
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of bicontinuous cubic poly(3,4-ethylene dioxythiophene) gyroid (PEDOT GYR) gels†
Whirang
Cho
a,
Jinghang
Wu‡
a,
Bong Sup
Shim§
a,
Wei-Fan
Kuan
b,
Sarah E.
Mastroianni
b,
Wen-Shiue
Young¶
b,
Chin-Chen
Kuo
a,
Thomas H.
Epps, III
ab and
David C.
Martin
*ac
aDepartment of Materials Science & Engineering, University of Delaware, 201 DuPont Hall, Newark, DE 19716, USA. E-mail: milty@udel.edu; Fax: +1-508-256-8352; Tel: +1-302-831-2062
bDepartment of Chemical and Biomolecular Engineering, University of Delaware, 150 Academy Street, Newark, DE 19716, USA
cDepartment of Biomedical Engineering, University of Delaware, 125 E. Delaware Avenue, Newark, DE 19716, USA
First published on 12th January 2015
Abstract
We describe the synthesis and characterization of bicontinuous cubic poly(3,4-ethylenedioxythiophene) (PEDOT) conducting polymer gels prepared within lyotropic cubic poly(oxyethylene)10 nonylphenol ether (NP-10) templates with Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d (gyroid, GYR) symmetry. The chemical polymerization of EDOT monomer in the hydrophobic channels of the NP-10 GYR phase was initiated by AgNO3, a mild oxidant that is activated when exposed to ultraviolet (UV) radiation. The morphology and physical properties of the resulting PEDOT gels were examined as a function of temperature and frequency using optical and electron microscopy, small-angle X-ray scattering (SAXS), dynamic mechanical spectroscopy, and electrochemical impedance spectroscopy (EIS). Microscopy and SAXS results showed that the PEDOT gels remained ordered and stable after the UV-initiated chemical polymerization, confirming the successful templated-synthesis of PEDOT in bicontinuous GYR nanostructures. In comparison to unpolymerized 3,4-ethylenedioxythiophene (EDOT) gel phases, the PEDOT structures had a higher storage modulus, presumably due to the formation of semi-rigid PEDOT-rich nanochannels. Additionally, the storage modulus (G′) for PEDOT gels decreased only modestly with increasing temperature, from ∼1.2 × 105 Pa (10 °C) to ∼7 × 104 Pa (40 °C), whereas G′ for the NP-10 and EDOT gels decreased dramatically, from ∼5.0 × 104 Pa (10 °C) to ∼1.5 × 102 Pa (40 °C). EIS revealed that the impedance of the PEDOT gels was smaller than the impedance of EDOT gels at both high frequencies (PEDOT ∼102 Ω and EDOT 2–3 × 104 Ω at 105 Hz) and low frequencies (PEDOT 103–105 Ω and EDOT ∼5 × 105 Ω at 10−1 Hz). These results indicated that PEDOT gels were highly ordered, mechanically stable and electrically conductive, and thus should be of interest for applications for which such properties are important, including low impedance and compliant coatings for biomedical electrodes.
d (gyroid, GYR) symmetry. The chemical polymerization of EDOT monomer in the hydrophobic channels of the NP-10 GYR phase was initiated by AgNO3, a mild oxidant that is activated when exposed to ultraviolet (UV) radiation. The morphology and physical properties of the resulting PEDOT gels were examined as a function of temperature and frequency using optical and electron microscopy, small-angle X-ray scattering (SAXS), dynamic mechanical spectroscopy, and electrochemical impedance spectroscopy (EIS). Microscopy and SAXS results showed that the PEDOT gels remained ordered and stable after the UV-initiated chemical polymerization, confirming the successful templated-synthesis of PEDOT in bicontinuous GYR nanostructures. In comparison to unpolymerized 3,4-ethylenedioxythiophene (EDOT) gel phases, the PEDOT structures had a higher storage modulus, presumably due to the formation of semi-rigid PEDOT-rich nanochannels. Additionally, the storage modulus (G′) for PEDOT gels decreased only modestly with increasing temperature, from ∼1.2 × 105 Pa (10 °C) to ∼7 × 104 Pa (40 °C), whereas G′ for the NP-10 and EDOT gels decreased dramatically, from ∼5.0 × 104 Pa (10 °C) to ∼1.5 × 102 Pa (40 °C). EIS revealed that the impedance of the PEDOT gels was smaller than the impedance of EDOT gels at both high frequencies (PEDOT ∼102 Ω and EDOT 2–3 × 104 Ω at 105 Hz) and low frequencies (PEDOT 103–105 Ω and EDOT ∼5 × 105 Ω at 10−1 Hz). These results indicated that PEDOT gels were highly ordered, mechanically stable and electrically conductive, and thus should be of interest for applications for which such properties are important, including low impedance and compliant coatings for biomedical electrodes.
1. Introduction
Poly(3,4-ethylenedioxythiophene) (PEDOT) is a well-known π-conjugated conductive polymer of interest for a wide variety of applications including antistatic layers, electroluminescent devices, hole injection layers in organic light emitting diodes, and charge transfer layers on biomedical electrodes.1–5 For biomedical electrodes, it is important to create structures that optimize biological interactions with surrounding cells while maintaining facile charge transport. In particular, to obtain reliable and successful chronic signals, electrode coatings must be biocompatible, have low impedance, and have high charge injection capacity.6 EDOT can be polymerized by chemical, electrochemical, or vapor-phase methods to form soft, low impedance, chemically stable, and biocompatible semiconducting films. The electrical impedance of PEDOT coatings can be minimized by maximizing the effective surface area between the electrode and the electrolyte.3–6Several previous studies have demonstrated that a variety of conducting polymers such as polythiophene, polyaniline, and polypyrrole can be deposited into hydrogels to create soft, electrically, and ionically conductive materials.7–12 However, these gels typically exhibit disordered morphologies and, correspondingly, poorly defined conductive pathways. We hypothesized that templating PEDOT gels within a self-assembled bicontinuous gyroid (GYR) phase might lead to particularly well-defined and isotropic nanostructures, thus improving control of electrical properties by providing charge diffusion pathways within the resulting nanoscale channels.
Previously, several groups13–18 have explored the use of amphiphilic surfactants as structure-directing templates for conjugated polymers and shown the control and lock-in of nanoscale self-assembly in conducting polymers transferred from the nanostructure of the surfactant molecules. Our group has electrochemically deposited ordered PEDOT using self-assembled non-ionic poly(oxyethylene)-oleyl ether (n ∼ 10) surfactants from a hexagonal mesophase, resulting in highly anisotropic microdomains.6
Bicontinuous cubic phases, such as the gyroid (GYR) with Iad symmetry, are structured materials containing three-dimensionally (3-D) interconnected nanochannels.19–27 In self-assembling amphiphilic systems, the GYR morphology has high interconnectivity of both hydrophobic (oily) and hydrophilic (watery) constituent phases within discrete channels.28–36 Possible applications of these materials include photonic crystals,37 mechanical actuators,38 drug delivery carriers,39 and selectively permeable membranes.40 Khiew et al. previously described the polymerization of polyaniline within a bicontinuous cubic non-ionic surfactant nanostructure.41 However, their results regarding the symmetry of the ordered cubic phase were inconclusive, and the physical properties of the materials were not examined in detail.
In this study, we report the successful preparation of PEDOT gels employing templates consisting of poly(oxyethylene)10 nonylphenol ether (NP-10) surfactants, in which EDOT monomer in octane was preferentially incorporated into the hydrophobic GYR bicontinuous channels (Fig. 1). NP-10 surfactant was used as the template for our experiments because of its relatively large lattice parameter (d ∼ 3–10 nm) as well as its precisely-defined gyroid symmetry near room temperature at concentrations of ∼63% by weight in water.41,42 UV-initiated polymerization of EDOT was performed slowly using the mild oxidant AgNO3 to insure that the NP-10 GYR phase would not be significantly disrupted by the polymerization. The resulting PEDOT structures were characterized by optical microscopy, thermogravimetric analysis (TGA), Fourier Transform infrared spectroscopy (FTIR), small-angle X-ray scattering (SAXS), and dynamic mechanical spectroscopy. Also, we have explored the temperature and frequency dependent conductivity of the samples using electrochemical impedance spectroscopy (EIS).
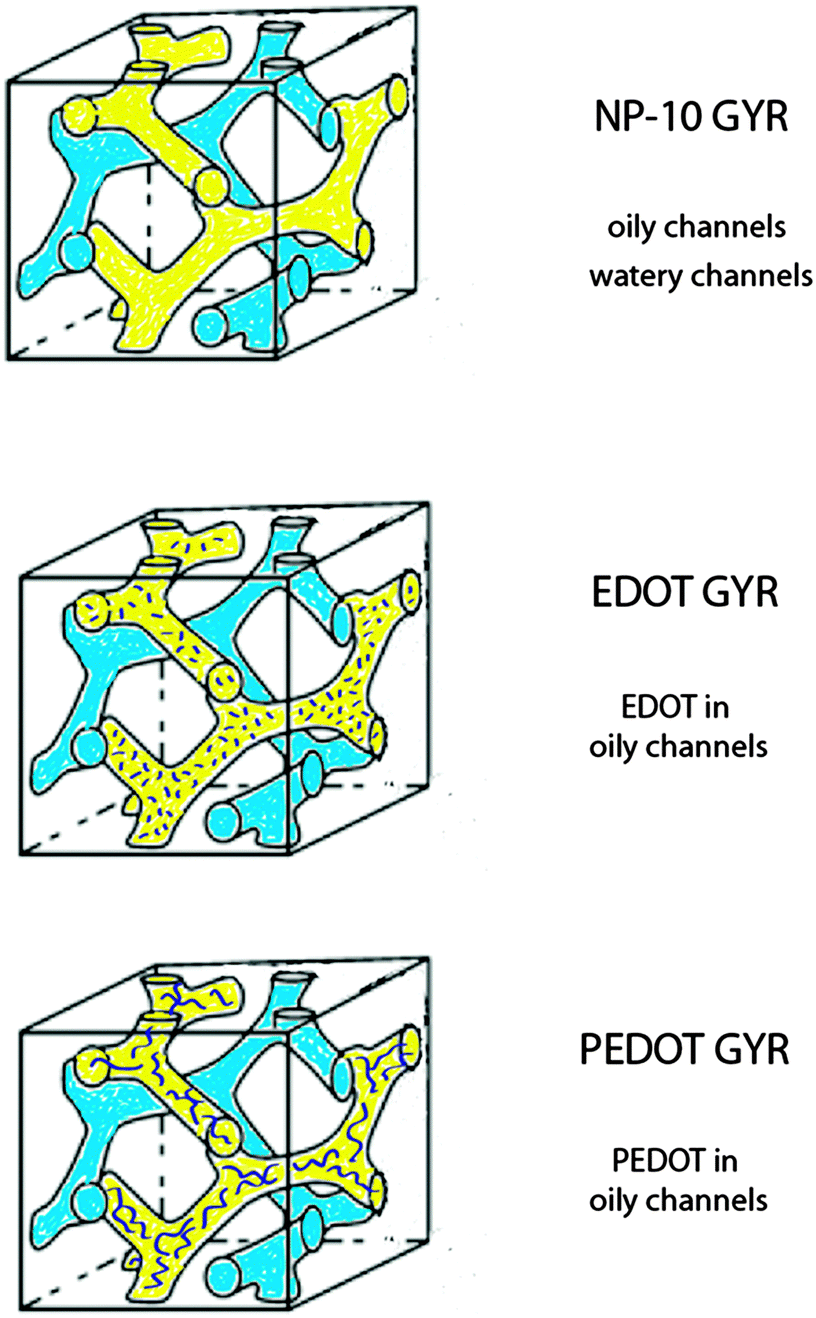 | ||
| Fig. 1 Schematic diagram of the bicontinuous cubic phases (gyroid, GYR, with Iad lattice symmetry) formed by chemical polymerization of EDOT in an ordered NP-10 surfactant mesophase. The original NP-10 GYR phase (top) is infiltrated with EDOT monomer in the oily channels (middle), and then polymerized to PEDOT (bottom). | ||
2. Experimental
2.1 Materials
Reagent grade non-ionic surfactant poly(oxyethylene)10 nonylphenol ether (NP-10, also known as Tergitol®), octane (98%), and silver nitrate (AgNO3, reagent grade, 99+%) were purchased from Sigma-Aldrich. 3,4-Ethylenedioxylthiophene (EDOT, >97%, Clevios™) monomer was purchased from Heraeus Conductive Polymers Division. All chemicals were used as received.2.2 Preparation of bicontinuous cubic phases
The molecular structure of EDOT and the NP-10 surfactant are shown in Fig. 2. The 3,4-dioxy substitution of the EDOT monomer prevents undesired α − β(β′) couplings within the PEDOT polymer, resulting in chemically stable conjugated molecular backbones that promote electrical conductivity, even in biological media.43,44 Bicontinuous cubic GYR phases were prepared following the methods of Wang et al. using 55 wt% NP-10, 22 wt% octane or octane–EDOT and 23 wt% DI-water.42 For initial studies, EDOT monomer was dissolved in octane (oil phase) at various concentrations (7.5, 10, and 12.5 wt%) to determine the concentration limit below which the desired GYR nanostructure was retained in the organic–aqueous mixture. This GYR phase was maintained after incorporation of EDOT into the oily phase of NP-10 surfactant. The symmetries of the different phases were determined by examining the characteristic textures in polarized optical microscopy (Fig. S1, ESI†). The GYR phase was readily identified as an optically clear phase (dark in polarized optical microscopy (POM)) that formed at NP-10 compositions intermediate to the hexagonal and lamellar (LAM) mesophases. Based on the results of these experiments and complimentary SAXS measurements, a 10 wt% EDOT in octane solution was used in subsequent templating studies.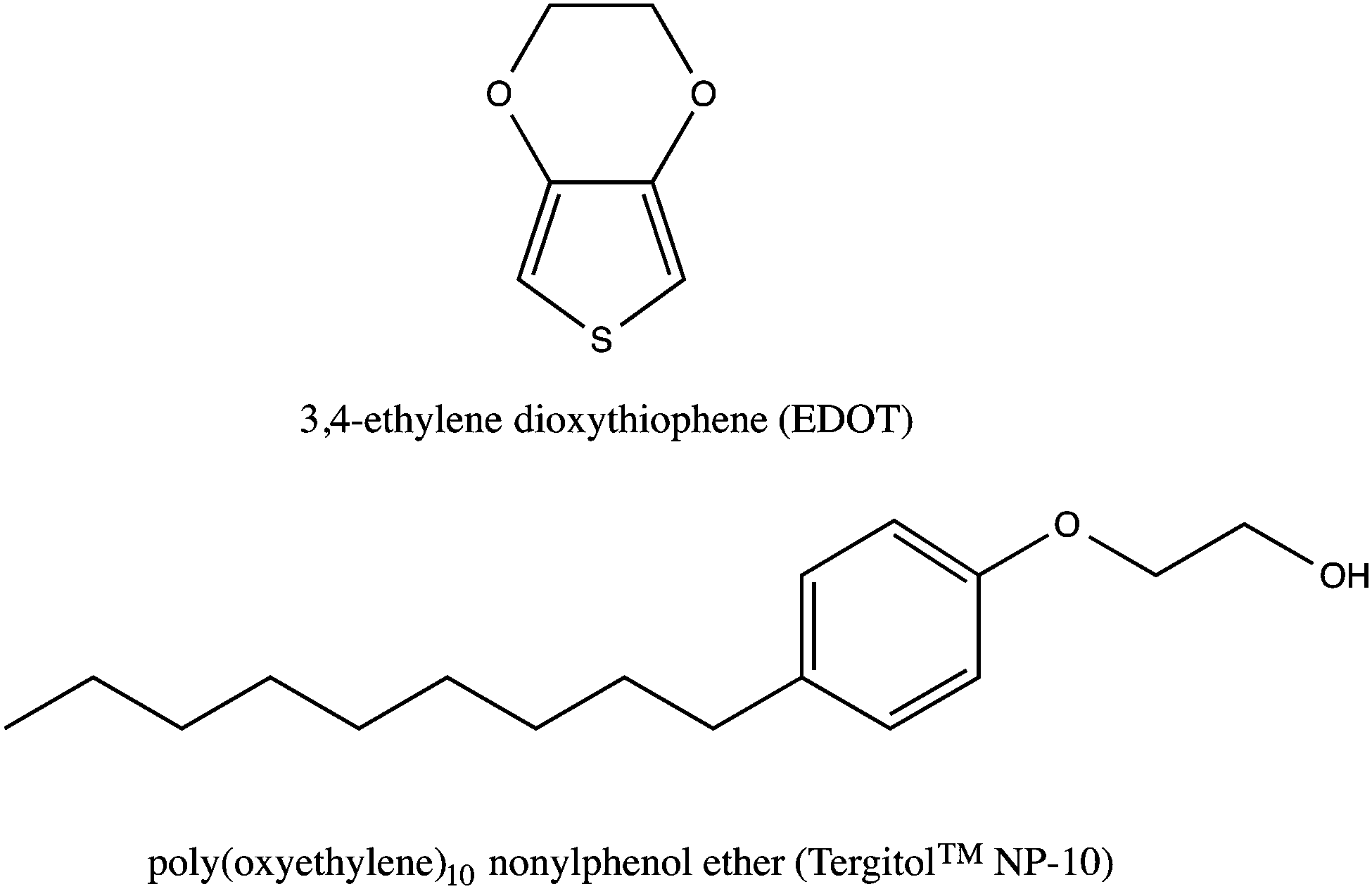 | ||
| Fig. 2 Chemical structures of 3,4-ethylenedioxythiophene (EDOT, top) and poly(oxyethylene)10 nonylphenol ether (NP-10, bottom). | ||
To form the nanotemplated PEDOT constructs, a 10 wt% EDOT–octane solution was added to the NP-10 surfactant and mixed using a vortex mixer for 1 min. Then, DI-water was added to the mixture of NP-10/EDOT–octane, and the resulting mixture was vortexed for ∼5 min. The mixture was refrigerated for 1 day to stabilize as a clear transparent sample (NP-10 surfactant gels and EDOT gels, respectively). Following stabilization, a 4-times molar excess of AgNO3 initiator relative to the EDOT was added and allowed to diffuse into the pre-formed mixture over 1 week. Subsequently, UV light (Spectroline® model EN-140L Long Wave [365 nm] UV-A/Ultraviolet lamp, Spectronics Corporation) was used to polymerize the EDOT phase (into PEDOT) at 4 °C, as the AgNO3 was reduced from silver ions to silver metal after exposure to the UV-source.45,46 The UV polymerization of the EDOT gel was done for 6 h (PEDOT gels), which was determined to be sufficient time for the absorption properties to reach steady-state. We chose AgNO3 as the initiator because we found that it caused the least amount of disruption to the GYR phase after being added to the mixture, in comparison to other initiators that we investigated (including FeCl3). AgNO3 also dissolved less exothermically in water than FeCl3, which helped insure that the ordered EDOT phases would not be thermally disrupted before their conversion to PEDOT.47
2.3 Characterization
UV-vis absorption spectra were obtained using a Shimadzu UV-3600 UV-Vis-NIR spectrophotometer. Fourier Transform Infrared (FT-IR) measurements were performed at room temperature using a PerkinElmer Spectrum 100 FT-IR spectrometer, using 20 scans in transmission mode. Thermogravimetric analysis (TGA) was performed using a TA model Q500 TGA instrument. The samples were heated from 25 °C to 500 °C at a heating rate of 10 °C min−1 under N2 flow.Synchrotron SAXS experiments were conducted on the X27C beamline at the National Synchrotron Light Source of Brookhaven National Laboratory (NSLS-SAXS) using an incident beam of wavelength λ = 1.371 Å and a 1823 mm sample-to-detector distance. Sample temperatures were controlled using a Linkam HFS91 CAP stage while acquiring in situ scattering data. One dimensional SAXS data are presented as azimuthally integrated logarithmic intensity versus the scattering vector, q = |q| = 4πλ−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(θ/2), where θ is the scattering angle.48 The in situ SAXS data were acquired at each temperature after annealing for at least 15 min, and acquisition times were ∼1 min.
sin(θ/2), where θ is the scattering angle.48 The in situ SAXS data were acquired at each temperature after annealing for at least 15 min, and acquisition times were ∼1 min.
An AR-2000 rheometer (TA instruments) with a 40 mm 1° cone-and-plate fixture was used to measure the storage/elastic modulus (G′) and loss/viscous modulus (G′′) of the samples. The sample temperature was controlled by a water bath that circulated through a Peltier plate. Temperature scans at a fixed frequency of 6.28 rad s−1 were carried out at a heating rate of 1 °C min−1 and the equilibration time was 5 min. The torque was fixed at 10 μN m for the NP-10 and EDOT samples and at 100 μN m for the PEDOT samples, which was small enough to ensure a linear viscoelastic response in all cases.
A custom built microindentor was used to measure the compressive modulus. A 1.6 mm indenter tip was driven by a piezoelectric actuator with a total moving range of 1 mm and a resolution of 60 nm. During indentation, a 50 g load cell recorded force. A ball head was used to make the sample perpendicular to the indenter tip. Subsequently, indenter tip was lowered manually until it makes solid contact with the sample, then 0.5–1 g preload was applied to ensure flat contact. Indentation was then performed at 10 μm s−1 to a total placement of 1000 μm.
A Princeton Applied Research PARSTAT 2273 frequency response analyzer (AC Impedance Spectrometer) with a built-in test cell on a Linkam HFS91 CAP stage was used to measure charge transport properties as a function of temperature. The morphology of each sample was confirmed at each temperature tested using simultaneous SAXS in a lab-scale Rigaku instrument with a 1.6 kW sealed-tube X-ray source (Cu Kα, λ = 1.54 Å) and 1200 mm sample-to-detector distance. Samples were sandwiched between two ion blocking aluminum foil electrodes using a 0.5 mm thick Teflon O-ring as a spacer. The contact area (A) between sample and each electrode was 0.32 cm2. The impedance measurements were conducted on heating between 10 °C and 50 °C. Two impedance measurements were taken at each temperature with 5 and 8 min annealing times. The AC frequency range and voltage amplitude were 10−1–105 Hz and 10 mV, respectively. The effective conductivities (σ) at both high (105 Hz, σh) and low (10−1 Hz, σl) frequencies were calculated using σ = L/(|Z|A), for which L (0.5 mm) is the sample (Teflon O-ring) thickness and |Z| is the amplitude of the impedance at that frequency.48
3. Results and discussion
The polymerization caused a dramatic change in the appearance of the sample from clear (for the EDOT GYR gels) to a deep, dark bluish black (for the PEDOT GYR gels) (Fig. 3). The UV/vis spectra showed a distinctive peak near 280 nm for the EDOT gel samples and relatively low absorption across the entire visible range of wavelengths (Fig. 4). After polymerization, the absorption increased dramatically across the entire visible spectrum, consistent with the formation of low energy absorption bands known to be characteristic of PEDOT molecules with extended backbone conjugation.49–51 The UV spectra of PEDOT gels showed a strong absorption band above 600 nm, indicating the formation of extended conjugation along the polymer backbone. The functional groups of the PEDOT sample were also confirmed by FT-IR (Fig. 5). The FTIR spectrum showed peaks at 820 cm−1 and 940 cm−1 corresponding to the C–S vibration of the thiophene and a peak at 1100 cm−1 indicative of C–O stretching in the ethylenedioxyl group.52 The absorption peaks at 1245, 1295, 1455, 1512, and 1610 cm−1 all correspond to thiophene ring stretches.52 The FT-IR spectra shows characteristic peaks corresponding to the C–S vibration of the thiophene, and the C–O stretching of the ethylenedioxyl group, confirming the polymerization of EDOT to PEDOT and limited functional group degradation after UV irradiation. | ||
| Fig. 3 The initially colorless and optically transparent EDOT gels (left) change to a deep, dark bluish black (right, PEDOT gel) after the UV-initiated polymerization. | ||
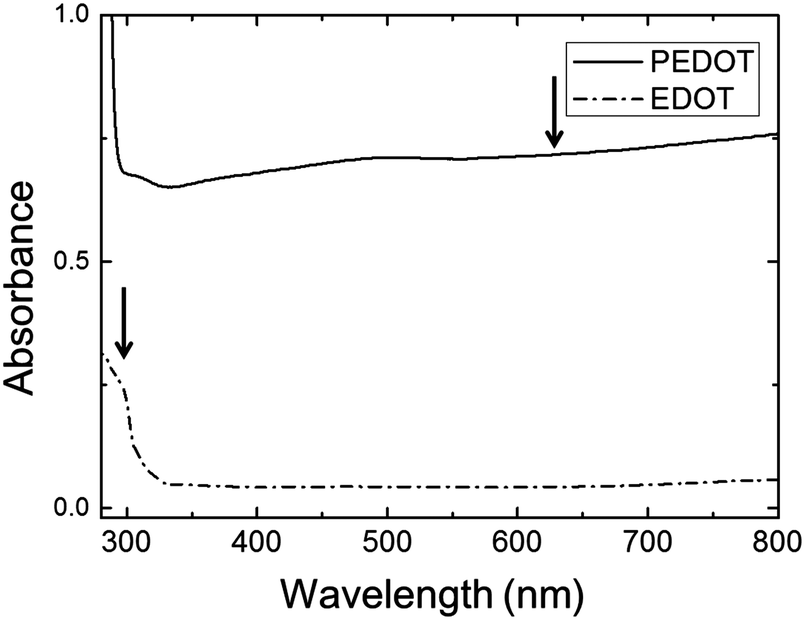 | ||
| Fig. 4 UV-vis absorbance spectrum of PEDOT gel (solid line) and EDOT gels (dot-dashed line). | ||
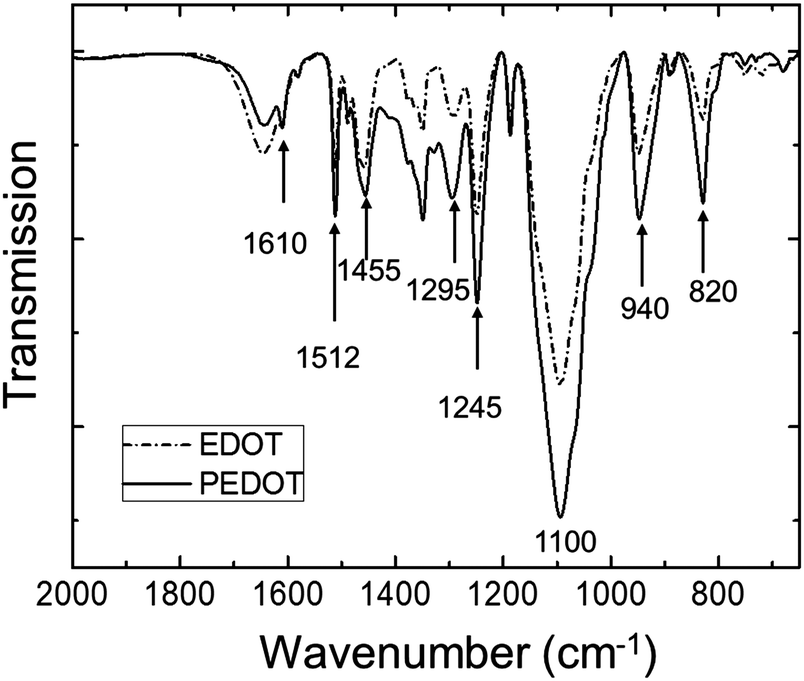 | ||
| Fig. 5 FT-IR spectrum of PEDOT gel (solid line) and EDOT gel (dot-dashed line). | ||
The thermal behavior of the gels was investigated by thermogravimetric analysis (TGA) as shown in Fig. 6. In all of samples, the mass loss below 100 °C was primarily attributed to the evaporation of water. From 100–150 °C the PEDOT samples retained just less than 70% of their initial mass, whereas the NP-10 and EDOT gels retained ∼60% of their initial mass over this same temperature range. The PEDOT samples lost ∼5% of their mass around 160 °C, perhaps from low molecular weight oligomers. The bulk of the mass was lost from 250–400 °C for all three gels. The PEDOT gels showed a residual mass of about 7% after heating to 500 °C, whereas for the EDOT and NP-10 gels the mass loss was essentially complete by that temperature.
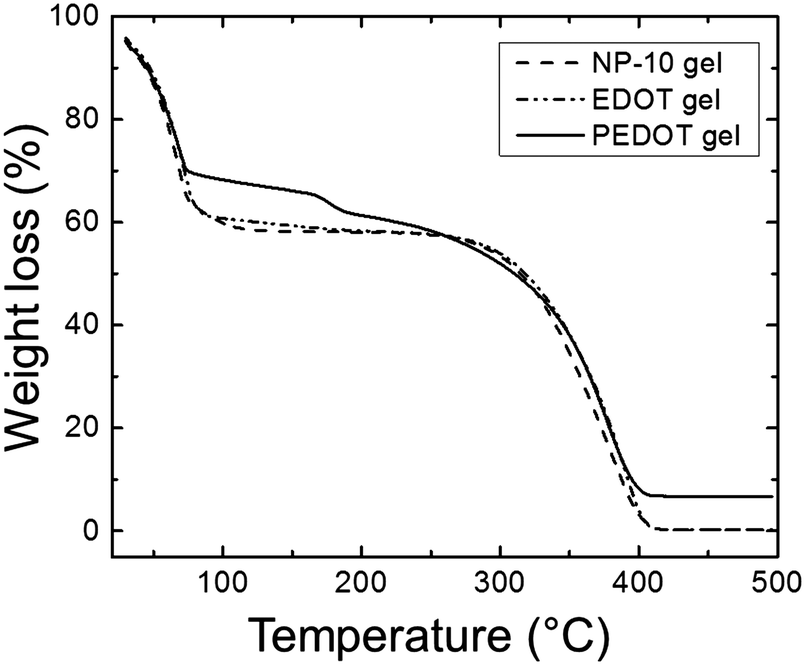 | ||
| Fig. 6 TGA spectrum of NP-10 gel (dashed line), EDOT gel (dot-dashed line), and PEDOT gel (solid line). | ||
The temperature-dependent variations in nanostructure were examined by in situ SAXS. Fig. 7 shows SAXS profiles of neat NP-10 surfactant gels (with no EDOT monomer, Fig. 7(a)), NP 10 surfactant gels containing 10 wt% EDOT monomer (Fig. 7(b)), and PEDOT gels (after polymerization, Fig. 7(c)). We chose 10 wt% EDOT monomer because it allowed us to introduce a significant amount of monomer into the NP-10 template yet still retain the desired bicontinuous cubic symmetry (Fig. S2, ESI†).
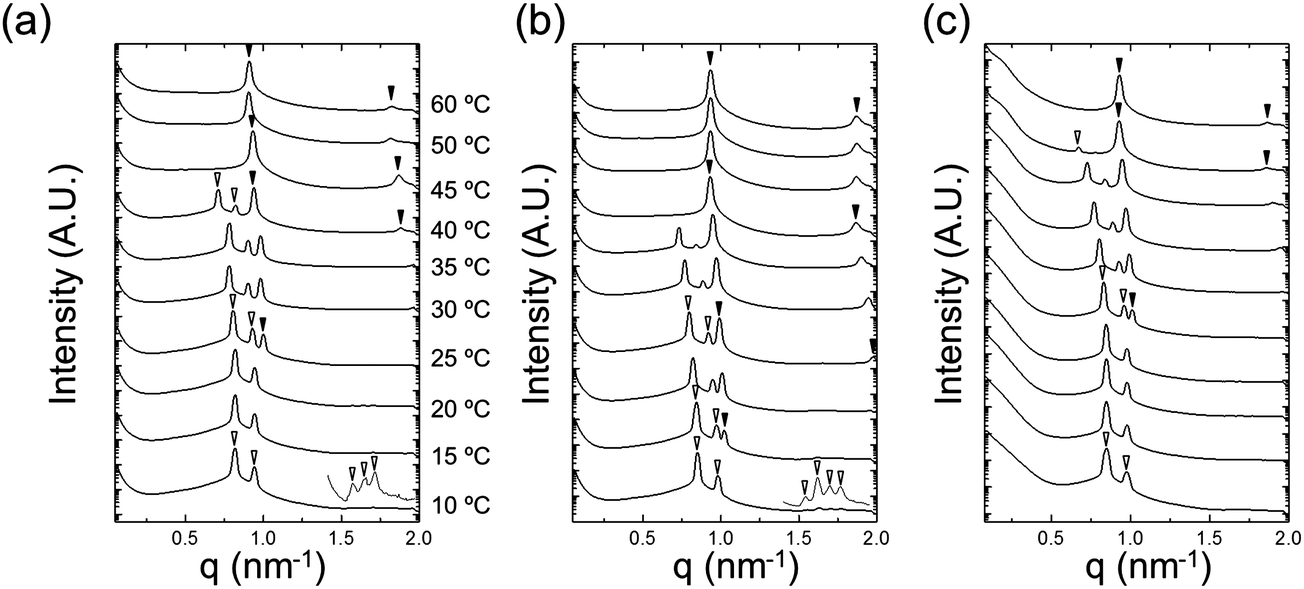 | ||
Fig. 7 Synchrotron SAXS patterns as a function of temperature for (a) NP-10 gel, (b) EDOT gel, and (c) PEDOT gel. The open triangles denote cubic (Iad) Bragg peaks (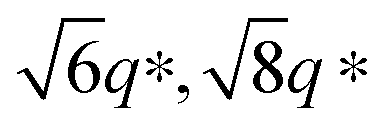 ) and the filled triangles indicate lamellar peaks (1:2). The scattering profiles are vertically offset for clarity. ) and the filled triangles indicate lamellar peaks (1:2). The scattering profiles are vertically offset for clarity. | ||
Characteristic Bragg peaks of phases with bicontinuous cubic (Iad) (GYR) symmetry were observed at 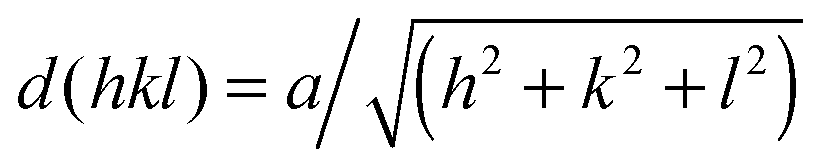 for which d(hkl) is the Bragg spacing, a is the unit cell size (lattice constant), and h, k, and l are the Miller indices.53 In the 10 °C profiles of NP-10 surfactant, EDOT monomer gels, and PEDOT gels, the ratio of the scattering vector moduli for the first two reflections was
for which d(hkl) is the Bragg spacing, a is the unit cell size (lattice constant), and h, k, and l are the Miller indices.53 In the 10 °C profiles of NP-10 surfactant, EDOT monomer gels, and PEDOT gels, the ratio of the scattering vector moduli for the first two reflections was 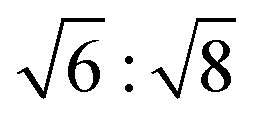 ; these are associated with the (211) and (220) spacings. In the NP-10 and EDOT gels, higher-order peaks at
; these are associated with the (211) and (220) spacings. In the NP-10 and EDOT gels, higher-order peaks at 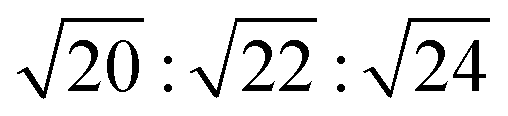 and in certain cases even
and in certain cases even 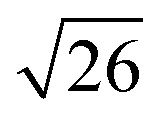 corresponding to the (420), (332), (422), and (431) spacings of the GYR structure also were observed. The lattice constant, a, at 10 °C for the neat NP-10, EDOT monomer-containing, and PEDOT-containing cubic mesophases was 18.7 nm, 18.1 nm, and 18.3 nm, respectively. The cubic structure of the neat NP-10 surfactant gel was stable through 20 °C. However, when the temperature was increased to 25 °C, an additional peak was evident (qL: 0.97 nm−1). At 30 °C, another peak at twice the value of the qL emerged. This second set of peaks with a relative peak ratio of 1:2 at 30 °C indicated the formation of a lamellar (LAM) morphology with a domain spacing d = 6.5 nm. The GYR peaks decreased in intensity with further increases in temperature (from 30–40 °C), with only the LAM mesophase remaining at 45 °C. Because of the narrow width of both the temperature and concentration region over which the GYR phase is stable in the NP-10–water system, the GYR phase is never far from a phase boundary with the LAM phase. The proximity of these phase boundaries evidently gives rise to mixed phase structures under these experimental conditions.54 Two-phase coexistence regions also have been reported at the boundaries of the cubic phase in several poly(oxyethylene)-alkyl ether surfactant–water systems.55–57 At 15 °C the neat NP-10 gel sample exhibited only the GYR mesophase, while at this temperature a LAM peak had already appeared in the EDOT samples. The incorporation of EDOT into the oily phase evidently allows for the LAM phase to appear at lower temperatures. This may be due to the relative ease of incorporating EDOT into the layered structure of the LAM phase, rather than the highly curved domains of the GYR sample. In the EDOT samples from 20 °C to 35 °C, the two mesophases continued to coexist, and the samples transitioned entirely to the LAM phase at 40 °C.
corresponding to the (420), (332), (422), and (431) spacings of the GYR structure also were observed. The lattice constant, a, at 10 °C for the neat NP-10, EDOT monomer-containing, and PEDOT-containing cubic mesophases was 18.7 nm, 18.1 nm, and 18.3 nm, respectively. The cubic structure of the neat NP-10 surfactant gel was stable through 20 °C. However, when the temperature was increased to 25 °C, an additional peak was evident (qL: 0.97 nm−1). At 30 °C, another peak at twice the value of the qL emerged. This second set of peaks with a relative peak ratio of 1:2 at 30 °C indicated the formation of a lamellar (LAM) morphology with a domain spacing d = 6.5 nm. The GYR peaks decreased in intensity with further increases in temperature (from 30–40 °C), with only the LAM mesophase remaining at 45 °C. Because of the narrow width of both the temperature and concentration region over which the GYR phase is stable in the NP-10–water system, the GYR phase is never far from a phase boundary with the LAM phase. The proximity of these phase boundaries evidently gives rise to mixed phase structures under these experimental conditions.54 Two-phase coexistence regions also have been reported at the boundaries of the cubic phase in several poly(oxyethylene)-alkyl ether surfactant–water systems.55–57 At 15 °C the neat NP-10 gel sample exhibited only the GYR mesophase, while at this temperature a LAM peak had already appeared in the EDOT samples. The incorporation of EDOT into the oily phase evidently allows for the LAM phase to appear at lower temperatures. This may be due to the relative ease of incorporating EDOT into the layered structure of the LAM phase, rather than the highly curved domains of the GYR sample. In the EDOT samples from 20 °C to 35 °C, the two mesophases continued to coexist, and the samples transitioned entirely to the LAM phase at 40 °C.
After PEDOT polymerization, the PEDOT GYR structure persisted below 25 °C and underwent a transition from GYR to LAM beginning at 30 °C. These results demonstrate that the stable temperature range of the GYR phase was wider in the polymerized sample compared to the neat NP-10 surfactant and EDOT monomer gels. In-house SAXS experiments were conducted to examine the effect of increased annealing time (several hours acquisition time) at each temperature (Fig. S3, ESI†). The GYR to LAM phase transition happened at lower temperatures in all cases, indicating the importance of the kinetics on this process. The thermal stability of GYR morphology in PEDOT gels relative to NP-10 gels and EDOT gels also was confirmed by relatively long-time (1 h) SAXS annealing experiments. The (211) d-spacing of the GYR structure (and hence the lattice parameter) increased with increasing temperature, as shown in Fig. 8(a). The d-spacings were relatively insensitive to temperature at low temperatures, but began to expand significantly at higher temperatures. The temperature corresponding to this change in behavior was higher for PEDOT gels (∼30 °C) than for the neat NP-10 surfactant (∼20–25 °C) and EDOT monomer containing gels (∼15 °C). The temperatures at which this transition occurs correspond with the appearance of LAM peaks in each sample. Also, the thermal onset of the GYR-to-LAM phase transition of the PEDOT gels was higher than that for EDOT monomer gels as shown in Fig. 8(b).
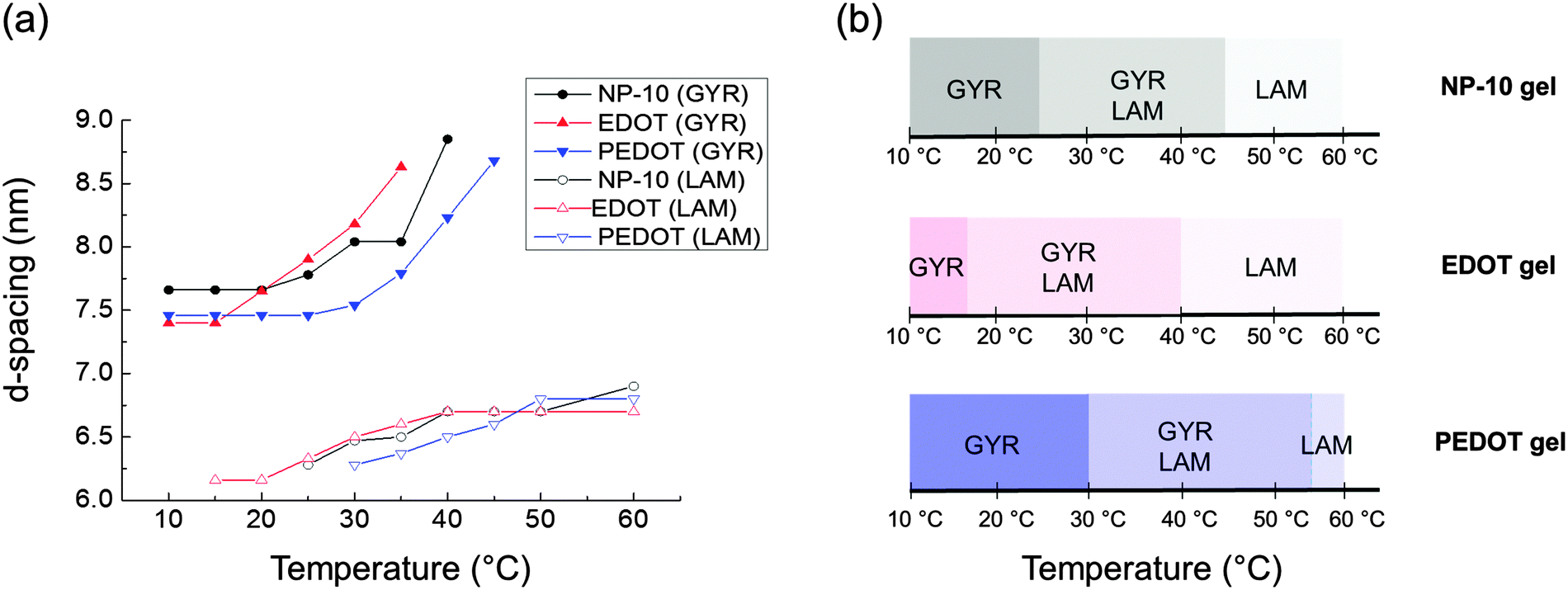 | ||
| Fig. 8 (a) Change of d-spacings determined from the primary (211) cubic peak q* and the lamellar peaks from each synchrotron SAXS pattern (Fig. 7), (b) nanostructure changes as a function of temperature. | ||
3.1 Rheological properties
Rheological properties were examined to determine the effect of PEDOT polymerization on the mechanical stability of the gels. Quantitative tests were performed in the frequency domain (oscillatory shear) to measure the elastic or storage modulus (G′) and the viscous or loss modulus (G′′) as functions of angular frequency (ω).58,59 The changes in G′ and G′′ as a function of ω at different temperatures for the EDOT and PEDOT samples during a heating sweep (10–60 °C) are shown in Fig. 9 and 10, respectively. For temperatures less than 50 °C, the EDOT sample (Fig. 9) exhibited a larger G′ than G′′, with both moduli essentially independent of ω between 0.1 s−1 and 100 s−1, indicating predominantly elastic (gel-like) behavior. At 60 °C, G′ was smaller than G′′, indicating viscous (liquid-like) behavior.60 Thus, the EDOT gel-to-sol transition temperature is between 50 °C and 60 °C. Both the G′ and G′′ of the unpolymerized NP-10 surfactant gel also were nearly independent of frequency within the frequencies tested up to 50 °C (see Fig. S4, ESI†). The PEDOT gel was stable over a wider temperature range as evidenced by the nearly frequency-independent G′ and G′′ curves up to 60 °C as shown in Fig. 10. The G′ values were greater than G′′ over the entire tested frequency range and showed a slight increase with frequency at low ω (long time scales) confirming its gel-like behavior. The small frequency dependence at long time scales (low frequencies) in the PEDOT gels compared to the NP-10 and EDOT gels is consistent with the finite relaxation time (τ) usually found in surfactant gels in contrast with the near infinite relaxation time of cross-linked gels.61,62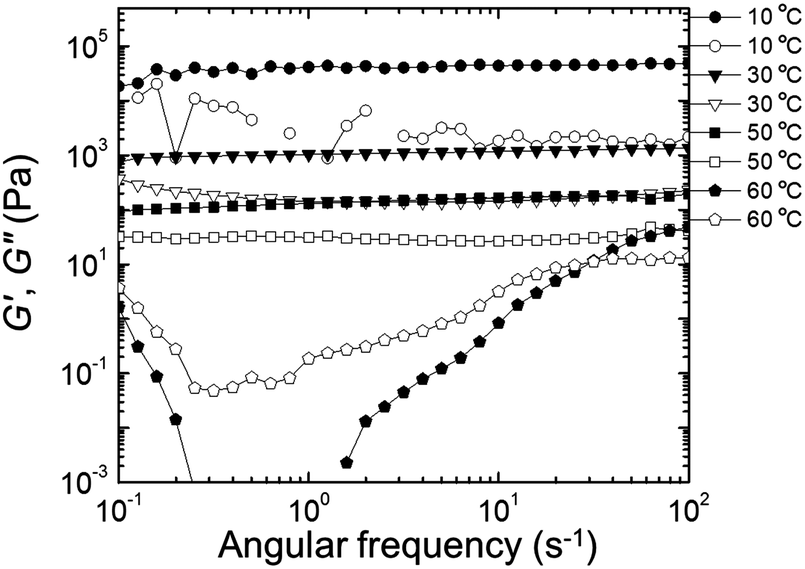 | ||
| Fig. 9 Storage modulus (G′, filled circles) and loss modulus (G′′, empty circles) as a function of frequency (ω) during heating sweep (from low to high frequency) of EDOT gels. | ||
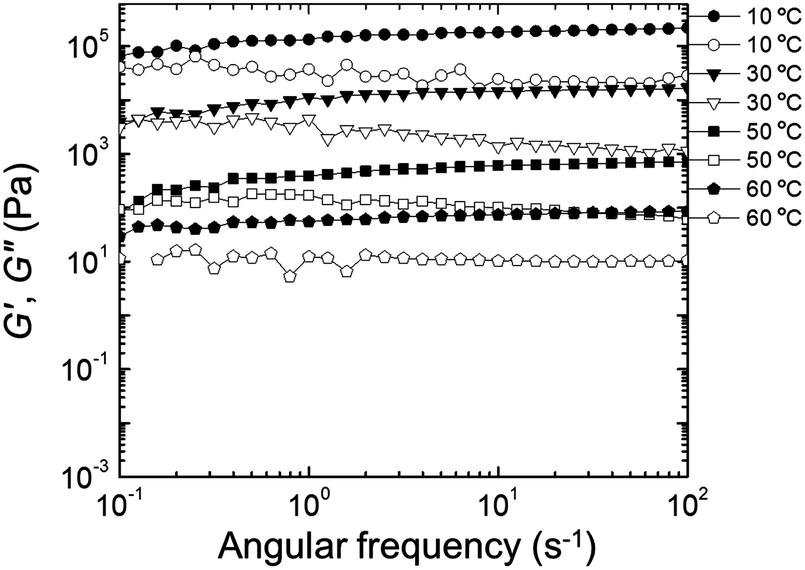 | ||
| Fig. 10 Storage modulus (G′, filled circles) and loss modulus (G′′, empty circles) as a function of frequency (ω) during heating sweep (from low to high frequency) of PEDOT gels. | ||
Fig. 11 presents the changes in G′ (closed symbols) and G′′ (open symbols) as a function of temperature during a dynamic temperature sweep at a constant frequency of 6.28 rad s−1 for the neat NP-10 surfactant gel, EDOT monomer containing gel, and PEDOT gel. The G′ and G′′ of the PEDOT gel exceeded those of both the NP-10 and EDOT monomer gels, confirming the substantial enhancement of stiffness by PEDOT polymerization. In comparison to unpolymerized NP-10 and EDOT cubic phases (with storage modulus G′ ∼ 104 Pa at 10 °C), the PEDOT cubic structures had a much higher storage modulus (G′ ∼ 105 Pa at 10 °C), presumably due to the formation of semi-rigid PEDOT-rich nanochannels. Also, no gel-to-sol crossover point of the G′ and G′′ curves59 occurred between 10 °C and 60 °C in the polymerized PEDOT gel. Additionally, the G′ for the polymeric PEDOT gels decreased only modestly with increasing temperature, from ∼1.2 × 105 Pa (10 °C) to ∼7 × 104 Pa (40 °C), whereas G′ for the NP-10 surfactant and monomeric EDOT phases decreased dramatically, from ∼5 × 104 Pa (10 °C) to ∼1.5 × 102 Pa (40 °C). The structural reinforcement of the PEDOT GYR gels also was confirmed by the one order of magnitude higher compressive modulus of PEDOT (∼105 Pa) compared to NP-10 surfactant gels and EDOT monomer gels (∼104 Pa) (see Fig. S5, ESI†).
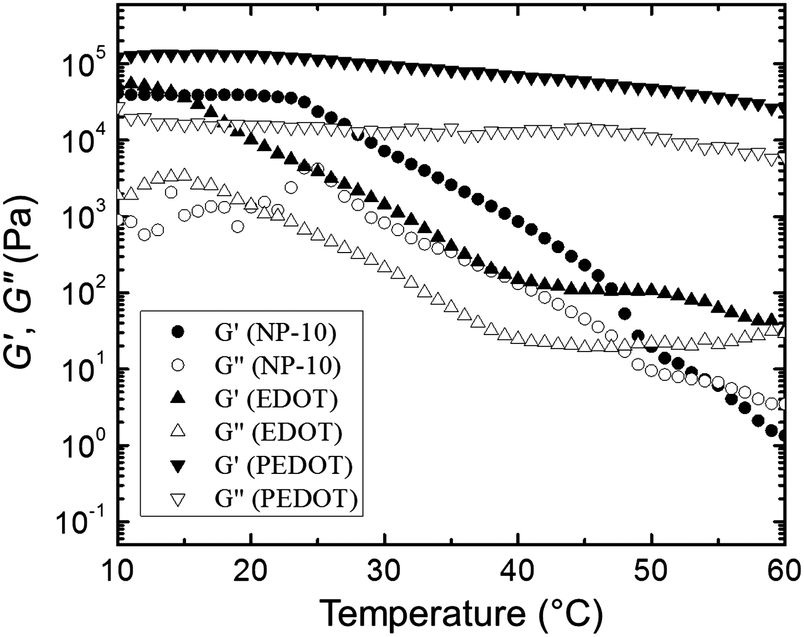 | ||
| Fig. 11 Changes in G′ and G′′ of NP-1, EDOT, and PEDOT gels as function of temperature during dynamic temperature sweeps with a fixed frequency of 6.28 rad s−1. | ||
3.2 Electrochemical properties
The amplitude of the AC impedance of EDOT monomer gels and PEDOT gels as a function of frequency (Bode plot) is shown in Fig. 12. The simultaneous presence of both electronic and ionic charge carriers makes it challenging to extract their relative contributions to the overall charge transport. However, as has been shown in other studies,63–65 we expected that the slower, diffusion-limited ion transport processes would dominate at low frequencies (∼10−1 Hz), whereas electron transport would be more important at higher frequencies (∼105 Hz). In the high frequency region, the impedance of PEDOT samples was much smaller (∼102 Ω) than that of EDOT materials (2–3 × 104 Ω) as can be seen in Fig. 13(a). This difference in impedances suggests that electron transport was facilitated by the development of continuous conductive PEDOT paths throughout the sample. In addition, as can be seen from Fig. 13(b), the impedances at 10−1 Hz of the PEDOT gels were also much smaller (103–105 Ω) than those of EDOT gels (5 × 105 Ω). The PEDOT samples showed decreasing impedance with temperature, and this effect was much stronger at the lower frequencies. On the other hand, the impedances of the EDOT samples were relatively independent of temperature at both frequencies. From these impedances we estimated effective high frequency and low frequency conductivities, these results are also shown in Fig. 14(a) and (b).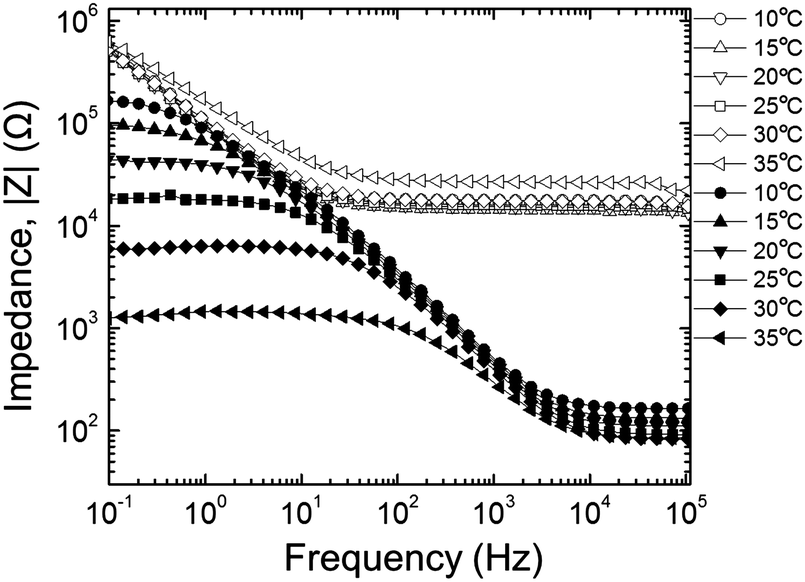 | ||
| Fig. 12 AC impedance of EDOT gels (open symbols) and PEDOT gels (filled symbols). | ||
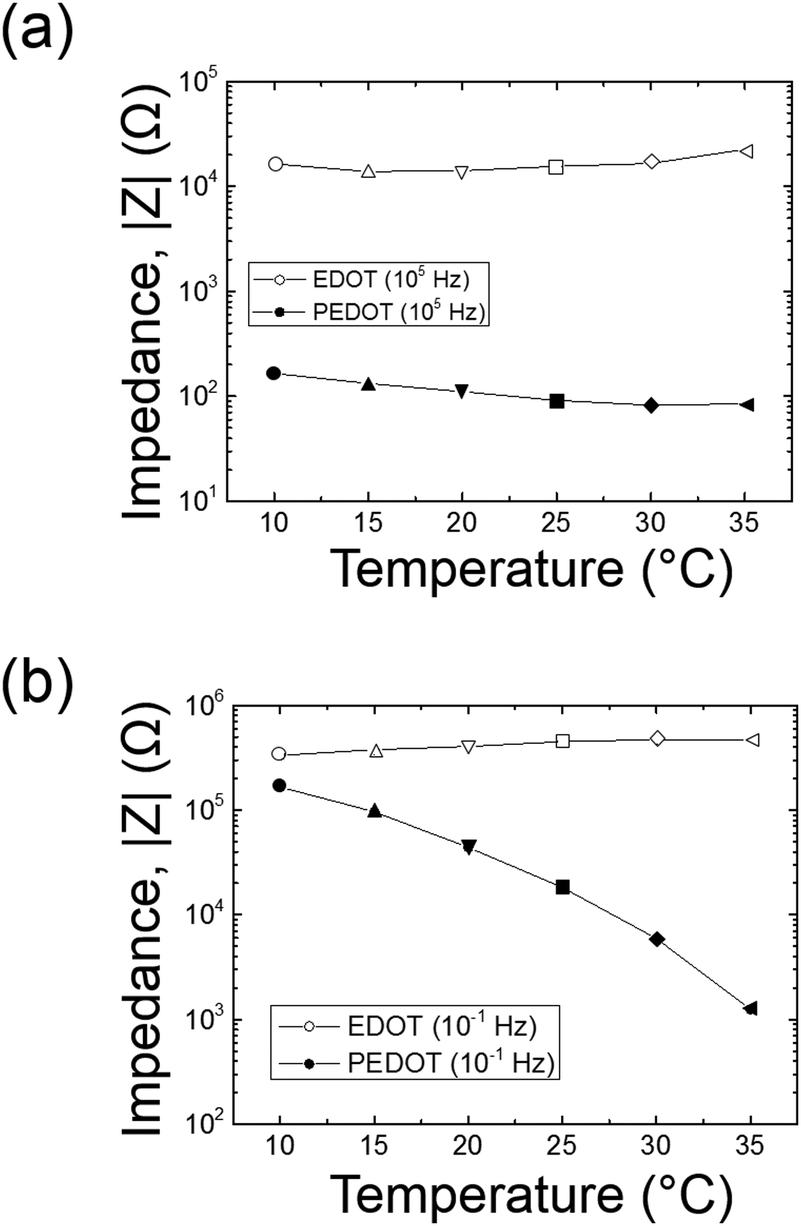 | ||
| Fig. 13 AC impedance comparison between EDOT and PEDOT gels at (a) 105 Hz and (b) 10−1 Hz. | ||
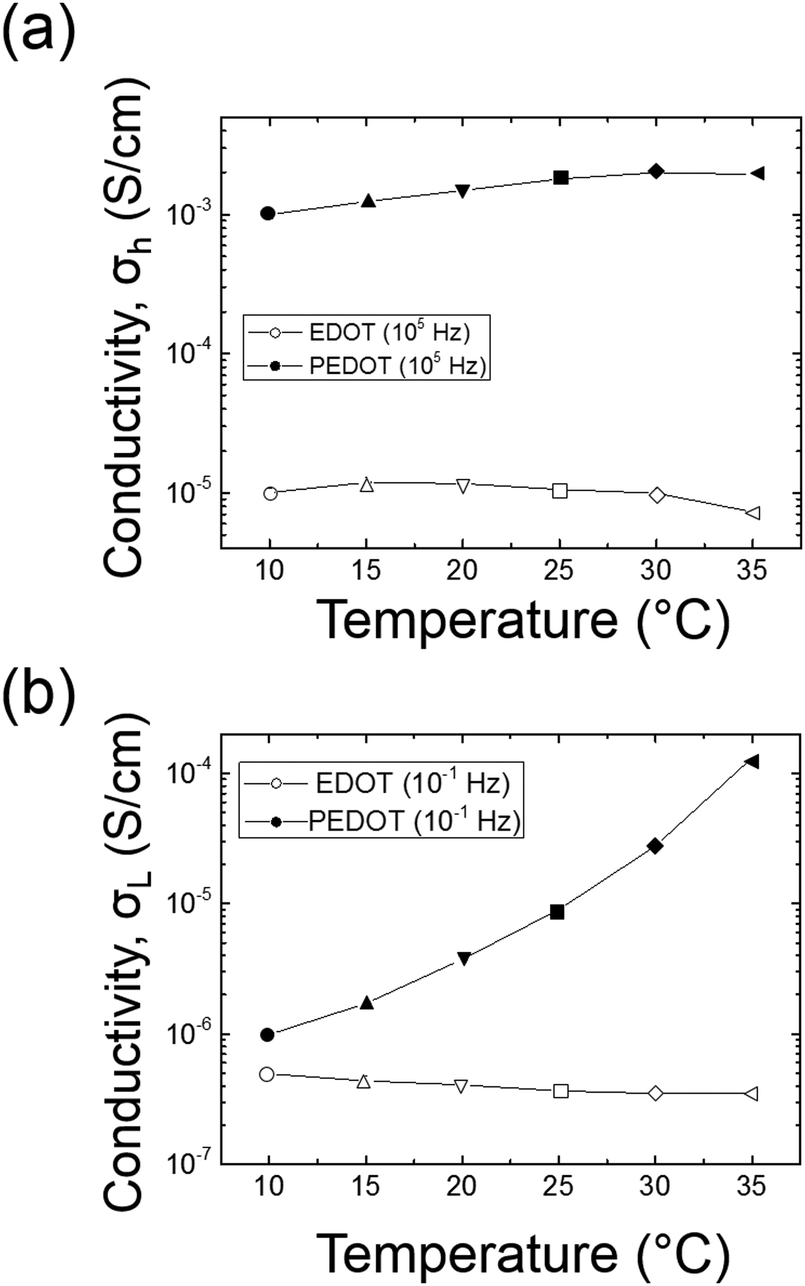 | ||
| Fig. 14 Effective conductivity comparison between EDOT and PEDOT gels at 105 Hz (top) and 10−1 Hz (bottom). | ||
These results clearly show that forming PEDOT GYR from the bicontinuous EDOT GYR nanotemplates enhances charge transport properties at both high and low frequencies. The data suggest that the more rapid electron transport and the slower ionic transport processes have both been enhanced by the formation of the ordered PEDOT interconnected domains. The increased high frequency (electron) transport likely is due to the formation of conjugated PEDOT chains that are known to accommodate easy movement of charge along the molecular backbone and by lateral hopping between molecules. The enhanced low frequency (ion) transport may be due to the increased rigidity of the locally porous structure provided by the PEDOT polymerization process (as was seen by mechanical testing), perhaps because the more rigid PEDOT framework is not able to dissipate local ion diffusion processes as efficiently as the unpolymerized EDOT. It may also be a synergistic effect related to the enhanced electronic conductivity of the PEDOT network. Further studies are underway to examine the detailed origin of these observations.
4. Conclusions
We have successfully developed chemical polymerization methods for the synthesis and characterization of ordered 3-D bicontinuous cubic gyroid (Iad) PEDOT GYR by exploiting the self-assembled structure-directing properties of the GYR phase in NP-10 surfactant systems. The PEDOT samples exhibited a morphological transition from bicontinuous cubic gyroid (Iad, GYR) to lamellar (LAM) mesophases as a function of temperature. The wider thermal stability of the PEDOT GYR samples, relative to EDOT GYR specimens, was evidenced by the nearly frequency-independent elastic modulus (G′) curves at temperatures up to 60 °C. Additionally, the AC impedance spectroscopy results demonstrated that substantially higher conductivities were seen in the PEDOT gels compared to the EDOT gels at both high and low frequencies. We expect that these cubic nanostructured, and conductive PEDOT GYR materials will be useful for a variety of applications such as neural electrodes and materials for controlled drug delivery.
Acknowledgements
This research was supported in part by the National Science Foundation (NSF) (DMR-1103027), the National Institutes of Health (Eureka-RO1EB010892), and the American Chemical Society Petroleum Research Fund to DCM (47371-AC7). WFK and WSY were supported by a Department of Defense AFOSR-PECASE grant award to THE (FA9550-09-1-0706). BSS acknowledges the funding support by NRF-2013R1A1A1076111. SEM was supported by an NSF grant awarded to THE (CBET-0930986). Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886. The authors thank B. Zimmerman for assistance with the compressive modulus measurements, which were done in the laboratory of Prof. Lucas Lu in Mechanical Engineering.Notes and references
- Y. Ha, N. Nikolov, S. K. Pollack, J. Mastrangelo, B. D. Martin and R. Shashidhar, Adv. Funct. Mater., 2004, 14, 615–622 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- M. Asplund, T. Nyberg and O. Inganäs, Polym. Chem., 2010, 1, 1374–1391 RSC.
- R. A. Green, N. H. Lovell, G. G. Wallace and L. A. Poole-Warren, Biomaterials, 2008, 29, 3393–3399 CrossRef CAS PubMed.
- X. Cui, V. A. Lee, Y. Raphael, J. A. Wiler, J. F. Hetke, D. J. Anderson and D. C. Martin, J. Biomed. Mater. Res., 2001, 56, 261–272 CrossRef CAS.
- X. Cui and D. C. Martin, Sens. Actuators, B, 2003, 89, 92–102 CrossRef CAS.
- K. Gilmore, A. Hodgson, B. Luan, C. Small and G. Wallace, Polym. Gels Networks, 1994, 2, 135–143 CrossRef CAS.
- S. Ghosh and O. Inganäs, Adv. Mater., 1999, 11, 1214–1218 CrossRef CAS.
- R. A. Green, S. Baek, L. A. Poole-Warren and P. J. Martens, Sci. Technol. Adv. Mater., 2010, 11, 014107 CrossRef.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- A. Guiseppi-Elie, Biomaterials, 2010, 31, 2701–2716 CrossRef CAS PubMed.
- D. Kim, M. Abidian and D. C. Martin, J. Biomed. Mater. Res., Part A, 2004, 71, 577–585 CrossRef PubMed.
- J. Yang, D. H. Kim, J. L. Hendricks, M. Leach, R. Northey and D. C. Martin, Acta Biomater., 2005, 1, 125–136 CrossRef PubMed.
- J. F. Hulvat and S. I. Stupp, Adv. Mater., 2004, 16, 589–592 CrossRef CAS.
- J. F. Hulvat and S. I. Stupp, Angew. Chem., Int. Ed., 2003, 42, 778–781 CrossRef CAS PubMed.
- G. Li, H. Peng, Y. Wang, Y. Qin, Z. Cui and Z. Zhang, Macromol. Rapid Commun., 2004, 25, 1611–1614 CrossRef CAS.
- H. Yoon, J. Hong and J. Jang, Small, 2007, 3, 1774–1783 CrossRef CAS PubMed.
- Y. Long, M. Li, C. Gu, M. Wan, J. Duvail, Z. Liu and Z. Fan, Prog. Polym. Sci., 2011, 36, 1415–1442 CrossRef CAS PubMed.
- V. Luzzati and P. Spegt, Nature, 1967, 215, 701–704 CrossRef CAS.
- D. W. Bruce, Acc. Chem. Res., 2000, 33, 831–840 CrossRef CAS PubMed.
- S. Kutsumizu, K. Morita, T. Ichikawa, S. Yano, S. Nojima and T. Yamaguchi, Liq. Cryst., 2002, 29, 1447–1458 CrossRef CAS.
- X. Zeng, G. Ungar and M. Impéror-Clerc, Nat. Mater., 2005, 4, 562–567 CrossRef CAS PubMed.
- K. Ozawa, Y. Yamamura, S. Yasuzuka, H. Mori, S. Kutsumizu and K. Saito, J. Phys. Chem. B, 2008, 112, 12179–12181 CrossRef CAS PubMed.
- B. Donnio, B. Heinrich, T. Gulik-Krzywicki, H. Delacroix, D. Guillon and D. W. Bruce, Chem. Mater., 1997, 9, 2951–2965 CrossRef CAS.
- P. Fuchs, C. Tschierske, K. Raith, K. Das and S. Diele, Angew. Chem., Int. Ed., 2002, 41, 628–631 CrossRef CAS.
- M. Ichihara, A. Suzuki, K. Hatsusaka and K. Ohta, Liq. Cryst., 2007, 34, 555–567 CrossRef CAS.
- T. Ichikawa, M. Yoshio, A. Hamasaki, S. Taguchi, F. Liu, X. Zeng, G. Ungar, H. Ohno and T. Kato, J. Am. Chem. Soc., 2012, 134, 2634–2643 CrossRef CAS PubMed.
- T. F. Jaramillo, S. H. Baeck, B. R. Cuenya and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 7148–7149 CrossRef CAS PubMed.
- E. J. Crossland, M. Kamperman, M. Nedelcu, C. Ducati, U. Wiesner, D. Smilgies, G. E. Toombes, M. A. Hillmyer, S. Ludwigs and U. Steiner, Nano Lett., 2008, 9, 2807–2812 CrossRef PubMed.
- H. Wang, C. Oey, A. Djurisic, M. Xie, Y. Leung, K. Man, W. Chan, A. Pandey, J. Nunzi and P. Chui, Appl. Phys. Lett., 2005, 87, 023507 CrossRef PubMed.
- T. Hashimoto, K. Tsutsumi and Y. Funaki, Langmuir, 1997, 13, 6869–6872 CrossRef CAS.
- S. Granick, S. K. Kumar, E. J. Amis, M. Antonietti, A. C. Balazs, A. K. Chakraborty, G. S. Grest, C. Hawker, P. Janmey and E. J. Kramer, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2755–2793 CrossRef CAS.
- B. H. Jones and T. P. Lodge, Chem. Mater., 2010, 22, 1279–1281 CrossRef CAS.
- S. G. Jang, B. J. Kim, C. J. Hawker and E. J. Kramer, Macromolecules, 2011, 44, 9366–9373 CrossRef CAS.
- B. Winther-Jensen, O. Winther-Jensen, M. Forsyth and D. R. MacFarlane, Science, 2008, 321, 671–674 CrossRef CAS PubMed.
- J. H. Pikul, H. G. Zhang, J. Cho, P. V. Braun and W. P. King, Nat. Commun., 2013, 4, 1732 CrossRef PubMed.
- A. M. Urbas, M. Maldovan, P. DeRege and E. L. Thomas, Adv. Mater., 2002, 14, 1850–1853 CrossRef CAS.
- K. Jeong, J. Jang, D. Kim, C. Nah, J. H. Lee, M. Lee, H. Sun, C. Wang, S. Z. Cheng and E. L. Thomas, J. Mater. Chem., 2011, 21, 6824–6830 RSC.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS.
- G. P. Sorenson, K. L. Coppage and M. K. Mahanthappa, J. Am. Chem. Soc., 2011, 133, 14928–14931 CrossRef CAS PubMed.
- P. Khiew, S. Radiman, N. Huang, C. Kan and M. S. Ahmad, Colloids Surf., A, 2004, 247, 35–40 CrossRef CAS PubMed.
- H. Wang, G. Zhang, S. Feng and X. Xie, Colloids Surf., A, 2005, 256, 35–42 CrossRef CAS PubMed.
- S. Kim, W. Oh, Y. S. Jeong and J. Jang, Adv. Funct. Mater., 2013, 23, 1947–1956 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- J. Wang, K. Neoh, E. Kang and K. Tan, J. Mater. Chem., 2000, 10, 1933–1938 RSC.
- X. Zhang and S. K. Manohar, J. Am. Chem. Soc., 2005, 127, 14156–14157 CrossRef CAS PubMed.
- D. C. Martin, Y. Shen, J. Wu, L. Povlich, M. Leach, S. Spanninga and C. Shaw, Proc. ACS Meeting, Salt Lake City, UT, 2009.
- W. Young and T. H. Epps III, Macromolecules, 2012, 45, 4689–4697 CrossRef CAS.
- J. M. Pringle, O. Ngamna, J. Chen, G. G. Wallace, M. Forsyth and D. R. MacFarlane, Synth. Met., 2006, 156, 979–983 CrossRef CAS PubMed.
- E. Park, O. S. Kwon, S. J. Park, J. S. Lee, S. You and J. Jang, J. Mater. Chem., 2012, 22, 1521–1526 RSC.
- F. Tran-Van, S. Garreau, G. Louarn, G. Froyer and C. Chevrot, J. Mater. Chem., 2001, 11, 1378–1382 RSC.
- C. Kvarnström, H. Neugebauer, A. Ivaska and N. Sariciftci, J. Mol. Struct., 2000, 521, 271–277 CrossRef.
- P. Garstecki and R. Hołyst, J. Chem. Phys., 2001, 115, 1095 CrossRef CAS PubMed.
- R. C. Hayward, P. C. Alberius, E. J. Kramer and B. F. Chmelka, Langmuir, 2004, 20, 5998–6004 CrossRef CAS.
- C. Fairhurst, M. Holmes and M. Leaver, Langmuir, 1997, 13, 4964–4975 CrossRef CAS.
- T. Inoue, M. Matsuda, Y. Nibu, Y. Misono and M. Suzuki, Langmuir, 2001, 17, 1833–1840 CrossRef CAS.
- L. Zheng, M. Suzuki and T. Inoue, Langmuir, 2002, 18, 1991–1998 CrossRef CAS.
- C. W. Macosko, Rheology: Principles, Measurements and Applications, VCH Publishers, New York, 1994 Search PubMed.
- R. G. Larson, The structure and rheology of complex fluids, Oxford University Press, New York, 1999 Search PubMed.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367 CrossRef CAS.
- S. R. Raghavan and J. F. Douglas, Soft Matter, 2012, 8, 8539–8546 RSC.
- H. Song, Y. Niu, Z. Wang and J. Zhang, Biomacromolecules, 2011, 12, 1087–1096 CrossRef CAS PubMed.
- S. Kim, Synthesis, characterization, and applications of conducting polymers with functional dopants, PhD Dissertation, Brown University, Providence, RI, 2011 Search PubMed.
- S. N. Patel, A. E. Javier, G. M. Stone, S. A. Mullin and N. P. Balsara, ACS Nano, 2012, 6, 1589–1600 CrossRef CAS PubMed.
- B. Chang and S. Park, Annu. Rev. Anal. Chem., 2010, 3, 207–229 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp04426f |
| ‡ Present address: The Dow Chemical Company, Midland, MI, USA. |
| § Present address: Department of Chemical Engineering, Inha University, Incheon 402-751, South Korea. |
| ¶ Present address: The Dow Chemical Company, Spring House, PA, USA. |
| This journal is © the Owner Societies 2015 |