
Nanostructure of [Li(G4)] TFSI and [Li(G4)] NO3 solvate ionic liquids at HOPG and Au(111) electrode interfaces as a function of potential
Ben
McLean
a,
Hua
Li
a,
Ryan
Stefanovic
a,
Ross J.
Wood
a,
Grant B.
Webber
a,
Kazuhide
Ueno
b,
Masayoshi
Watanabe
b,
Gregory G.
Warr
c,
Alister
Page
a and
Rob
Atkin
*a
aDiscipline of Chemistry, The University of Newcastle, Callaghan, Australia. E-mail: Rob.Atkin@newcastle.edu.au
bDepartment of Chemistry and Biotechnology, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
cSchool of Chemistry, The University of Sydney, Australia
First published on 31st October 2014
Abstract
Atomic force microscopy (AFM) force measurements have been used to study the solvate ionic liquid (IL) double layer nanostructure at highly ordered pyrolytic graphite (HOPG) and Au(111) electrode surfaces as a function of potential. Two solvate ILs are investigated, [Li(G4)] TFSI and [Li(G4)] NO3. Normal force versus apparent separation data indicates that both solvate ILs adopt a multilayered morphology at the electrode interface, similar to conventional ILs. Calculations of adsorption free energies indicate that at 0 V the ion layer in contact with the electrode surface is enriched in the more surface active cations. When a positive or negative surface bias is applied, the concentration of counterions in the innermost layer increases, and higher push-through forces are required to displace near surface layers, indicating a stronger interfacial structure. Generally, [Li(G4)] TFSI has a better defined structure than [Li(G4)] NO3 on both electrode surfaces due to stronger cohesive interactions within layers. Interfacial structure is also better defined for both solvate ILs on HOPG than Au(111) due to the greater surface roughness of Au(111). Further, at all negative potentials on both surfaces, a small final step is observed, consistent with either compression of the complex cation adsorbed structure or desolvation of the glyme from the Li+.
Introduction
Ionic liquids (ILs) are salts with melting points below 100 °C.1 ILs have attracted widespread scientific interest for electrochemical applications due to their remarkable physical properties such as negligible vapour pressure, high ionic conductivity, and non-flammability.2–6 Recently, equimolar mixtures of glymes (oligoethers of the form CH3(OCH2CH2)nOCH3, abbreviated Gn) with alkali metal salts have been classified as “solvate ILs” using characteristics proposed by Angell et al.7,8 Solvate ILs, particularly those containing a Li salt, have great potential for applications as electrolytes in Li secondary batteries and capacitors due to the high Li+ concentration and high Li transference number.9,10In solvate ILs, partial donation of lone pair electrons from the glyme molecule to the alkali metal ion leads to the formation of a complex cation. Whether the resulting liquid is a “good” or “poor” solvate IL8 depends on the relative affinities of the glyme ligand and the anion for the metal ion. For example, mixing lithium bis(trifluoromethylsulfonyl)imide (LiTFSI) with tetraglyme (G4) produces [Li(G4)] TFSI. [Li(G4)] TFSI is a good solvate IL because the weakly basic TFSI− anion does not compete effectively for Li+, so stable complex cations ([Li(G4)]+) result. Conversely, mixing lithium nitrate (LiNO3) with G4 results in a mixture of [Li(G4)]+, NO3−, Li+ and free glyme, in equilibrium, due to the strongly basic NO3− anion competing to solvate Li+.11 The presence of free glyme and LiNO3 salt means that this liquid is classified as a poor solvate IL.8
Whether a solvate IL is good or poor is revealed by the diffusion coefficients of the Li+ and the glyme. Pulsed-field gradient NMR11 has been used to show that for [Li(G4)] TFSI the ratio of the glyme and Li+ diffusion coefficients is ∼1. This means that the glyme and the Li+ diffuse together as a stable, long lived complex whereas in [Li(G4)] NO3 the ratio of the diffusion coefficients is ∼1.5, indicating the presence of free glyme molecules.8,11
The bulk structure of conventional ILs has been extensively examined, with many shown to have distinct sponge-like bulk nanostructures consisting of charged and non-polar domains percolating throughout the liquid.12,13 Near a solid surface, the isotropic symmetry of the bulk structure is broken and a flatter, but related, nanostructure results. To date, only one paper has studied the ion arrangements of solvate ILs in the bulk,14 and the structure of solvate ILs near a solid electrode surface is completely unexplored. This means that the electrical double layer structure of solvate ILs is poorly understood despite its key role in electrochemical applications. Understanding the ion arrangements near the electrode surface will pave the way for optimisation of the solvate IL electrical double layer in the same way as for molecular solvents15–18 and conventional ILs.19–25
The electrical double layer structure has a significant impact on the performance of electronic devices such as batteries and capacitors.26 The double layer structure of conventional ionic liquids has been extensively studied by a range of techniques including atomic force microscopy (AFM) (force curves19,20,22,27–35 and images19,20,36–38), scanning tunnelling microscopy (STM),19,27,39–41 X-ray reflectivity,42,43 electrochemical impedance spectroscopy (EIS),44–47 and molecular dynamics simulations,43,48,49 amongst others.50,51
Distinct regions can be identified at the IL–solid interface.52 The interfacial (innermost) layer is composed of ions that are in contact with the solid surface. IL structure, or ordering of ions, is most pronounced in this interfacial layer. Next to the innermost layer are near-surface liquid layers that have nanostructure different to that of the bulk liquid. These layers are referred to as the transition zone. Through the transition zone, which is typically a few nanometres across, the pronounced interfacial layer structure decays into the bulk morphology.12,13 Over a series of papers19,20,28,29 we have demonstrated that, when the interfacial IL structure is better defined, more steps often with higher push through forces, are present in AFM force distance curves, due to higher liquid cohesion within layers. Other authors have reported similar results from AFM force curves21,35,53 and other techniques.54–56 The level of definition between near surface layers, often referred to as the strength of the structure, is a consequence of the level of ion enrichment in the interfacial layer, as this templates structure in subsequent layers, and the surface roughness; smooth surfaces produce better defined IL layers.20
The ion composition of the interfacial layer is largely determined by the properties of the surface. For negatively charged mineral oxide surfaces like mica and silica, the IL cation neutralises surface charge sites and is therefore enriched in the interfacial layer.20,22,29,34 For electrode surfaces, the composition of the interfacial layer is determined by the surface properties at open circuit potential (OCP), and (predominantly) by the surface polarity when a potential is applied.28 For example, for Au(111) at OCP, which is typically about −0.2 V,28 the interfacial layer is slightly cation rich. The interfacial layer becomes enriched in anions at positive potentials and in cations at negative potentials. For highly ordered pyrolytic graphite (HOPG), at OCP the interfacial layer is enriched in cations due to solvophobic attractions between cation alkyl chains and the surface.33,57 However, as with Au(111) electrodes, when a positive potential is applied the interfacial layer becomes enriched in anions.
In this paper AFM force curves are used to probe the structures of [Li(G4)] TFSI, a good solvate IL, and [Li(G4)] NO3, a poor solvate IL, at HOPG and Au(111) surfaces as a function of applied potential. The structures and key physical properties of these liquids are shown in Table 1. To our knowledge, this is the first structural study of solvate IL–electrode interfaces. Force curve analysis enables the ion arrangements near the substrate to be ascertained. Clear structural differences between good and poor solvate ILs are elucidated for both the carbonaceous and metal electrodes as a function of potential.
| Species | Structure | η (mPa s) | ρ (g cm−3) | σ (mS cm−1) |
|---|---|---|---|---|
| [Li(G4)] TFSI |

|
81 | 1.40 | 1.6 |
| [Li(G4)] NO3 |
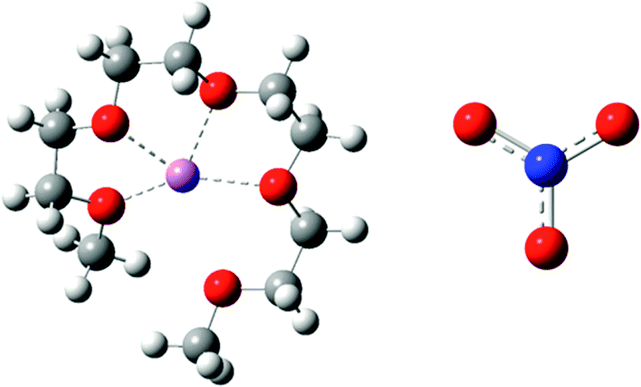
|
172 | 1.17 | 0.26 |
Materials and methods
Tetraglyme (99.99%), LiNO3 (99.995%) and LiTFSI (99.995%) were purchased from Sigma Aldrich. Tetraglyme was stored in a desiccator in a sealed bottle and the Li salts were dried and stored in a vacuum desiccator at an elevated temperature prior to use. The solvate ILs were synthesised by mixing stoichiometric quantities of Li salt and glyme in a sealed bottle at 80 °C for 4–6 hours. The resulting solvate ILs were stored in a sealed bottle within a desiccator. Prior to use, the solvate ILs were dried under vacuum at 40 °C and were found to have less than 1 ppm water by Karl Fischer titration.All AFM normal force measurements were performed using a Multimode 9 AFM (Bruker Instruments, USA) with a vertical engage E-scanner in contact mode. Four silicon cantilevers with sharp tips were used throughout the study and were cleaned immediately prior to use by rinsing with Milli-Q water and ethanol and irradiating with ultraviolet light for at least 20 minutes. The spring constant of the AFM tips was calculated using the thermal noise method.58
The AFM electrochemical cell was assembled as described previously.28 HOPG surfaces were freshly cleaved before each experiment, while Au(111) surfaces were rinsed with Milli-Q water and irradiated with ultraviolet light for 30 minutes. These surfaces acted as the working electrode and the solid substrate for force measurements during their respective experiments. A thin cylindrical strip of Cu metal and 0.25 mm Pt wire were used as the counter and pseudo-reference electrodes, respectively. Once a potential was applied, the system was allowed to reach equilibrium at that potential for 30 minutes. Cyclic voltammograms were collected in situ to ensure measurements were within each solvate IL's electrochemical window.
Normal force data was collected with ramp size of 30 nm and ramp rate of 0.1 Hz. The measured cantilever deflection as a function of piezo (surface) movement was converted to normal force vs. apparent separation data through standard methods.59 As the measurements and experiments were repeated, the features of the normal forces remained qualitatively similar. More than 50 force curves were collected for all experimental conditions; a force curve that most closely resembles the typical data is presented.
The interaction between [Li(G4)] TFSI and [Li(G4)] NO3 and HOPG was analysed using density functional theory (DFT). Truhlar's M06-2X functional60 in conjunction with a 6-31G(d) basis set was employed. The accuracy of the M06-2X functional in the context of conventional ILs, both in the bulk and adsorbed on HOPG has been demonstrated previously,61,62 and this is due in part to its description of long-range/non-covalent interactions. Adsorption free energies of component moieties on a model HOPG surface have been calculated. Adsorption free energy ΔGads of a species X is defined here as:
| ΔGads = ΔG(X + HOPG) − [ΔG(X) + ΔG(HOPG)] | (1) |
Results and discussion
Normal force profiles for [Li(G4)] TFSI and [Li(G4)] NO3 confined between an AFM tip and an HOPG or Au(111) electrode surface were measured at varying applied potentials within electrochemical window of the solvate ILs. These data are presented in Fig. 1. | ||
| Fig. 1 Typical normal forces versus apparent separation data for [Li(G4)] TFSI and [Li(G4)] NO3 on HOPG (A–E, K–O) and Au(111) (F–J, P–T) as a function of potential. | ||
The force data presented for each system was selected by determining the average number of steps and the average push-through forces for each step for more than 50 force curves. A representative force curve was then selected that most closely matched the average values. We favour presenting a single force curve over an average,21 because averages tend to obscure features that, although consistent, occur at slightly different separations and small normal forces, such as the small steps (oscillations) at wide separations at 0 V in Fig. 1.
The steps in these AFM force curves result from interfacial liquid layers being expelled from the space between the substrate and the AFM tip. This “push-through” force required to rupture the layer is a function of the strength of cohesive interactions between the ions within a layer. Inferences about the composition of these layers can be made by comparing the step width to the expected dimension of the solvate IL ion pair, the individual cations and anions, and the lithium and glyme molecule that form the complex cation. Steps are apparent in the normal force-distance profiles for all systems at all potentials. In general, forces and step widths are higher for the good solvate IL than the poor solvate IL, and on HOPG compared to Au(111). The dimensions of the [Li(G4)] TFSI and [Li(G4)] NO3 ion pairs were calculated using the molecular weight and the density of the solvate IL, assuming a cubic packing geometry:37
 | (2) |
Preliminary SAXS/WAXS measurements of [Li(G4)] TFSI and [Li(G4)] NO3 show that the “pre-peak”12,13,71 that is indicative of bulk amphiphilic nanostructure in conventional ILs is absent in these systems. This suggests that these solvate ILs do not have any such long-range bulk nanostructures, and can be treated as simple molten salts,72,73 consistent with neither the cation nor anion having amphiphilic structure. A more complete picture will emerge from our recently-completed, multiply isotopically-substituted neutron diffraction experiments for [Li(G4)] TFSI and [Li(G4)] NO3, to be described in detail in a forthcoming article. This suggests that the observed steps in these AFM force curves should correspond to the dimensions of individual ionic species or ion pairs, and not to any larger-scale structures.20,56
In any AFM experiment the absolute separation between the tip and the surface is unknown. For force curves the zero position is denoted by the region in the raw force data when the tip and the surface are in constant compliance. This does not mean that the tip is necessarily in contact with the surface but rather that the tip is pushing against matter that it cannot displace. Hence the distance axis is referred to as the “apparent separation.” Data obtained for a wide variety of ILs on different substrates has consistently revealed that the zero of apparent separation occurs when the AFM tip is pushing against an ion layer bound to the surface that it cannot displace.28,32,33,57 True contact only occurs in cases such as strongly sterically hindered ion layers.34
Fig. 1 shows typical normal force versus apparent separation data of [Li(G4)] TFSI on HOPG as a function of electrode potential. At 0 V, when the tip and surface are more than 1 nm apart the measured force is 0 nN as the tip moves through the bulk liquid. At an apparent separation of 0.84 nm, the tip encounters a liquid layer and pushes against it. The force increases sharply while the separation decreases slightly, indicating slight compression of the layer, until a force of ∼2 nN is reached. At this point the tip ruptures the layer and jumps by 0.37 nm into contact with the layer next nearest the surface at a separation of 0.47 nm. Both the compression of, and the force required to rupture, the layer nearest the surface is much higher for this layer. These steps are too narrow to be caused by expulsion of ion pairs. However, the width of the outer (0.37 nm) and inner (0.47 nm) layers are consistent with the diameters of the complex cation and TFSI− anion, respectively (Table 2), indicating the formation of discrete sub-layers on HOPG.
From the force curve alone it is impossible to discern whether at 0 apparent separation the AFM tip is in contact with the HOPG surface or a layer of strongly bound ions. To resolve this issue, DFT was used to determine the adsorption free energies of solvate IL components to the HOPG substrate, in the absence of potential (cf.Fig. 2 and Table 3). These calculations reveal that the complex cation binds to the HOPG surface with an adsorption free energy of −46.9 kJ mol−1 at 298 K (primarily due to dispersive interactions). Compared to the complex cation, TFSI− adsorbs very weakly to HOPG, with ΔGads = −0.8 kJ mol−1.57 ΔGads for free Li+ on HOPG is −190.4 kJ mol−1, which suggests that the strong Li+–HOPG cation–π interaction is muted in the [Li(G4)]+ complex. This is consistent with the conformational change between the free and complexed glyme on the HOPG surface (cf.Fig. 2). Despite the high affinity of Li+ for the graphite surface, it is thermodynamically unlikely that appreciable concentrations of free Li+ ions are present in the surface-adsorbed ion layer, since the [Li(G4)]+ binding free energy is much greater at −432.3 kJ mol−1 (here the [Li(G4)]+ binding free energy has been calculated in a manner analogous to eqn (1), i.e. ΔG([Li(G4)+]) − [ΔG(Li+) + ΔG(G4)]). Thus at an apparent separation of 0 nm we expect that the tip is pushing against a layer of complex cations bound to the HOPG surface that it cannot displace. The next layer is expected to be anion rich, which is consistent with the step width in the force curve. Overall there are three layers detected at the [Li(G4)] TFSI–HOPG interface: a complex cation rich layer strongly bound to the surface, followed by an anion rich layer and then another cation rich layer. The decrease in push through force with distance indicates the level of enrichment is decreasing.
 | ||
| Fig. 2 Structure and adsorption free energies of G4 glyme on HOPG (a) with and (b) without complexed Li+ ion. G4 complexes Li+ with 4 of its 5 O atoms, resulting in the G4 only partially adsorbing to the graphite surface. The 4 Li+–O bonds (dashed lines, clockwise from top) are 2.00, 2.08, 2.04 and 2.08 Å, respectively. The presence of Li+ contracts the G4 by 2.2 Å and enhances adsorption significantly. Grey, red, white and purple spheres represent C, O, H and Li atoms, respectively. | ||
| [LiG4]+ | TFSI−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 57 |
NO3− | Li+ | G4 | |||||
|---|---|---|---|---|---|---|---|---|---|
| ΔGads | ΔEads | ΔGads | ΔEads | ΔGads | ΔEads | ΔGads | ΔEads | ΔGads | ΔEads |
| −46.9 | −104.5 | −0.8 | −13.0 | +25.7 | −23.7 | −190.4 | −235.9 | +8.9 | −61.0 |
It should be noted that the free energy values determined for the adsorption of these ions to the graphite surface are markedly lower than the 0 K adsorption energies, which are also shown in Table 3. This highlights the importance of including thermal and entropic effects in calculations of binding affinities for ions to solid surfaces, particularly for species with molecular flexibility.
When a bias is applied to the surface (either positive or negative) the features of the force curves change markedly. A greater number of steps are present, and the step size also increases to a value consistent with expulsion of layers of solvate IL ion pairs, except for a small step nearest the surface at negative potentials. Steps in the force curve are detected further from the surface, consistent not only with more layers, but more pronounced layer structure. This is consistent with results for a number of conventional ILs at electrode interfaces,21,28,32,55,74 where applying a potential led to greater enrichment of one ion over the other in the interfacial layer. As the composition of the innermost layer became more uniform in these ILs, it more effectively templated subsequent near surface layers which increased the thickness of the transition zone between the surface and the bulk liquid structure.
When a positive bias is applied to the HOPG surface (Fig. 1A and B) the complex cation will be electrostatically repelled from, and the anion attracted to, the surface. As the electrostatic attraction between the anion and the surface will be markedly stronger than the dispersive attraction between the complex cation and the surface minus the electrostatic repulsion, the innermost layer will consequently become enriched in anions. Thus for positive potentials at 0 apparent separation the tip is likely to be pushing against an anion-rich layer that it cannot displace.
At both +0.5 V and +1.0 V, the steps in the force curve are ∼0.85 nm wide, consistent with the ion pair diameter. On electrostatic grounds, it is not obvious why cation and anion sub-layers should be present at 0 V but ion pair layers are present at the higher potential, as the adsorption energies are such that the surface layer should consist only of cations even at 0 V. At 0 V the cations are adsorbed and generate an “inner Helmholtz”-like plane of effectively fixed charge, obliging (allowing) the IL to form sub-layers. At negative applied potentials the cations neutralise the charge in the electrode so that the effective potential adjacent to the adsorbed cations is smaller in magnitude and formation of sub-layers harder to achieve. The same argument would apply to positive potentials, where the anion layer reduces potential.
At +0.5 V the highly enriched, and relatively smooth, innermost anion layer more effectively templates subsequent near surface structure. This means that near surface layers are better defined and cohesive attractions within a layer are stronger. Stronger cohesive interactions mean that when the liquid is compressed between the tip and the surface complex cations and anions are more likely to leave as an ion pairs than separately.
When the potential is increased to +1 V the innermost layer becomes more strongly enriched in anions, and thus more effectively templates near surface layers. This is consistent with the higher push-through force for the near surface layer. Similar to +0.5 V, the steps in the force curve are consistent with the ion pair dimension within error. The origin of the increased width and compressibility of this layer is less clear. One possibility is that rather than the complex cations and anions being relatively well mixed within a layer (consistent with the cubic packing structure) they adopt a better-defined structure with ions arranged in fashion that maximizes charge density near the surface. Alternatively, the higher potential could decrease the mobility of near surface ions, leading to an increase in the liquid viscosity immediately adjacent to the surface. The increase in normal force with separation for the step nearest the surface would then be fluid dynamic in origin.
The striking difference between the data obtained at positive potentials and negative potentials (Fig. 1A, B and D, E) is that the last detectable layer at negative potentials is only ∼0.25 nm thick. Moon et al. have recently shown that for the [Li(G3)] TFSI–HOPG interface Li+ is (reversibly) desolvated from the complex cations and intercalated between graphene sheets via step edges.75 This raises two possibilities that could explain the small step in these force curves. The first is that when the complex cation rich innermost layer is compressed by the AFM tip the negative surface potential strongly binds the Li+, holding it in place while simultaneously weakening Li+–glyme bonds. This could allow the glyme molecule is displaced. Calculations predict the glyme would desorb from the surface as ΔGads = +8.9 kJ mol−1 for free (molecular) G4. This would mean that at 0 separation the tip is pushing against a layer of Li+ bound to the HOPG surface. Alternatively, the glyme remains bound to the Li+, but compression of the portion of the glyme molecule that naturally extends away from the surface (i.e. out of the plane of the lithium ion in Fig. 2A) produces the small step in the force curve. In either case, the width of subsequent steps is approximately equal to the packing dimension of the ion pair, as per the results obtained at positive potentials. The underlying reason is most likely the same: the enriched, innermost layer effectively templates subsequent near surface layers and the resulting strong cohesive attractions between ions leads to cations and anions being expelled as ion pairs.
Fig. 1F–J shows typical force data for the [Li(G4)] TFSI–Au(111) electrode interface as a function of potential. Comparison of this data with that obtained for HOPG reveals the influence of the electrode type on solvate IL interfacial structure. In general, forces are lower on Au(111) compared to HOPG, which is attributed to increased surface roughness.19 For both molecular solvents76 and conventional ILs19,20 at a solid interface, greater surface roughness decreases interfacial nanostructure.
At 0 V the width of the steps is identical within error for [Li(G4)] TFSI at the Au(111) and HOPG interfaces. However, the magnitude of the push-through forces is lower on Au(111) due to the higher surface roughness disrupting the formation of interfacial layers. DFT calculations cannot be completed for the Au(111) system because the required harmonic vibration frequencies means that computation times are impractical. However, the open circuit potential for both liquids on Au(111) is slightly negative (as per graphite) which suggests that the adsorption affinity of the cation for Au(111) is higher than the anion. When combined, these results suggest layers occur in the same order on the Au(111) and HOPG surfaces at 0 V, but are less well defined at the rougher metal electrode. When the potential is increased to +0.5 V and +1 V the force required to push-through the final detectable layer increases, and there is some evidence for an additional layer at wide separations. Both results are consistent with stronger interfacial structure at higher potentials. The push-through forces on Au(111) are smaller than for HOPG at the same potential, meaning that interfacial structure is less pronounced.
At negative Au(111) surface biases, a thin ∼0.21 nm step is once again measured, similar to that found at the HOPG interface at the same potentials. Here the complex cation will be electrostatically attracted to the Au(111) surface and, as per the HOPG electrode, the narrow interfacial layer is due either compression of the glyme in the complex cation, or the pressure applied by the AFM tip desolvating glyme molecules from Li+. In this case the Li+ ions would either remain adsorbed to the Au(111) surface, or alloyed into it. Alloying requires an electrochemical reaction that would produce a peak in the cyclic voltammogram. The absence of such a peak at negative potentials reveals that Li+ is adsorbed rather than alloyed.
At −0.5 V all subsequent steps are less than the ion pair dimension indicating relatively weak interfacial ordering compared to the same potential on HOPG, attributed to increased surface roughness. However, when the potential is set to −1 V the width of the step adjacent to the thin layer increases to a value consistent with an ion pair, indicative of stronger near surface structure. That is, the order induced by the elevated negative bias is sufficient to overcome the disruptive effect of roughness, and the interfacial structure (and force data) is similar to that on HOPG at −1 V.
Fig. 1K–T show force data for [Li(G4)] NO3 on HOPG and Au(111) substrates, respectively. [Li(G4)] NO3 is a poor solvate IL which means that a significant population of free glyme molecules, as well as LiNO3 salt, are present in addition to the complex cation and anion. This liquid is thus more complex than the good solvate IL examined in Fig. 1A–J, which makes assigning features in the force curve more difficult. Nonetheless, general features can be commented upon, particularly given that it may reasonably be expected that the complex cation and anion will be more strongly attracted to the electrode surface than neutral species, especially when a potential is applied.
Force data for the [Li(G4)] NO3–HOPG interface at 0 V is presented in Fig. 1 M. It is similar in form to that obtained for the [Li(G4)] TFSI–HOPG interface, except that the separation at which the force first begins to increase is reduced to 0.74 nm. This is a consequence of the packing diameter of NO3− being 0.1 nm less than TFSI−. DFT calculations predict that NO3− will desorb from the HOPG surface at 298 K due to entropic and thermal corrections, with ΔGads = +25.7 kJ mol−1, cf.Table 3. This is significantly less than the complex cation ΔGads, −46.9 kJ mol−1. Based on ΔGads, there is a much greater chance of free Li+ ions being adsorbed to the HOPG substrate in [Li(G4)] NO3, compared to [Li(G4)] TFSI, as it is a poor solvate IL. This means that the innermost layer at 0 V (that the AFM tip cannot displace) will be cation rich, followed by the measured anion rich and cation rich layers. The force required to rupture corresponding layers is reduced for [Li(G4)] NO3 compared to [Li(G4)] TFSI (compare Fig. 1C and M) despite the viscosity of the [Li(G4)] NO3 being higher. This is likely a consequence of the presence of neutral species in the interfacial layers, which become trapped in the near surface region by successively more-structured ion layers, but which do not adsorb to the HOPG substrate, cf.Fig. 2.
When the potential is increased to +0.5 V and +1 V the cation is electrostatically repelled from the surface and the innermost layer becomes anion rich. The subsequent measured layers are then cation rich and anion rich. The forces required to displace these layers are reduced compared to 0 V, suggesting an anion-rich innermost layer templates structure in subsequent layers less effectively than a cation rich one. A similar effect is noted at the [Li(G4)] TFSI–HOPG interface. When the potential is made negative (−0.5 V and −1 V) a thin final layer is measured similar to that seen at the [Li(G4)] TFSI–HOPG interface, and attributed to the same cause: complex cation compression or desolvation of the glyme from the Li+ ion due to the pressure applied by the AFM tip. The forces measured for [Li(G4)] NO3 are lower than [Li(G4)] TFSI due to the presence of neutral species.
Force data for the [Li(G4)] NO3–Au(111) interface are presented in Fig. 1P–T. As for [Li(G4)] TFSI, the Au(111) surface roughness leads to rupture forces being lower at 0 V. When the bias is made positive (+0.5 V and +1 V) the innermost layer is enriched in anions. The innermost step observed is thin at ∼0.2 nm. A similar step was present at the [Li(G4)] TFSI–Au(111) interface at negative potentials and was attributed to compression of the adsorbed complex cation structure or desolvation of the glyme from Li+ ions in the cation rich near surface layer. However, at positive potentials, this is unlikely to occur as the innermost layer is enriched in anions. It is most likely that for [Li(G4)] NO3 neutral species are present that could produce the thin step. However the absence of a similar thin step for the [Li(G4)] NO3–Au(111) interface at 0 V favours the desolvation mechanism, as does the fact that the glyme binds to the Li+ less strongly in the poor solvate IL. For the [Li(G4)] NO3–Au(111) interface at negative potentials (−0.5 V and −1 V) data similar in form to that obtained at the HOPG interface is obtained, but the push-through forces are lower due to the increased roughness of the Au(111) substrate. The innermost layer must be cation rich on electrostatic grounds and, like for all other systems at negative potentials, the final measured layer is thin at ∼0.2 nm. As per the [Li(G4)] NO3–HOPG system at the same potentials, this final step could be due to desolvation of the glyme from the complex cation resulting in the presence of glyme molecules in the near surface layer.
Conclusions
The interfacial nanostructure of the solvate ILs [Li(G4)] TFSI and [Li(G4)] NO3 has been investigated using AFM normal force curve measurements on both HOPG and Au(111) surfaces. Similar to conventional ILs, ions arrange into ion or ion pair layers near the interface. An applied surface potential is able to modify the interfacial structure and attract counterions to the surface. As the magnitude of the bias increases, the near surface layers require a larger push-through force to be ruptured. This is a consequence of better templating of interfacial structure by an increased concentration of counterions within the interfacial layer at higher potentials. Generally, the good solvate IL [Li(G4)] TFSI has better defined interfacial structure than the poor solvate IL [Li(G4)] NO3 on both surfaces, with stronger interactions between the complex cation and the anion leading to the displacement of ion pair layers instead of discrete cation and anion sub-layers. The interfacial structure of both solvate ILs is better defined on HOPG than on Au(111) due to the increased roughness of the Au(111) surface.The force curve measurements on both HOPG and Au(111) electrode surfaces suggest the presence of a strongly bound interfacial layer which is unable to be displaced by the AFM tip. For 0 V, DFT calculations show this interfacial layer is enriched in complex cations as the adsorption free energy of the complex cation is significantly greater than for either anion on both surfaces. For negative and positive potentials, the strongly bound interfacial layer is enriched in counterions. At negative potentials, a narrow innermost step is observed. This small step is consistent with either compression of the complex cation structure or desolvation of the Li+ from the glyme.
Acknowledgements
This research was supported by an ARC Discovery Project (DP120102708) and an ARC Future Fellowship (FT120100313) for RA, and by JST-ALCA and NEDO of Japan for MW. The authors thank Paul FitzGerald for measuring the SAXS/WAXS patterns of [Li(G4)] TFSI and [Li(G4)] NO3.References
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed
.
- F. Endres, ChemPhysChem, 2002, 3, 144 CrossRef CAS
- M. Gorlov and L. Kloo, Dalton Trans., 2008, 2655 RSC
- S. Z. E. Abedin and F. Endres, Acc. Chem. Res., 2007, 40, 1106 CrossRef PubMed
- M. Galiński, A. Lewandowski and I. Stępniak, Electrochim. Acta, 2006, 51, 5567 CrossRef
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed
- C. Austen Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9 RSC
- T. Mandai, K. Yoshida, K. Ueno, K. Dokko and M. Watanabe, Phys. Chem. Chem. Phys., 2014, 16, 8761 RSC
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121 CrossRef CAS PubMed
- K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Electrochem. Soc., 2012, 159, A1005 CrossRef CAS
- K. Ueno, K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, J. Phys. Chem. B, 2012, 116, 11323 CrossRef CAS PubMed
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 3237 RSC
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 13544 RSC
- S. Tsuzuki, W. Shinoda, S. Seki, Y. Umebayashi, K. Yoshida, K. Dokko and M. Watanabe, ChemPhysChem, 2013, 14, 1993 CrossRef CAS PubMed
- D. L. Chapman, Philos. Mag., 1913, 25, 475 CrossRef
- G. Gouy, Compt. Rend., 1910, 149, 654 Search PubMed
- O. Stern, Z. Elektrochem., 1924, 30, 508 CAS
- H. L. von Helmholtz, Wied. Ann., 1879, 7, 337 CrossRef
- R. Atkin, S. Z. E. Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113, 13266 CAS
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162 CAS
- J. M. Black, D. Walters, A. Labuda, G. Feng, P. C. Hillesheim, S. Dai, P. T. Cummings, S. V. Kalinin, R. Proksch and N. Balke, Nano Lett., 2013, 13, 5954 CrossRef CAS PubMed
- R. Hayes, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2010, 12, 1709 RSC
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545 CrossRef CAS PubMed
- Y. Lauw, M. D. Horne, T. Rodopoulos and F. A. M. Leermakers, Phys. Rev. Lett., 2009, 103, 117801 CrossRef CAS PubMed
- K. B. Oldham, J. Electroanal. Chem., 2008, 613, 131 CrossRef CAS
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978 CrossRef CAS PubMed
- R. Atkin, N. Borisenko, M. Druschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849 RSC
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855 CAS
- R. Hayes, S. Z. El Abedin and R. Atkin, J. Phys. Chem. B, 2009, 113, 7049 CrossRef CAS PubMed
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, J. Phys. Chem. C, 2014, 118, 13998 CAS
- A. Labuda and P. Grütter, Langmuir, 2012, 28, 5319 CrossRef CAS PubMed
- H. Li, F. Endres and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 14624 RSC
- H. Li, R. J. Wood, F. Endres and R. Atkin, J. Phys.: Condens. Matter, 2014, 26, 284115 CrossRef PubMed
- D. Wakeham, R. Hayes, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2009, 113, 5961 CrossRef CAS PubMed
- X. Zhang, Y.-X. Zhong, J.-W. Yan, Y.-Z. Su, M. Zhang and B.-W. Mao, Chem. Commun., 2012, 48, 582 RSC
- A. Elbourne, J. Sweeney, G. B. Webber, E. J. Wanless, G. G. Warr, M. W. Rutland and R. Atkin, Chem. Commun., 2013, 49, 6797 RSC
- R. G. Horn, D. F. Evans and B. W. Ninham, J. Phys. Chem., 1988, 92, 3531 CrossRef CAS
- J. J. Segura, A. Elbourne, E. J. Wanless, G. G. Warr, K. Voitchovsky and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 3320 RSC
- N. Borisenko, S. Zein El Abedin and F. Endres, J. Phys. Chem. B, 2006, 110, 6250 CrossRef CAS PubMed
- T. Carstens, R. Hayes, S. Z. E. Abedin, B. Corr, G. B. Webber, N. Borisenko, R. Atkin and F. Endres, Electrochim. Acta, 2012, 82, 48 CrossRef CAS
- F. Endres, N. Borisenko, S. Z. El Abedin, R. Hayes and R. Atkin, Faraday Discuss., 2012, 154, 221 RSC
- D. Wakeham, A. Nelson, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 20828 RSC
- H. Zhou, M. Rouha, G. Feng, S. S. Lee, H. Docherty, P. Fenter, P. T. Cummings, P. F. Fulvio, S. Dai, J. McDonough, V. Presser and Y. Gogotsi, ACS Nano, 2012, 6, 9818 CrossRef CAS PubMed
- M. T. Alam, M. M. Islam, T. Okajima and T. Ohsaka, J. Phys. Chem. C, 2008, 112, 2601 CAS
- V. Lockett, M. Horne, R. Sedev, T. Rodopoulos and J. Ralston, Phys. Chem. Chem. Phys., 2010, 12, 12499 RSC
- V. Lockett, R. Sedev, J. Ralston, M. Horne and T. Rodopoulos, J. Phys. Chem. C, 2008, 112, 7486 CAS
- C. Nanjundiah, S. F. McDevitt and V. R. Koch, J. Electrochem. Soc., 1997, 144, 3392 CrossRef CAS
- D. Dragoni, N. Manini and P. Ballone, ChemPhysChem, 2012, 13, 1772 CrossRef CAS PubMed
- R. S. Payal and S. Balasubramanian, J. Phys.: Condens. Matter, 2014, 26, 284101 CrossRef PubMed
- C. Aliaga and S. Baldelli, J. Phys. Chem. C, 2008, 112, 3064 CAS
- C. Romero and S. Baldelli, J. Phys. Chem. B, 2006, 110, 6213 CrossRef CAS PubMed
-
R. Hayes, D. Wakeham and R. Atkin, Interfaces of Ionic Liquids, John Wiley & Sons, Inc., 2012, vol. 2, p. 51 Search PubMed
- J. Hoth, F. Hausen, M. H. Müser and R. Bennewitz, J. Phys.: Condens. Matter, 2014, 26, 284110 CrossRef PubMed
- M. Mezger, H. Schroder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schoder, V. Honkimaki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424 CrossRef CAS PubMed
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243 RSC
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572 RSC
- T. Carstens, R. Gustus, O. Höfft, N. Borisenko, F. Endres, H. Li, R. J. Wood, A. J. Page and R. Atkin, J. Phys. Chem. C, 2014, 118, 10833 CAS
- J. L. Hutter and J. Bechhoefer, Rev. Sci. Instrum., 1993, 64, 1868 CrossRef CAS
- J. Ralston, I. Larson, M. W. Rutland, A. A. Feiler and M. Kleijn, Pure Appl. Chem., 2005, 77, 2149 CrossRef CAS
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS
- M. A. Addicoat, S. Fukuoka, A. J. Page and S. Irle, J. Comput. Chem., 2013, 34, 2591 CrossRef CAS PubMed
- S. Chen, R. Vijayaraghavan, D. R. MacFarlane and E. I. Izgorodina, J. Phys. Chem. B, 2013, 117, 3186 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. D01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- R. Lim, S. F. Y. Li and S. J. O'Shea, Langmuir, 2002, 18, 6116 CrossRef CAS
- R. Lim and S. J. O'Shea, Phys. Rev. Lett., 2002, 88, 246101 CrossRef PubMed
- R. Lim and S. J. O'Shea, Langmuir, 2004, 20, 4916 CrossRef CAS PubMed
- A. Maali, T. Cohen-Bouhacina, G. Couturier and J. P. Aime, Phys. Rev. Lett., 2006, 96, 086105 CrossRef PubMed
- S. J. O'Shea, M. E. Welland and J. B. Pethica, Chem. Phys. Lett., 1994, 223, 336 CrossRef
- H. D. B. Jenkins and K. P. Thakur, J. Chem. Educ., 1979, 56, 576 CrossRef CAS
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef
- T. Murphy, R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2014, 16, 13182 RSC
- S. Biggin and J. E. Enderby, J. Phys. C: Solid State Phys., 1982, 15, L305 CrossRef CAS
- M. Revere and M. P. Tosi, Rep. Prog. Phys., 1986, 49, 1001 CrossRef
- Y.-Z. Su, Y.-C. Fu, J.-W. Yan, Z.-B. Chen and B.-W. Mao, Angew. Chem., 2009, 121, 5250 CrossRef
- H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2014, 118, 20246 CAS
-
J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, London, 1992 Search PubMed
| This journal is © the Owner Societies 2015 |