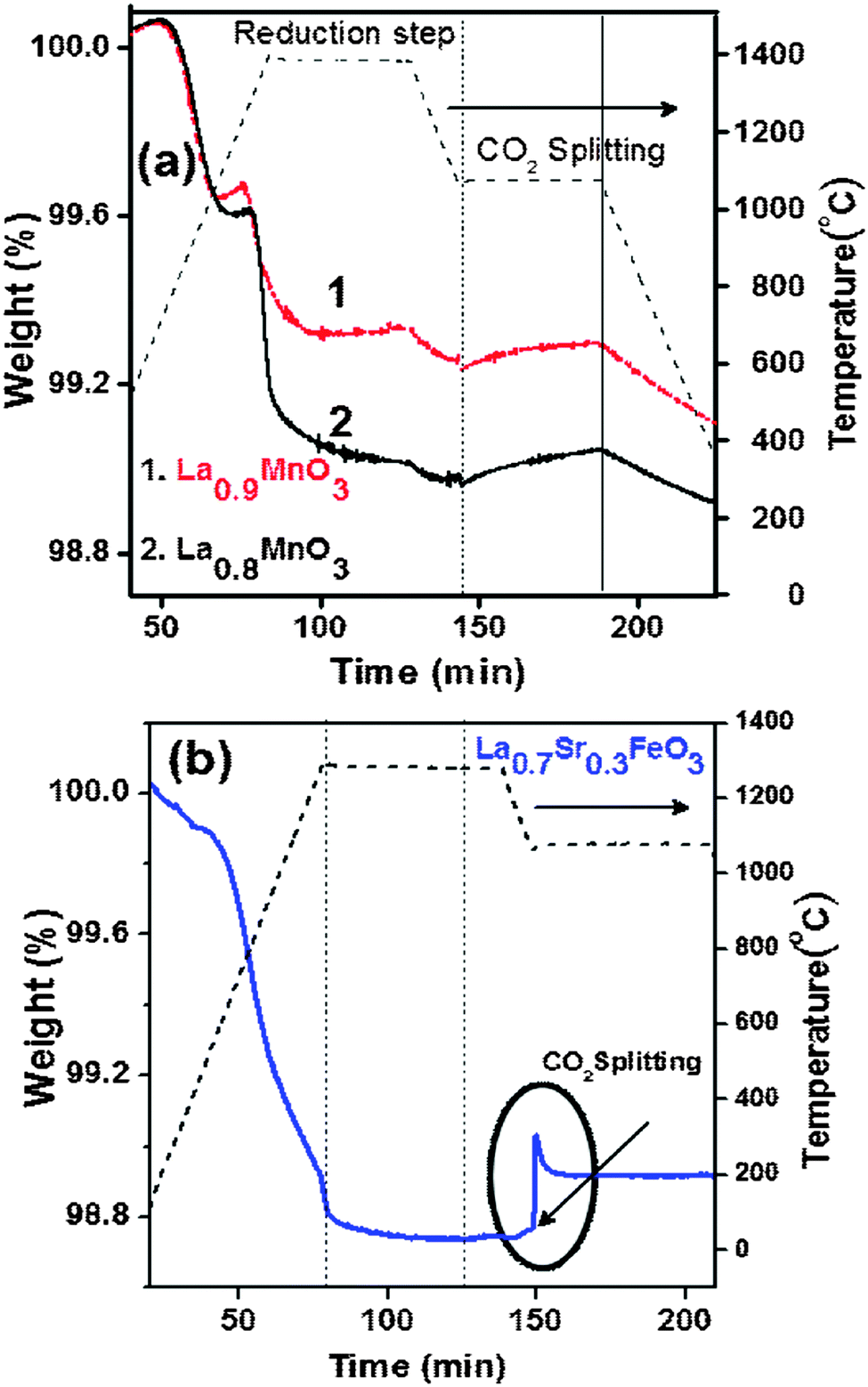 | ||
| Fig. 6 Representative TGA of thermochemical CO2 splitting of (a) La1−xMnO3 (x = 0.1, 0.2; performed in the temperature range of 1400–1100 °C) and (b) La0.7Sr0.3FeO3 (performed in the temperature range of 1300–1100 °C). | ||
Besides having different structures, LCM and LSM compositions possess Mn3+ and Mn4+ ions in different electronic states. While two Mn ions with distinctly different charges are present in LCMs, there is fast electron transfer from Mn3+ to Mn4+ in LSMs giving rise to magnetism and mettalicity.19
Conclusions
The present results show that La1−xCaxMnO3 is an excellent material for the thermochemical splitting of both H2O and CO2. The performance of La1−xCaxMnO3 is superior to that of CeO2 as well as that of the corresponding La1−xSrxMnO3 composition. The amount of O2 and CO evolved by La0.5Ca0.5MnO3 is 1.6 times and 5 times more than that evolved by La0.5Sr0.5MnO3 and the current state-of-the-art material ceria, respectively. Our investigation also indicates the superior performance of orthorhombic perovskite over the rhombohedral one in thermochemical fuel production. In conclusion, it would be profitable to attempt high temperature fuel production using LCM perovskites.Acknowledgements
SD thanks CSIR for a fellowship. BSN acknowledges DST for a fellowship.Notes and references
- E. Bilgen and C. Bilgen, Int. J. Hydrogen Energy, 1982, 7, 637 CrossRef CAS.
- S. Abanades, P. Charvin, F. Lemont and G. Flamant, Int. J. Hydrogen Energy, 2008, 33, 6021 CrossRef CAS PubMed.
- A. Steinfeld, Int. J. Hydrogen Energy, 2002, 27, 611 CrossRef CAS.
- Y. Tamaura, A. Steinfeld, P. Kuhn and K. Ehrensberger, Energy, 1995, 20, 325 CrossRef CAS.
- T. Kodama, N. Gokon and R. Yamamoto, Sol. Energy, 2008, 82, 73 CrossRef CAS PubMed.
- H. Kaneko, T. Kodama, N. Gokon, Y. Tamaura, K. Lovegrove and A. Luzzi, Sol. Energy, 2004, 76, 317 CrossRef CAS PubMed.
- Y. Tamaura, N. Kojima, N. Hasegawa, M. Inoue, R. Uehara, N. Gokon and H. Kaneko, Int. J. Hydrogen Energy, 2001, 26, 917 CrossRef CAS.
- W. C. Chueh, C. Falter, M. Abbott, D. Scipio, P. Furler, S. M. Haile and A. Steinfeld, Science, 2010, 330, 1797 CrossRef CAS PubMed; S. G. Rudisill, L. J. Venstrom, N. D. Petkovich, T. Quan, N. Hein, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2013, 117, 1692 Search PubMed.
- H. Kaneko, T. Miura, H. Ishihara, S. Taku, T. Yokoyama, H. Nakajima and Y. Tamaura, Energy, 2007, 32, 656 CrossRef CAS PubMed; A. L. Gal and S. Abanades, J. Phys. Chem. C, 2012, 116, 13516 Search PubMed; M. Kang, X. Wu, J. Zhang, N. Zhao, W. Wei and Y. Sun, RSC Adv., 2014, 4, 5583 RSC.
- A. L. Gal, S. Abanades, N. Bion, T. L. Mercier and V. Harle, Energy Fuels, 2013, 27, 6068 CrossRef CAS; A. L. Gal, S. Abanades and G. Flamant, Energy Fuels, 2011, 25, 4836 CrossRef; J. R. Scheffe, R. Jacot, G. R. Patzke and A. Steinfeld, J. Phys. Chem. C, 2013, 117, 24104 Search PubMed; S. Abanades and G. Flamant, Sol. Energy, 2006, 80, 1611 CrossRef PubMed.
- J. R. Scheffe, D. Weibel and A. Steinfeld, Energy Fuels, 2013, 27, 4250 CrossRef CAS.
- A. Demont, S. Abanades and E. Beche, J. Phys. Chem. C, 2014, 118(24), 12682 CAS.
- C. K. Yang, Y. Yamazaki, A. Aydin and S. M. Haile, J. Mater. Chem. A, 2014, 2, 13612 CAS.
- A. H. McDaniel, E. C. Miller, D. Arifin, A. Ambrosini, E. N. Coker, R. O. Hayre, W. C. Chueh and J. H. Tong, Energy Environ. Sci., 2013, 6, 2424 CAS.
- S. Faaland, K. D. Knudsen, M. A. Einarsrud, L. Rormark, R. Hoiera and T. Grande, J. Solid State Chem., 1998, 140, 320 CrossRef CAS.
- P. M. Woodward, T. Vogt, D. E. Cox, A. Arulraj, C. N. R. Rao, P. Karen and A. K. Cheetham, Chem. Mater., 1998, 10, 3652 CrossRef CAS.
- A. Arulraj, R. Mahesh, G. N. Subbanna, R. Mahendiran, A. K. Raychaudhuri and C. N. R. Rao, J. Solid State Chem., 1996, 127, 87 CrossRef CAS.
- L. Nalbandian, A. Evdou and V. Zaspalis, Int. J. Hydrogen Energy, 2009, 34, 7162 CrossRef CAS PubMed.
- C. N. R. Rao, J. Phys. Chem. B, 2000, 104, 5877 CrossRef CAS; V. B. Shenoy and C. N. R. Rao, Philos. Trans. R. Soc., A, 2008, 366, 63 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of perovskite synthesis, reactivity testing methods and additional figures. See DOI: 10.1039/c4cp04578e |
| This journal is © the Owner Societies 2015 |