
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetics of stabilised Criegee intermediates derived from alkene ozonolysis: reactions with SO2, H2O and decomposition under boundary layer conditions
Mike J.
Newland
a,
Andrew R.
Rickard
b,
Mohammed S.
Alam
a,
Luc
Vereecken
c,
Amalia
Muñoz
d,
Milagros
Ródenas
d and
William J.
Bloss
*a
aUniversity of Birmingham, School of Geography, Earth and Environmental Sciences, Birmingham, UK. E-mail: W.J.Bloss@bham.ac.uk
bWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, York, UK, and National Centre for Atmospheric Science, University of York, York, UK
cMax Planck Institute for Chemistry, Atmospheric Sciences, J.-J.-Becher-Weg 27, Mainz, Belgium
dInstituto Universitario CEAM-UMH, EUPHORE Laboratories, Avda/Charles R. Darwin. Parque Tecnologico, Valencia, Spain
First published on 6th January 2015
Abstract
The removal of SO2 in the presence of alkene–ozone systems has been studied for ethene, cis-but-2-ene, trans-but-2-ene and 2,3-dimethyl-but-2-ene, as a function of humidity, under atmospheric boundary layer conditions. The SO2 removal displays a clear dependence on relative humidity for all four alkene–ozone systems confirming a significant reaction for stabilised Criegee intermediates (SCI) with H2O. The observed SO2 removal kinetics are consistent with relative rate constants, k(SCI + H2O)/k(SCI + SO2), of 3.3 (±1.1) × 10−5 for CH2OO, 26 (±10) × 10−5 for CH3CHOO derived from cis-but-2-ene, 33 (±10) × 10−5 for CH3CHOO derived from trans-but-2-ene, and 8.7 (±2.5) × 10−5 for (CH3)2COO derived from 2,3-dimethyl-but-2-ene. The relative rate constants for k(SCI decomposition)/k(SCI + SO2) are −2.3 (±3.5) × 1011 cm−3 for CH2OO, 13 (±43) × 1011 cm−3 for CH3CHOO derived from cis-but-2-ene, −14 (±31) × 1011 cm−3 for CH3CHOO derived from trans-but-2-ene and 63 (±14) × 1011 cm−3 for (CH3)2COO. Uncertainties are ±2σ and represent combined systematic and precision components. These values are derived following the approximation that a single SCI is present for each system; a more comprehensive interpretation, explicitly considering the differing reactivity for syn- and anti-SCI conformers, is also presented. This yields values of 3.5 (±3.1) × 10−4 for k(SCI + H2O)/k(SCI + SO2) of anti-CH3CHOO and 1.2 (±1.1) × 1013 for k(SCI decomposition)/k(SCI + SO2) of syn-CH3CHOO. The reaction of the water dimer with CH2OO is also considered, with a derived value for k(CH2OO + (H2O)2)/k(CH2OO + SO2) of 1.4 (±1.8) × 10−2. The observed SO2 removal rate constants, which technically represent upper limits, are consistent with decomposition being a significant, structure dependent, sink in the atmosphere for syn-SCI.
1. Introduction
Atmospheric oxidation processes are central to understanding trace gas atmospheric composition, the abundance of air pollutants harmful to human health, crops and ecosystems, and the removal of reactive greenhouse gases such as methane. The principal atmospheric oxidants responsible for initiating the gas-phase degradation of volatile organic compounds (VOCs), NOx and SO2 are OH, NO3 and O3, with additional contributions from other species such as halogen atoms. Recent field measurements1 in a boreal forest have identified the presence of an additional oxidant species, removing SO2 and producing H2SO4. The additional SO2 oxidation observed was substantial (comparable to that due to OH radicals alone), and attributed to a product of alkene ozonolysis – the boreal forest environment being one in which substantial biogenic alkene emissions occur – suggested to be the stabilised Criegee intermediate (SCI). The gas-phase oxidation of SO2 in the atmosphere is of interest to climate research as it leads to formation of H2SO4, contributing to new particle formation and sulphate aerosol loading, in competition with condensed phase oxidation.2 A missing mechanism for the conversion of SO2 to H2SO4 could lead to model underestimation of the sulphate aerosol burden and affect radiative forcing calculations,3 with corresponding implications for climate predictions. Enhanced SO2 oxidation in alkene–ozone systems was first reported by Cox & Penkett4 over forty years ago, however the precise reaction mechanism giving rise to this effect remains elusive. The “Criegee” ozonolysis reaction mechanism was first postulated in the 1940s.5 It is now accepted that the ozone molecule adds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond via a concerted cycloaddition to form a primary ozonide, followed by cleavage of the C–C bond and one of the O–O bonds forming a carbonyl molecule and a carbonyl oxide, or ‘Criegee intermediate’ (CI).6 Ozonolysis derived SCIs are formed with a broad internal energy distribution, to yield chemically activated and stabilised SCIs. SCIs can have sufficiently long lifetimes to undergo bimolecular reactions with H2O and SO2 amongst other species. Chemically activated SCIs may also undergo collisional stabilisation, unimolecular decomposition or isomerisation (Scheme 1). For substituted alkenes, SCIs can undergo a 1,4-H-shift rearrangement through a vinyl-hydroperoxide (VHP) via the so-called “hydroperoxide channel” and decompose to yield OH and a vinoxy radical – a substantial non-photolytic source of atmospheric oxidants.7,8 This is the favoured channel for SCIs formed in the syn-configuration. Time resolved studies9 show that the VHP may persist for appreciable timescales under boundary layer conditions, giving rise to the observed pressure dependence of OH radical yields,10 and opening the possibility for bimolecular reactions of this species to occur. SCIs formed in the anti-configuration are thought to primarily undergo rearrangement and decomposition via a dioxirane intermediate (“the acid/ester channel”), producing a range of daughter products and contributing to the observed overall HOx radical yield.6,11
C double bond via a concerted cycloaddition to form a primary ozonide, followed by cleavage of the C–C bond and one of the O–O bonds forming a carbonyl molecule and a carbonyl oxide, or ‘Criegee intermediate’ (CI).6 Ozonolysis derived SCIs are formed with a broad internal energy distribution, to yield chemically activated and stabilised SCIs. SCIs can have sufficiently long lifetimes to undergo bimolecular reactions with H2O and SO2 amongst other species. Chemically activated SCIs may also undergo collisional stabilisation, unimolecular decomposition or isomerisation (Scheme 1). For substituted alkenes, SCIs can undergo a 1,4-H-shift rearrangement through a vinyl-hydroperoxide (VHP) via the so-called “hydroperoxide channel” and decompose to yield OH and a vinoxy radical – a substantial non-photolytic source of atmospheric oxidants.7,8 This is the favoured channel for SCIs formed in the syn-configuration. Time resolved studies9 show that the VHP may persist for appreciable timescales under boundary layer conditions, giving rise to the observed pressure dependence of OH radical yields,10 and opening the possibility for bimolecular reactions of this species to occur. SCIs formed in the anti-configuration are thought to primarily undergo rearrangement and decomposition via a dioxirane intermediate (“the acid/ester channel”), producing a range of daughter products and contributing to the observed overall HOx radical yield.6,11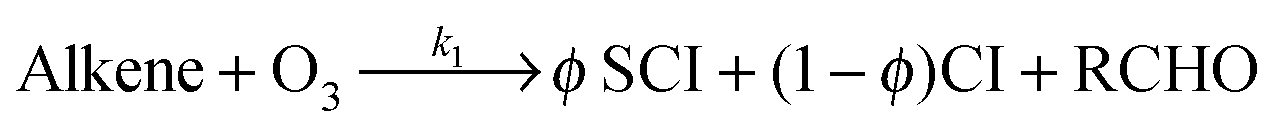 | (R1) |
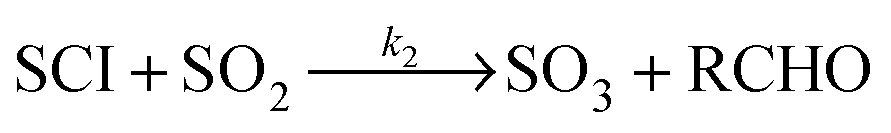 | (R2) |
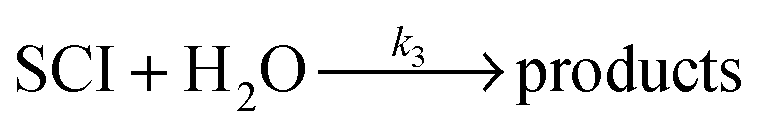 | (R3) |
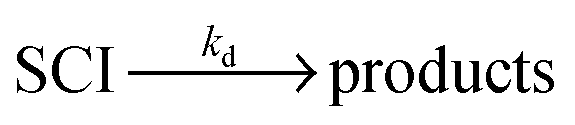 | (R4) |
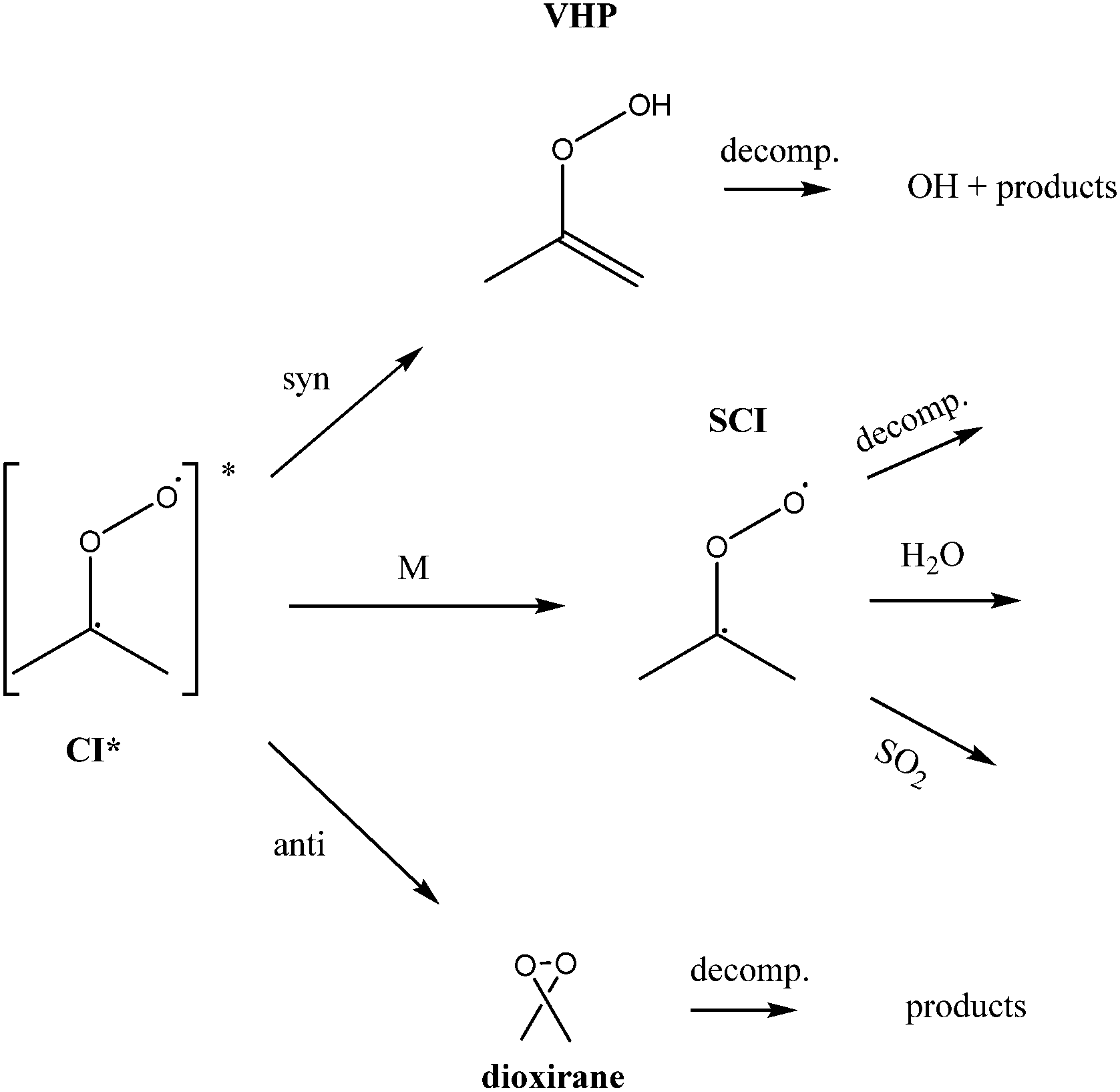 | ||
| Scheme 1 Simplified mechanism for the reaction of Criegee Intermediates (SCIs) formed from alkene ozonolysis. | ||
Until recently, it has been thought that the predominant atmospheric fate for SCIs was reaction with water vapour12,13 – leading to a significant source of organic acids and hydroperoxides, suggesting that bimolecular reaction with SCIs is an unimportant oxidation mechanism for trace gas species. This view was recently challenged by direct observation14 and kinetic studies15–17 of the CH2OO and CH3CHOO SCIs.
Taatjes and co-workers,15 directly observing CI kinetics for the first time, found reactions of CH2OO with SO2 and NO2 to be substantially faster than previously thought, pointing to a potentially important role for this species in atmospheric SO2 and NO2 oxidation; subsequent measurements have identified SO3 (ref. 16) and NO3 (ref. 18) as products of these reactions. The key to whether SCIs are indeed significant contributors to gas-phase atmospheric SO2 oxidation is the ratio of the rate constants for reaction of the SCI with SO2 (k2), to that with H2O (k3) and decomposition (kd). In laboratory studies where SCIs were produced by the 248 nm laser photolysis of alkyl iodide precursors at 4 Torr total pressure, with the SCI decay monitored by VUV photoionisation in the presence of excess co-reactants, this ratio has recently been reported to be 103–104 for the smallest two SCIs (CH2OO15,17 and CH3CHOO16), with k2 on the order of 10−11 cm3 s−1 and k3 on the order of 10−15 cm3 s−1. In contrast, alternative studies of SO2 oxidation in alkene–ozone systems, performed at atmospheric pressure through detection of the H2SO4 product, find much smaller SCI + SO2 rate coefficients (by ca. two orders of magnitude).19 Explanations for this apparent discrepancy may include the lifetime of the secondary ozonide (SOZ) adduct formed from the SCI + SO2 reaction, collisionally stabilised at atmospheric pressure,20 effects of the presence of multiple SCI conformers with differing reactivity, or contributions from other oxidant species, formed within the ozonolysis system, to SO2 reaction. Understanding the behaviour of the ozone–alkene–SO2 system in the presence of water vapour is critical to quantifying the impact of SCI chemistry upon atmospheric SO2 oxidation.21
An additional, potentially important, fate of SCI under atmospheric conditions is unimolecular decomposition (denoted kd in (R4)). For CH2OO, rearrangement via a ‘hot’ acid species represents the lowest accessible decomposition channel, but due to lack of alkyl substituents, the theoretically predicted 298 K rate constant is rather low, 0.3 s−1.22 Previous studies have identified the hydroperoxide rearrangement as dominant for SCIs with a syn configuration, determining their overall unimolecular decomposition rate.7,8 For syn-CH3CHOO recent experimental23 work yielded a decomposition rate of 3–30 s−1. Theoretical work24 has predicted a decomposition rate coefficient of 24.2 s−1 for syn-CH3CHOO and 67.2 s−1 for anti-CH3CHOO (for which only the ester channel exists), owing to the potential energy release from the higher energy anti-conformer.23 An upper limit to total CH3CHOO loss (decomposition and heterogeneous wall losses combined) of <250 s−1 has been reported by Taatjes et al.16 Earlier experimental work reported decomposition rate constants of ≤20 s−1 for CH3CHOO derived from cis-but-2-ene ozonolysis,25 and 76 s−1 (accurate to within a factor of three) for CH3CHOO derived from trans-but-2-ene ozonolysis.26 For (CH3)2COO (derived from 2,3-dimethyl-but-2-ene ozonolysis) a total loss rate of 3.0 s−1 has recently been determined experimentally.19 This value (which represents an upper limit for kd) is somewhat smaller than but comparable in magnitude to an earlier measurement of 6.4 s−1 (determined at 100 Torr).27 Theoretical estimates of (CH3)2COO decomposition rates are higher, at up to 250 s−1.22 Photolysis loss rates have also recently been reported for CH2OO28 and CH3CHOO29 of 1 s−1 and 0.2 s−1 respectively, calculated for actinic flux values at midday, SZA = 0°.
Here, we present results of a series of experiments in which the oxidation of SO2 during the ozonolysis of ethene, cis-but-2-ene, trans-but-2-ene and 2,3-dimethyl-but-2-ene (tetramethylethylene, TME), was investigated in the presence of varying amounts of water in the European Photochemical Reactor facility (EUPHORE), Valencia, Spain.
2. Experimental
2.1 EUPHORE
EUPHORE is a 200 m3 simulation chamber used for studying reaction mechanisms under atmospheric boundary layer conditions. In general, experiments comprised time-resolved measurement of the removal of SO2 in the presence of an alkene–ozone system, as a function of humidity. SO2 and O3 abundance were measured using conventional fluorescence and UV absorption monitors, respectively; alkene abundance was determined via FTIR spectroscopy. The precision of the SO2 and O3 monitors were 0.25 and 0.47 ppbv respectively (evaluated as 2 standard deviations of the measured value prior to SO2 or O3 addition). The chamber is fitted with large horizontal and vertical fans to ensure rapid mixing (three minutes). Further details of the chamber setup and instrumentation are available elsewhere.30,31 Experiments were performed in the dark (i.e. with the chamber housing closed; j(NO2) ≤ 10−6 s−1), at atmospheric pressure (ca. 1000 mbar) and temperatures between 296 and 303 K, on timescales of ca. 20–30 minutes. Chamber dilution was monitored via the first order decay of an aliquot of SF6, added prior to each experiment. Cyclohexane (ca. 75 ppmv) was added at the beginning of each experiment to act as an OH scavenger, such that SO2 reaction with OH was calculated to be ≤1% of the total chemical SO2 removal in all experiments.2.2 Experimental approach
Experimental procedure, starting with the chamber filled with clean air, comprised addition of SF6 and cyclohexane, followed by water vapour, O3 (ca. 500 ppbv) and SO2 (ca. 50 ppbv). A gap of five minutes was left prior to addition of the alkene, to allow complete mixing. The reaction was then initiated by addition of the alkene (ca. 500 ppbv for ethene, 200 ppbv for cis- and trans-but-2-ene and 400 ppbv for TME). The chamber was monitored for an hour subsequent to the addition of ethene and forty five minutes for cis- and trans-but-2-ene and TME. The rate of alkene–ozone consumption is dependent on k1. Roughly 25% of the ethene was consumed after an hour, while for cis- and trans-but-2-ene and TME 90% of the alkene was consumed within roughly 25 minutes, 20 minutes and 6 minutes respectively. Each experiment was performed at a constant humidity, which was increased in a step-wise manner for consecutive runs to cover the range 1.5–21% RH. Measured increases in [SO2] agreed with measured volumetric addition across the SO2 and humidity range used in the experiments.2.3 Analysis approach
The following sections describe (i) a common analysis applied to all systems, but which features several approximations, and (ii) more detailed consideration of each chemical system in turn, to address potential contributions from the water dimer, and multiple criegee species within each system (e.g. contrasting reactivity of different SCI conformers).From the chemistry presented in reactions (R1)–(R4) it is assumed that SCI will be produced in the chamber from the reaction of the alkene with ozone at a given yield, ϕ. The SCI produced can then react with SO2, with H2O, with other species or decompose under the experimental conditions applied. The rate at which SO2 is lost, compared with the total production of SCI, is determined by the fraction, f, of the total SCI produced which reacts with SO2, compared to the sum of the total loss processes of the SCI (eqn (E1)):
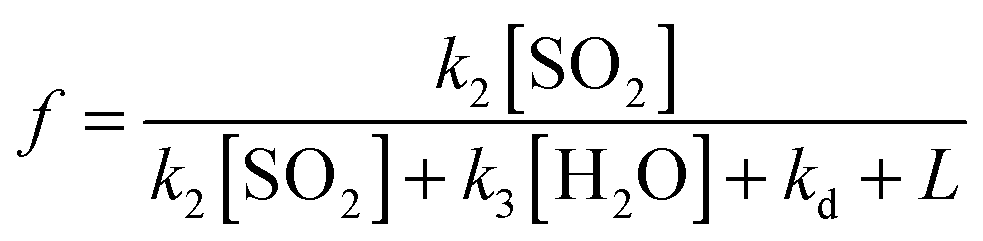 | (E1) |
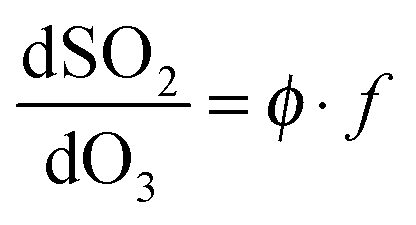 | (E2) |
Here, L is the sum of any other pseudo-first order chemical loss processes for SCI in the chamber, after correction for dilution. (E2) neglects other (non-alkene) chemical sinks for O3, such as reaction with HO2 – also produced directly during alkene ozonolysis,11 but indicated through model calculations to account for <2% of ozone loss under all the experimental conditions of this work. Eqn (E1) and (E2) treat the SCIs formed as a single species – this is the case for (e.g.) ethene and TME, but is an approximation for the 2-butenes; considered further below.
The lower limit criterion applies as in reality f will be less than one, at experimentally accessible SO2 levels, as a small fraction of the SCI will still react with any H2O present, or undergo decomposition. The actual yield, ϕ, was determined by combining the results from the high-SO2 experiments with those from the series of experiments performed at lower SO2, as a function of [H2O], to determine k3/k2 and kd/k2 (see Section 2.3.2), through an iterative process to determine the single unique value of ϕSCI which fits both datasets. It is important to note that the SCI yield is to an extent an operationally defined quantity – for example, OH formation from alkene ozonolysis is known to proceed over at least hundreds of milliseconds9 following the alkene–ozone reaction, and so the corresponding CI population must also be evolving with time. In this work, SCI yields reflect the amount of SCI available to oxidise SO2 on timescales of seconds to minutes.
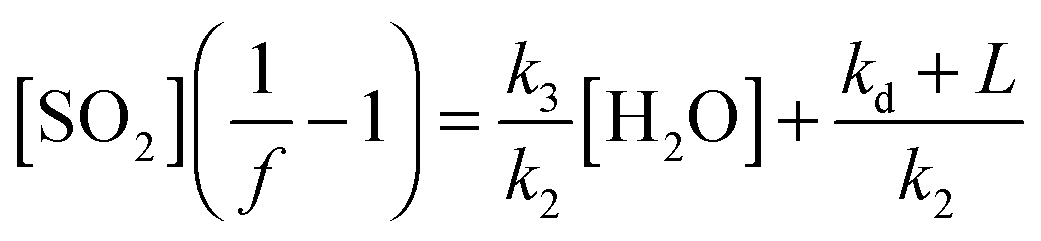 | (E3) |
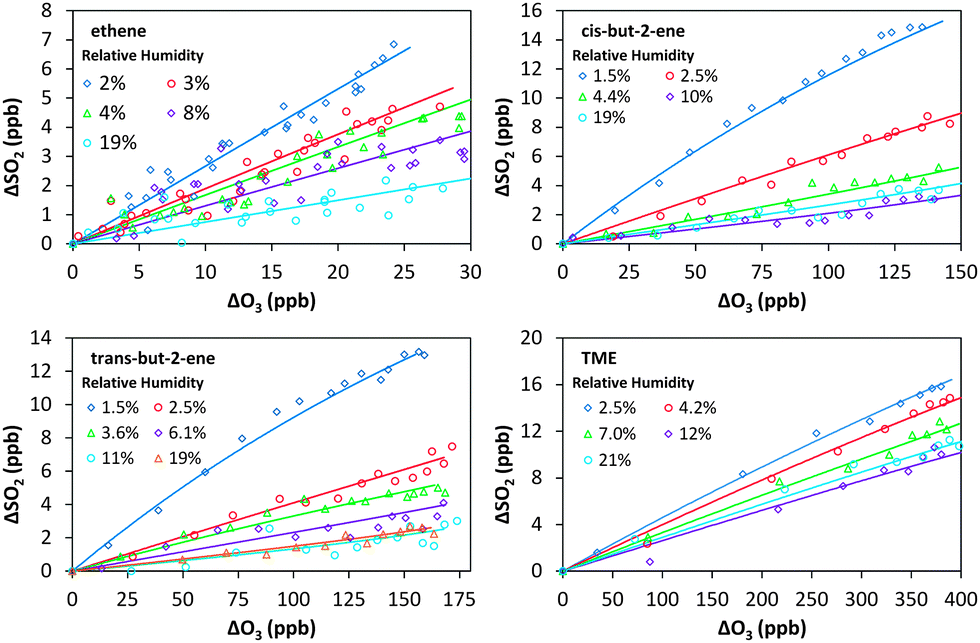 | ||
| Fig. 1 Cumulative consumption of SO2 and O3, ΔSO2versus ΔO3, for the ozonolysis of four alkenes in the presence of SO2 at a range of relative humidities from 1.5–21%. Open symbols are experimental data, corrected for chamber dilution. Solid lines are smoothed fits to the experimental data. | ||
 | ||
| Fig. 2 ΔSO2vs. ΔO3 during the excess SO2 experiments, to determine the minimum SCI yield for the four alkenes. | ||
The value [SO2]((1/f) − 1) can then be regressed against [H2O] for each experiment to give a plot with a gradient of k3/k2 and an intercept of (kd + L)/k2 (eqn (E3)). Our data cannot determine absolute rate constants (i.e. values of k2, k3, kd) in isolation, but is limited to assessing their relative values, which may be placed on an absolute basis through use of an (external) reference value.
Eqn (E1)–(E3) as presented above assume that only a single SCI species is present in each ozonolysis system. While this is the case for the ethene and TME systems, for the but-2-ene systems this is an approximation as two conformers of the CH3CHOO SCI (syn and anti) are produced. Further analysis is performed in Section 3.3.2 in which the SO2 loss in the but-2-ene systems is treated as having two components, related to the different SCI.
3. Results and discussion
3.1 Introduction
Table 1 shows the resulting SCI yields obtained for the ozonolysis of ethene, cis-but-2-ene, trans-but-2-ene and tetramethylethylene (TME); uncertainties are ±2σ, and represent the combined systematic (estimated measurement uncertainty) and precision components. Also shown are past literature values, obtained under various conditions using a range of different SCI scavengers.| SCI | φ SCI | Ref. |
|---|---|---|
| a http://mcm.leeds.ac.uk/MCM/. b At 700–760 Torr (low pressure limit of 0.15 at 0 Torr). | ||
| CH2OO | 0.37 (±0.04) | This work |
| 0.37 | MCMv3.2a (IUPAC)33 | |
| 0.35 (±0.05) | Niki et al.34 | |
| 0.39 (±0.053) | Hatakeyama et al.35 | |
| 0.47 (±0.05) | Horie and Moortgat36 | |
| 0.50 (±0.04) | Neeb et al.37 | |
| 0.39 (±0.11) | Hasson et al.38 | |
| 0.54 (±0.15) | Alam et al.31 | |
| CH3CHOO | 0.38 (±0.05) | This work |
| (C2B) | 0.19 | Rickard et al.39 |
| 0.18 | Niki et al.40 | |
| 0.43 | Cox and Penkett41 | |
| CH3CHOO | 0.28 (±0.03) | This work |
| (T2B) | 0.49 (±0.22) | Berndt et al.32 |
| 0.53 (±0.24) | Berndt et al.19 | |
| 0.45 | Cox and Penkett41 | |
| 0.19 (±0.03) | Hatakeyama et al.35 | |
| 0.42 | Horie and Moortgat36 | |
| 0.24 (±0.07) | Hasson et al.38 | |
| 0.13 | Rickard et al.39 | |
| (CH3)2COO | 0.32 (±0.02) | This work |
| 0.45 (±0.20) | Berndt et al.32 | |
| 0.62 (±0.28) | Berndt et al.19 | |
| ca. 0.65b | Drozd et al.10 | |
| 0.10 (±0.03) | Hasson et al.38 | |
| 0.30 | Niki et al.40 | |
| 0.11 | Rickard et al.39 | |
The yield of CH2OO from ethene ozonolysis obtained in this work is 0.37 (±0.04). This yield has been investigated in many previous studies, with values ranging from 0.34–0.50 determined – a more detailed review is available elsewhere.31 The yield obtained in this work is at the lower end but within the envelope of these estimates, and is in excellent agreement with the current IUPAC recommendation of 0.37.
The yield of CH3CHOO from cis-but-2-ene ozonolysis obtained in this work is 0.38 (±0.05), with that from trans-but-2-ene being 0.28 (±0.03). These values fall within the range of reported literature values of 0.18–0.43 and 0.13–0.53 for cis-but-2-ene and trans-but-2-ene respectively (Table 1). cis and trans-but-2-ene both yield (differing) mixtures of syn and anti conformers of CH3CHOO, the relative amounts of which are not well known, and which are treated here initially as a single SCI species (this approximation is discussed further in Section 3.3). Berndt et al.32 recently reported a yield of 0.49 (±0.22) for the CH3CHOO produced from trans-but-2-ene ozonolysis (also treating both syn and anti conformers as a single SCI species).
The yield of (CH3)2COO from TME ozonolysis obtained in this work is 0.32 (±0.02). This again falls within the (wide) range in the literature of 0.10–0.65, with Berndt et al.32 most recently reporting a yield of 0.45 (±0.20).
Fig. 1 shows the cumulative consumption of SO2 relative to that of O3, ΔSO2versus ΔO3 (after correction for dilution), as a function of [H2O] for each experiment for the four alkenes studied. A fit to each experiment, extrapolating the experimental data to evaluate dSO2/dO3 at t = 0 (start of each experimental run) for use in eqn (E1)–(E3), is also shown. The overall change in SO2, ΔSO2, is seen to decrease substantially with increasing humidity (over a relatively narrow range of RH (1.5–20%)) for all four alkenes. This trend would be expected from the understood chemistry ((R1)–(R4)), as there is competition between SO2, H2O, and decomposition for reaction with the SCI.
Fig. 3 shows a fit of eqn (E3) to the data for each alkene, giving a slope of k3/k2, and an intercept of (kd + L)/k2. The results appear to show a generally linear relationship; however, for cis- and trans-but-2-ene and TME, the data point at the highest relative humidity accessible in this work ([H2O] = 1.5–2.0 × 1017 cm−3) appears to deviate from this relationship. These data points lie outside the 95% confidence intervals defined by all the other (lower relative humidity) data for each alkene. For the analysis to determine k3/k2 and (kd + L)/k2 presented in Table 2, the points at the highest RH are excluded and the kinetic parameters are derived from a linear fit to the measurements from all other experiments. Extended analyses to account for the non-linearity observed for CH3CHOO and (CH3)2COO are presented in the following sections.
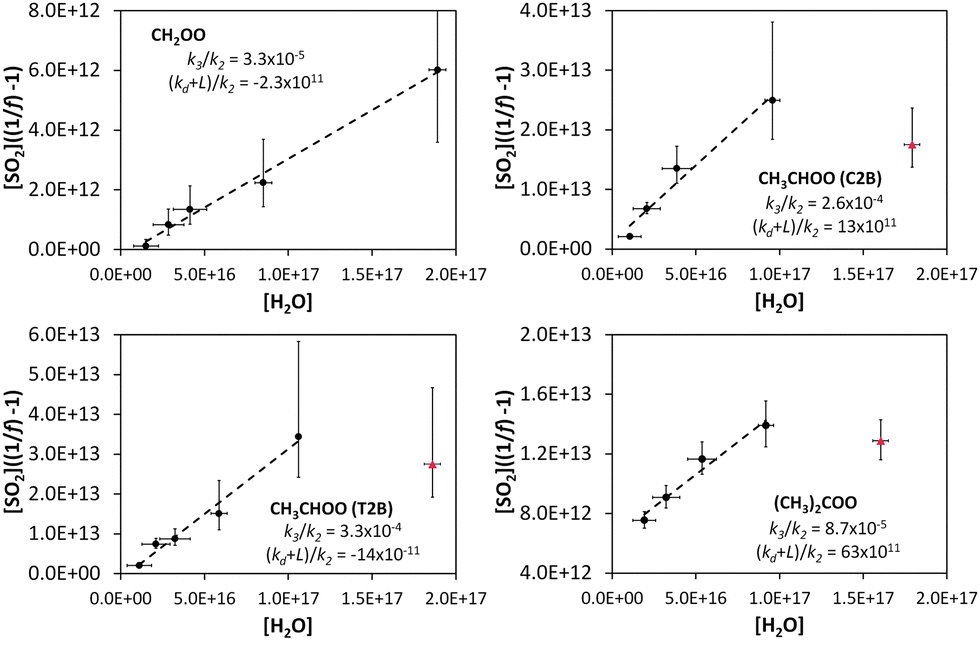 | ||
| Fig. 3 Application of eqn (E3) to derive rate constants for reaction of CH2OO, CH3CHOO (derived from cis-but-2-ene; C2B), CH3CHOO (derived from trans-but-2-ene; T2B), and (CH3)2COO with H2O (k3) and decomposition (kd), relative to that for reaction with SO2, k3/k2 and (kd + L)/k2 – see text. | ||
| SCI | 105k3/k2 | 10−11 cm−3kd/k2 |
|---|---|---|
| CH2OO | 3.3 (±1.1) | −2.3 (±3.5) |
| CH3CHOO (C2B) | 26 (±10) | 13 (±43) |
| CH3CHOO (T2B) | 33 (±10) | −14 (±31) |
| (CH3)2COO | 8.7 (±2.5) | 63 (±14) |
One potential explanation for the observed curvature in the CH3CHOO and (CH3)2COO data is measurement error. ΔSO2 is relatively small at high [H2O] compared to the precision of the measurements; however, even allowing for associated uncertainties, the points at high RH do not fit the linear relationship successfully applied to the remaining data points. Moreover, any systematic error in the measurement of O3, SO2 or H2O would also be expected to affect the results for the ethene system (and to a greater extent, given the slow ethene–ozone reaction rate and consequent lower overall chemical SO2 loss observed), suggesting that the cause lies in contrasting chemical behaviour. In terms of experimental factors, H2O was measured using multiple approaches (two dew-point hygrometers in addition to a solid state probe) with no evidence for any divergence with RH. SO2 monitors can exhibit humidity-dependent interferences (quenching of the SO2 signal), commonly of the order of a few percent, observed at very high RH, and corrected through incorporation of a nafion dryer (fitted in this case); in addition the monitor-derived SO2 concentration increments were in agreement with those calculated from the measured SO2 addition and chamber volume, across the relative humidity range studied.
It should be noted that the kd values reported here represent upper limits, as a consequence of possible further chemical losses for the SCI within our experimental system (as represented by L in eqn (E3), notwithstanding the approach of extrapolating to the start of each experiment to minimise these). Other potential fates for SCIs include reaction with ozone,42,43 other SCI,43 carbonyl products,44 acids,45 or with the parent alkene43 itself. Sensitivity analyses indicate that the reaction with ozone could be significant, as predicted by theory42,43 with a possible contribution of up to 10% of SCI loss for (CH3)2COO at 2% RH, while total losses from reaction with SCI (self-reaction), carbonyls and alkenes are calculated to account for <1% of the total SCI loss under the experimental conditions applied.
3.2 CH2OO
These relative rates can be placed on an absolute basis using absolute measurements of k2(SCI + SO2). In Table 3 we apply the absolute k2 values reported by Welz et al.,15 obtained using direct methods at reduced pressure (4 Torr), to the relative rates shown in Table 2. Using this method, the value obtained for k3(CH2OO + H2O) is 1.3 (±0.4) × 10−15 cm3 s−1. This is consistent with the recent determination by Welz et al.,15 that k3 < 4 × 10−15 cm3 s−1, but is (ca. 14–50 times) greater than the recent estimates of Ouyang et al.18 (k3 = 2.5 (±1) × 10−17 cm3 s−1) and Stone et al.17 (k3 < 9 × 10−17 cm3 s−1).
| SCI | 105k3/k2 | 1015k3 (cm3 s−1) | 10−11kd/k2 (cm−3) | k d (s−1) | Ref. | Method | Conditionsa |
|---|---|---|---|---|---|---|---|
| a Experiments were conducted at atmospheric pressure unless stated otherwise. b Assuming k2 = 3.9 × 10−11 cm3 s−1 (Welz et al.15). c Assuming k2 = 4.55 × 10−11 cm3 s−1. Average of k2 for syn and anti CH3CHOO conformers from Taatjes et al.16 d Assuming k2 = 2.4 × 10−11 cm3 s−1. k2 for syn-CH3CHOO from Taatjes et al.16 | |||||||
| CH2OO | 3.3 (±1.1) | 1.3 (±0.4)b | −2.3 (±3.5) | −8.8 (±13)b | This work | Ethene ozonolysis | 298–303 K |
| <4 | Welz et al.15 | Alkyl iodide photolysis | 4 Torr; 298 K | ||||
| <0.09 | Stone et al.17 | Alkyl iodide photolysis | 200 Torr; 295 K | ||||
| CH3CHOO | 26 (±10) | 12 (±4.5)c | 13 (±43) | 59 (±196)c | This work | C2B ozonolysis | 296–302 K |
| 33 (±10) | 15 (±4.5)c | −14 (±31) | −64 (±141)c | This work | T2B ozonolysis | 297–302 K | |
| anti 10 (±4); syn <4 | Taatjes et al.16 | Alkyl iodide photolysis | 4 Torr; 298 K | ||||
| 8.8 (±0.4) | 12 (±0.1) | Berndt et al.32 | T2B ozonolysis | 293 K | |||
| 76 (25–228) | Fenske et al.26 | T2B ozonolysis | 296 K | ||||
| (CH3)2COO | 8.7 (±2.5) | 2.1 (±0.6)d | 63 (±14) | 151 (±35)d | This work | TME ozonolysis | 298–299 K |
| <0.4 | 42 (±3) | Berndt et al.32 | TME ozonolysis | 293 K | |||
The derived (kd + L) value for CH2OO using this method is −8.8 (±13) s−1, i.e. zero within uncertainty. Theoretical work22 has predicted kd(CH2OO) to be small (∼0.3 s−1), in agreement with the experimentally derived value reported here.
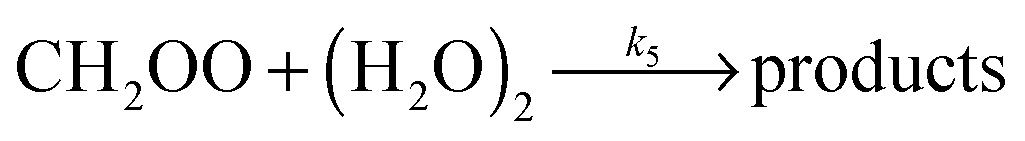 | (R5) |
The CH2OO data from this study appear to be well described by a linear fit under the experimental conditions applied (a fast reaction of CH2OO with (H2O)2 would be manifested as a significant upward curvature in Fig. 3). However, this does not mean the results are inconsistent with reaction of CH2OO with (H2O)2.
In Fig. 4, eqn (E4) (an expanded version of eqn (E3), including the SCI + (H2O)2 reaction (R5)) is applied to the data, now expressed in terms of (H2O)2, calculated for each RH via the equilibrium constant.48
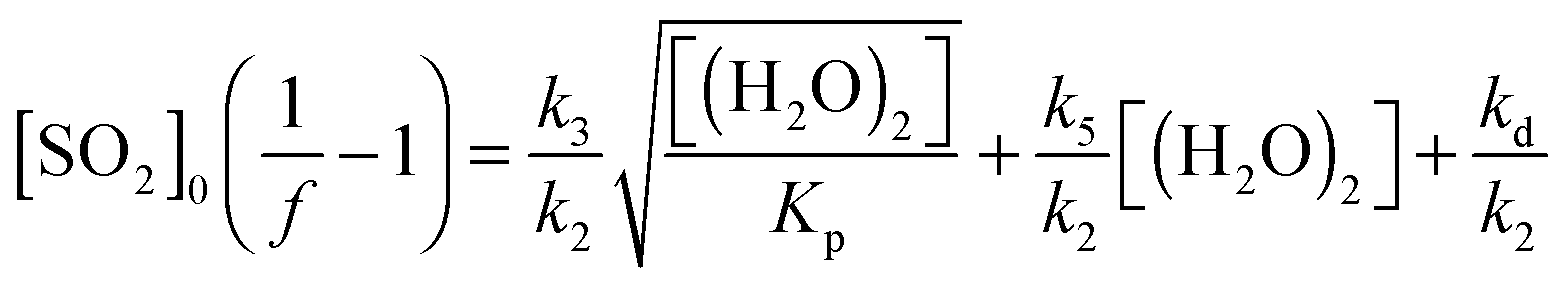 | (E4) |
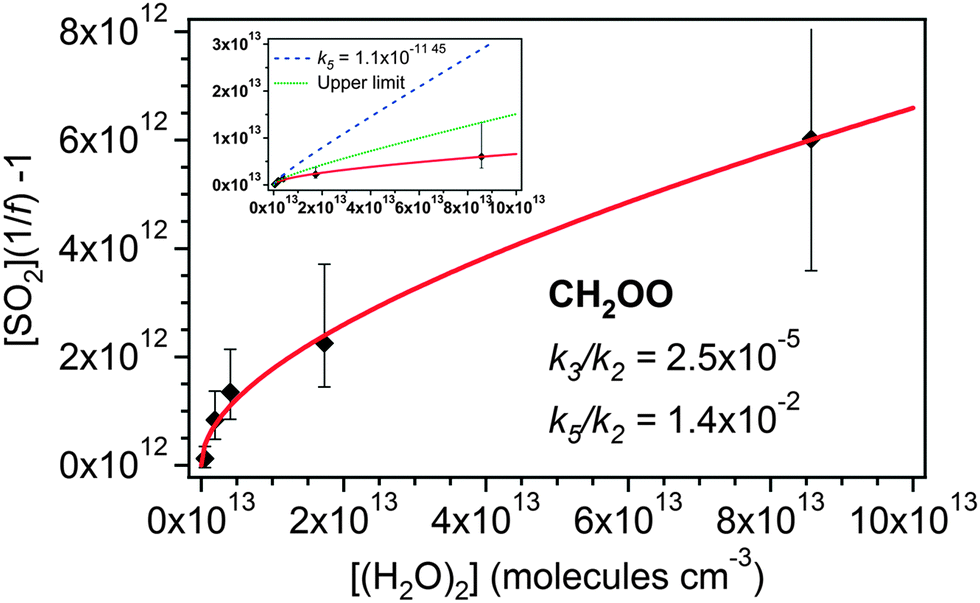 | ||
| Fig. 4 Application of eqn (E4) to derive rate constants for reaction of CH2OO with H2O (k3/k2) and (H2O)2 (k5/k2) relative to that of CH2OO with SO2. Inset: eqn (E4) as shown in the main figure (red line), (E4) applied using the dimer reaction rate (k5) reported by Berndt et al.45 (1.1 × 10−11 cm3 s−1) (dashed line) and a fit of (E4) to the upper limits of the uncertainties in the ethene data (solid green line). | ||
The value for k3/k2 (water monomer) derived from the fit shown in Fig. 4 is 2.5 (±0.7) × 10−5. It is seen that this value is rather insensitive to the inclusion of the (H2O)2 term in eqn (E4) as the value is within the uncertainties of the linear fit to the data presented in Fig. 3 – see also Table 2. Converting this value to an absolute value using the k2 from Welz et al.15 gives k3 = 9.9 (±2.9) × 10−16 cm3 s−1. The derived value of kd/k2 is −6.4 (±66) × 1010 cm−3, which, using the Welz et al.15k2 gives an absolute value for kd of −1.5 (±16) s−1. This is again indistinguishable from zero, within uncertainty, as is the kd determined from eqn (E3) (Fig. 3). Note the large uncertainties in kd, resulting from allowing three parameters to vary in the optimisation; consequently kd was fixed to zero in eqn (E4) to determine the k3/k2 and k5/k2 values.
The resulting value of k5/k2 (water dimer) is 1.4 (±1.8) × 10−2. Converting this to an absolute value using the k2 from Welz et al.15 gives k5 = 5.6 (±7.0) × 10−13 cm3 s−1. This is roughly a factor of twenty smaller than the value derived by Berndt et al.,46 but within a factor of two of the upper limit for k5 deduced by Welz et al. (<3 × 10−13 cm3 s−1) (ref. 45) from the data presented by Stone et al.17 The inset plot in Fig. 4 also shows two additional fits generated using eqn (E4) with k3/k2 fixed to 9.9 × 10−16 and kd fixed to zero. One fit line uses the k5 value reported by Berndt et al.46 (blue dashed line). This is seen to overestimate the presented data. The green dotted line shows a fit to the upper limits of the uncertainties of the measured data. This yields a k5/k2 value of 0.10 (±0.01), giving an upper limit k5 value of 3.9 (±0.39) × 10−12 cm3 s−1.
The contribution of (H2O)2 to the removal of CH2OO increases in relation to that of H2O as [H2O] increases. Hence at typical atmospheric [H2O] (∼2.5–7.5 × 1017 molecules per cm−3), greater than was accessible in this study, reaction with (H2O)2 could become the dominant sink for CH2OO. In this case using just the H2O monomer kinetics in models would considerably underestimate the total effect of water on removal of CH2OO in the atmosphere.
3.3 CH3CHOO
The relative rate constants (Table 2) can be placed on an absolute basis using the measurements of k2(SCI + SO2) reported by Taatjes et al.16 (derived using the same methodology as for CH2OO) (Table 3). As eqn (E3) treats the SCI produced as a single SCI, we use an average of the syn and anti conformer rates presented in Taatjes et al.,16 4.55 × 10−11 cm3 s−1. Using this method, the value obtained for k3(CH3CHOO + H2O) from cis-but-2-ene ozonolysis is 12 (±4.5) × 10−15 cm3 s−1 and from trans-but-2-ene ozonolysis is 15 (±4.5) × 10−15 cm3 s−1. Taking a mean of the k3 values reported for the two CH3CHOO conformers by Taatjes et al.16 gives a value of 7.0 × 10−15 cm3 s−1, while Sheps et al.49 give a mean value of 12 × 10−15 cm3 s−1. The values obtained for (kd + L) are 59 (±196) s−1 from cis-but-2-ene and −64 (±141) s−1 from trans-but-2-ene. Clearly there is a large uncertainty associated with the kd determined from this analysis. Fenske et al.26 have reported kd(CH3CHOO) from trans-but-2-ene ozonolysis to be 76 s−1 (accurate to within a factor of three).
One explanation for the observed non-linearity at high RH apparent in Fig. 3 is the differing reactivities of the syn- and anti-conformers of CH3CHOO produced in the ozonolysis of cis- and trans-but-2-ene. It has been predicted50 that the anti-conformer reacts with H2O several orders of magnitude faster than the syn-conformer, while the rate constant for the SCI reaction with SO2 has been determined experimentally16 to be about a factor of three greater for the anti-conformer than the syn-conformer. The fraction of each conformer that is lost to reaction with SO2 can be considered in the same way as illustrated in eqn (E2), leading to eqn (E5) and (E6) below, plus simplifications outlined in the following text. The total loss of SO2 to CH3CHOO is then the sum of the fractional loss to each conformer, multiplied by the relative SCI yield (γ) (i.e. φsyn/φ) of that conformer (eqn (E7)).
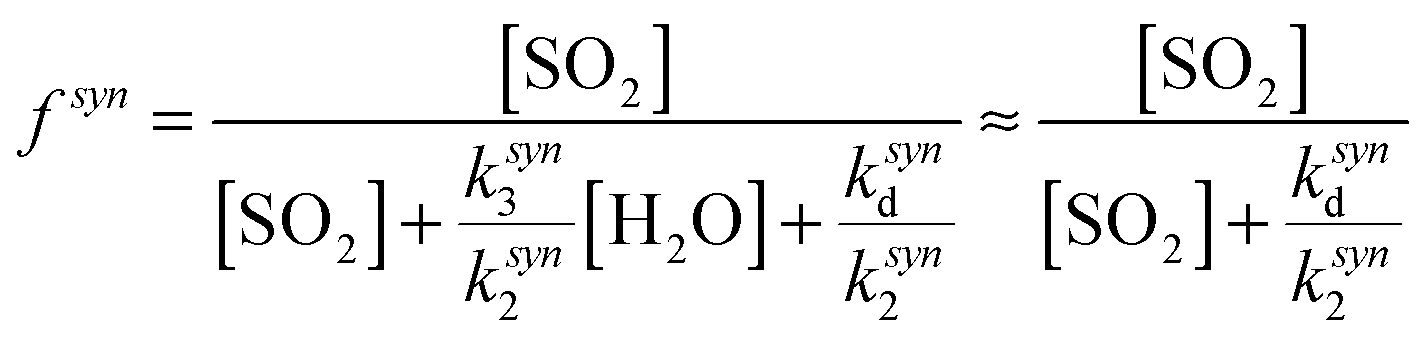 | (E5) |
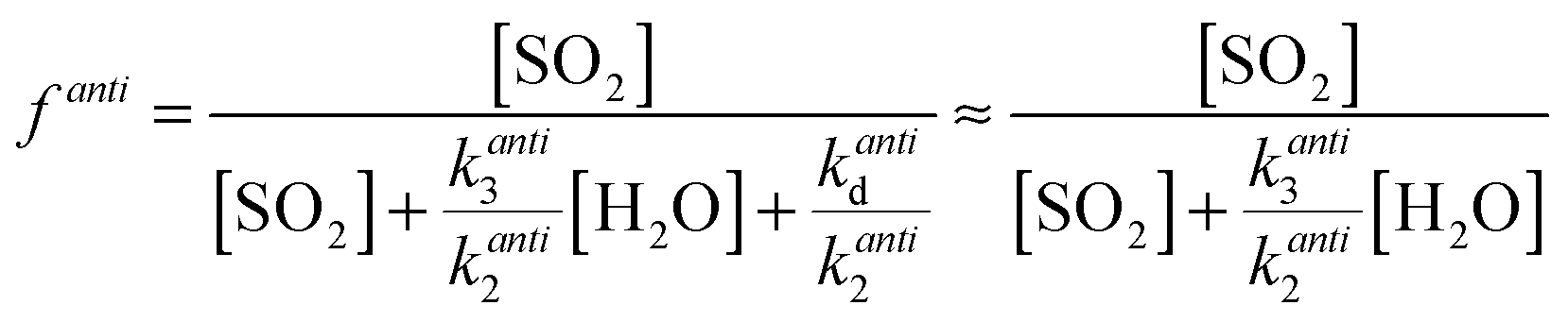 | (E6) |
| f = γsynfsyn + γantifanti | (E7) |
Eqn (E8) can then be fitted to the data presented in Fig. 3 for cis- and trans-but-2-ene (Fig. 5).
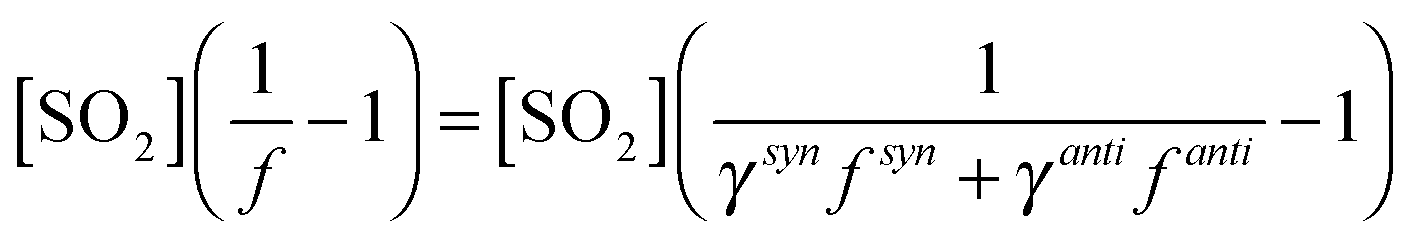 | (E8) |
Here we make two assumptions to reduce the degrees of freedom and hence make the problem tractable with the dataset available. First it is assumed that ksyn3[H2O] may be neglected, in keeping with theoretical predictions24 predicting ksynd to be over three orders of magnitude greater than ksyn3[H2O]. Further theoretical work50 predicts a rate constant for syn-CH3CHOO + H2O of 2.39 × 10−18 cm3 s−1, and recent experimental work49 yields an upper limit for ksyn3 of <2 × 10−16 cm3 s−1. Hence with ksynd expected to be relatively fast, given the decomposition rate presented here for (CH3)2COO (a syn-conformer) and the facile decomposition route available via the hydroperoxide mechanism for the syn-conformer, it can be assumed that ksynd ≫ ksyn3[H2O] at the experimental conditions reported here. Second it is assumed that kantid ≪ kanti3[H2O] under the experimental conditions used. If the kinetics derived from treating the but-2-ene data in Fig. 3 as representing a single SCI are dominated by the anti-conformer then the kd derived from these kinetics, which is indistinguishable from zero within the uncertainties, suggests that kantid is small. Taatjes et al.16 report kanti3 to be 1.0 (±0.4) × 10−14 cm3 s−1, while Sheps et al.49 report a value of 2.4 (±0.4) × 10−14 cm3 s−1. Thus, even at the lowest [H2O] considered here (∼1 × 1016 cm−3), loss of anti-CH3CHOO to H2O would be >100 s−1, and decomposition negligible in comparison (including a kantid of 50 s−1 changes the derived kanti3 and ksynd values by <5%).
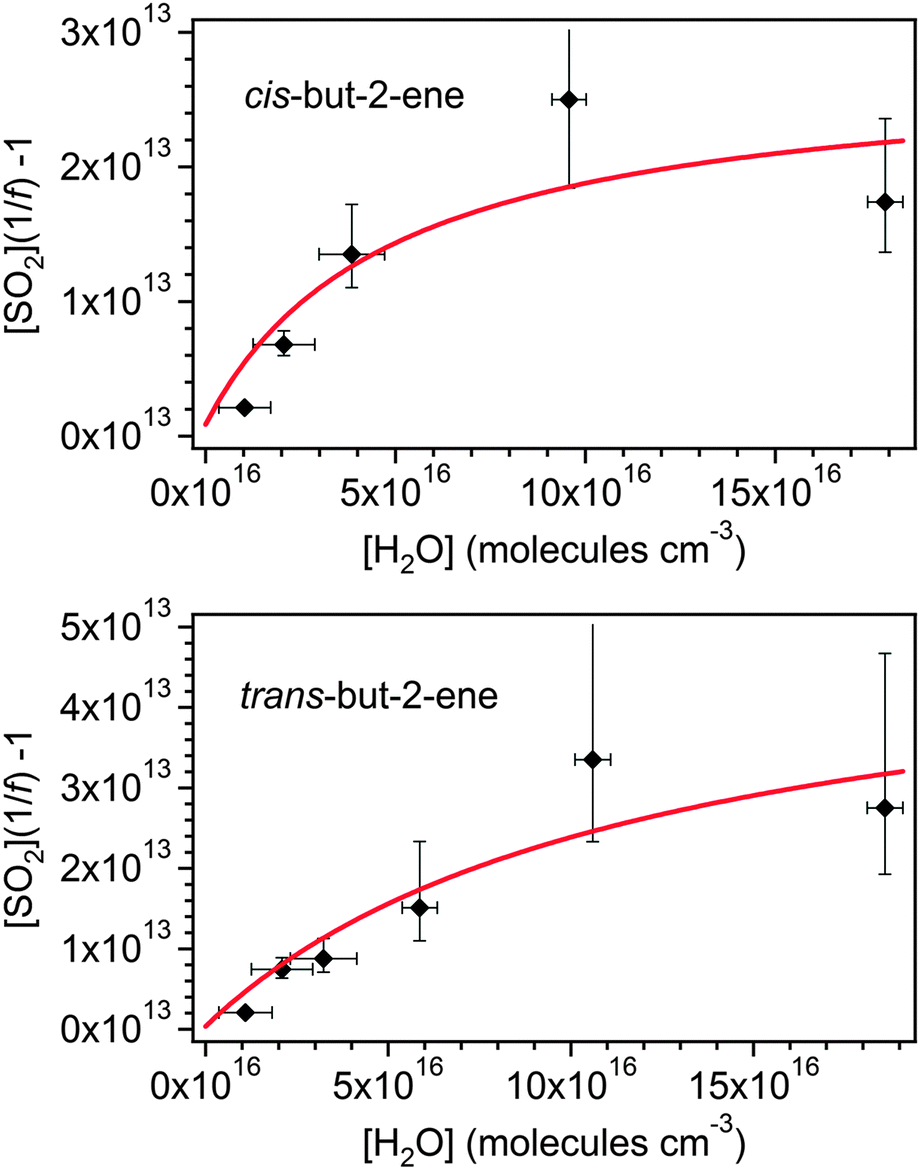 | ||
| Fig. 5 Fits of eqn (E8) to the cis-but-2-ene and trans-but-2-ene data shown in Fig. 3. γsyn and γanti are 0.45 and 0.55 for cis-but-2-ene and 0.25 and 0.75 for trans-but-2-ene (see Fig. 6). | ||
Fitting eqn (E8) to the data shown in Fig. 5 derives a range of values for kanti3 and ksynd dependent on the values of γanti and γsyn used. As the CIs, once formed and thermalised, are expected to show the same kinetic behaviour in these experiments irrespective of their precursor alkene, the measurements from the cis-but-2-ene and trans-but-2-ene experiments can be used in combination to constrain kanti3, ksynd and also γsyn and γanti from each alkene. Fig. 6 plots the kanti3vs. ksynd determined at different γsyn and γanti from cis-but-2-ene and trans-but-2-ene. Where these two lines intercept represents the unique solution for both kanti3 and ksynd and for γsyn and γanti (Table 4).
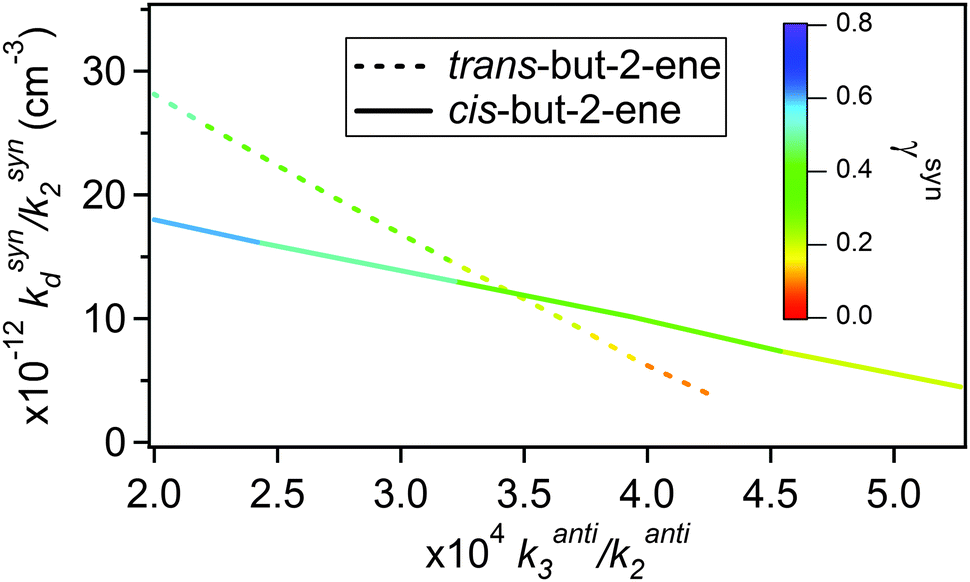 | ||
| Fig. 6 The ranges of kanti3/kanti2 and ksynd/ksyn2 determined from fitting eqn (E8) to the but-2-ene data (Fig. 5). The colour legend shows the fraction of the total CH3CHOO formed that is syn-CH3CHOO (γsyn). | ||
| CH3CHOO | 104k3/k2 | 1014k3 (cm3 s−1) | 10−13kd/k2 (cm−3) | k d (s−1) | γ | φ | Ref. | Conditionsb | ||
|---|---|---|---|---|---|---|---|---|---|---|
| T2B | C2B | T2B | C2B | |||||||
| a γ syn = φsyn/φ. b Experiments were conducted at atmospheric pressure unless stated otherwise and temperatures from 293 to 303 K. | ||||||||||
| syn | — | — | 1.2 (±1.1) | 288 (±275) | 0.25 | 0.45 | 0.07 | 0.17 | This work | — |
| <0.4 | Taatjes et al.16 | 4 Torr | ||||||||
| <0.02 | Sheps et al.49 | 200 Torr | ||||||||
| 20 (3–30) | Novelli et al.23 | 735 Torr | ||||||||
| anti | 3.5 (±3.1) | 2.3 (±2.1) | — | — | 0.75 | 0.55 | 0.21 | 0.21 | This work | — |
| 1.0 (±0.4) | Taatjes et al.16 | 4 Torr | ||||||||
| 2.4 (±0.4) | Sheps et al.49 | 200 Torr | ||||||||
Fig. 6 determines kanti3/kanti2 to be 3.5 (±3.1) × 10−4 and ksynd/ksyn2 to be 1.2 (±1.1) × 1013 cm−3. The 2σ uncertainties presented are, unsurprisingly, large as there are two free parameters. Using the relevant values of k2 for the syn and anti-CH3CHOO conformers from Taatjes et al.16 to place the relative rate constants on an absolute basis gives a value for kanti3 of 2.3 (±2.1) × 10−14 cm3 s−1 and for ksynd of 288 (±275) s−1. This kanti3 is comparable to (a factor of two greater than) that reported by Taatjes et al.16 (1.0 (±0.4) × 10−14). Novelli et al.23 have recently reported ksynd to be an order of magnitude smaller (3–30 s−1) based on direct observation of OH formation during trans-but-2-ene ozonolysis at atmospheric pressure.
The point of interception in Fig. 6 also determines the relative yields of the two conformers, γsyn and γanti (which in turn has been used to derive the optimised fits shown in Fig. 5). For cis-but-2-ene these are determined as 0.45 and 0.55 for γsyn and γanti respectively. For trans-but-2-ene they are determined as 0.25 for γsyn and 0.75 for γanti. The analysis performed in this section has implications for the determination of the SCI yield. Using the relative rate constant k3/k2(anti-CH3CHOO) obtained, as shown in Table 4, it is calculated that ∼90% of the anti-CH3CHOO produced in the SCI yield experiments would react with SO2. From the determined kd/k2(syn-CH3CHOO) it is calculated that ∼67% of the syn-CH3CHOO produced in the SCI yield experiments would react with SO2. Applying these (with the corresponding syn and anti yields shown in Table 4) corrections to φmin determines total SCI yields of 0.29 for trans-but-2-ene and 0.42 for cis-but-2-ene. These values both lie within the uncertainties in the SCI yields presented in Table 1 for the two but-2-ene systems.
It is not practicable to assess the possible contribution of the water dimer to the SCI loss for CH3CHOO because of the number of free parameters that would result for a small dataset. However, theoretical predictions47 suggest that this may be less important for CH3CHOO than for CH2OO, indicating k(H2O)2/k(H2O) to be two orders of magnitude smaller for anti-CH3CHOO than for CH2OO.
3.4 (CH3)2COO
Berndt et al.32 have recently reported the k3/k2 ratio for (CH3)2COO to be <0.4 × 10−5 (i.e. approximately a factor of 22 lower than the relative rate reported in this study). Theoretical predictions50 also suggest k3 to be very slow, 3.9 × 10−17 cm3 s−1. No measured values have been reported for kd ((CH3)2COO), but a more facile overall decomposition than for CH2OO or the mean of the CH3CHOO isomers might be anticipated as the vinyl-hydroperoxide isomerisation channel26 is always available.
Eqn (E9) (below) is an expanded version of (E2), in which we consider the contribution from a second SO2 oxidant, making the approximation that this species does not react appreciably with water vapour. In eqn (E9), f is the sum of fSCI (the fraction of SCI reacting with SO2) and fx, each multiplied by the relative yield of the total oxidant (i.e. SCI + X) γSCI and γx. Following the assumption of negligible H2O reactivity, (dSO2/dO3)x in eqn (E9) can be derived from the SO2 loss at the highest RH experiments (i.e. when all the SO2 loss is attributed to X + SO2) of ∼10 ppbv. Therefore, loss of SO2 from reaction with X, relative to the loss of O3 (dSO2/dO3)x is approximately 0.025. φ represents the total oxidant yield (i.e. φSCI + φx). Assuming that γx is not dominant (<0.5), then φ, as calculated from correcting φmin as in Section 3.1, changes little (0.31–0.34) from the value presented in Table 1. Eqn (E10) is then an expanded version of eqn (E3) that includes the additional oxidant term.
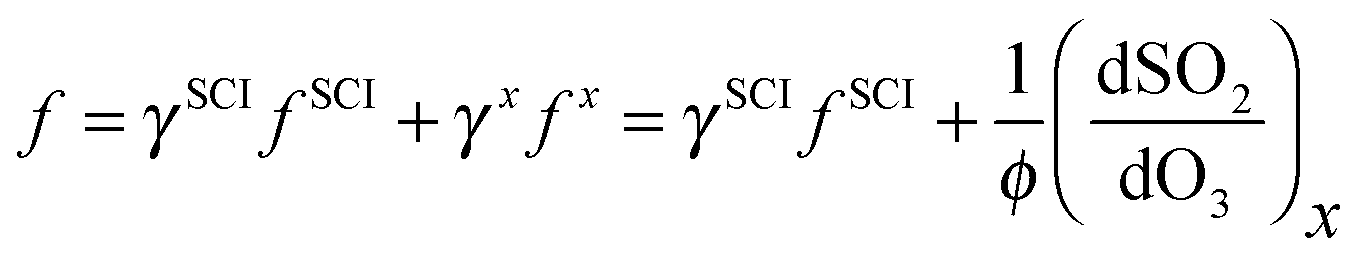 | (E9) |
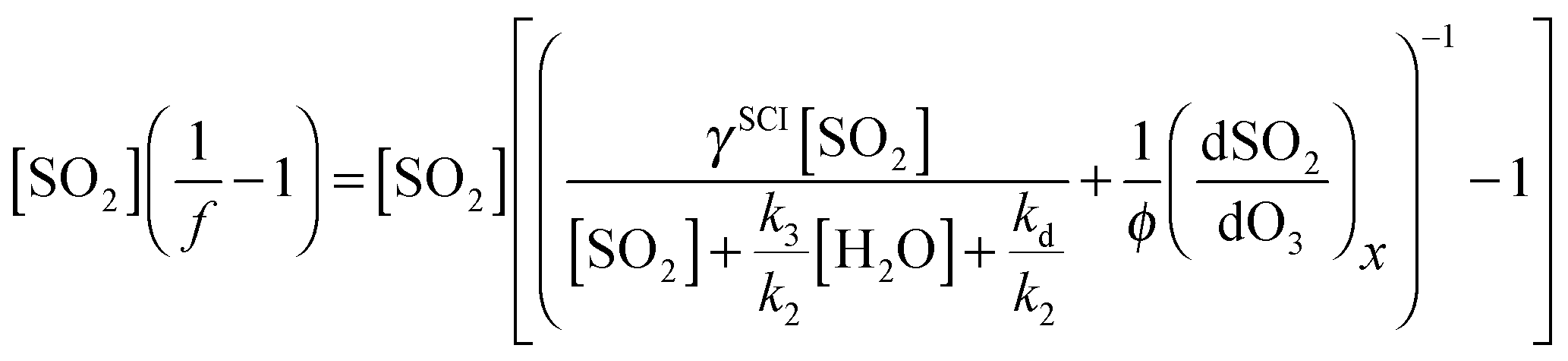 | (E10) |
Fig. 7 shows eqn (E10) fitted to the TME data from Fig. 3. It is not possible to determine unique values for the parameters included in eqn (E10) due to the degrees of freedom vs. the limited data set. The fit shown in Fig. 7 uses values of φ = 0.34, γSCI = 0.88, k3/k2 = 6.7 × 10−4 and kd/k2 = 1.2 × 1012 cm−3. However, Fig. 7 does demonstrate that a two-oxidant system, as represented by eqn (E10), is able to describe the data within uncertainty.
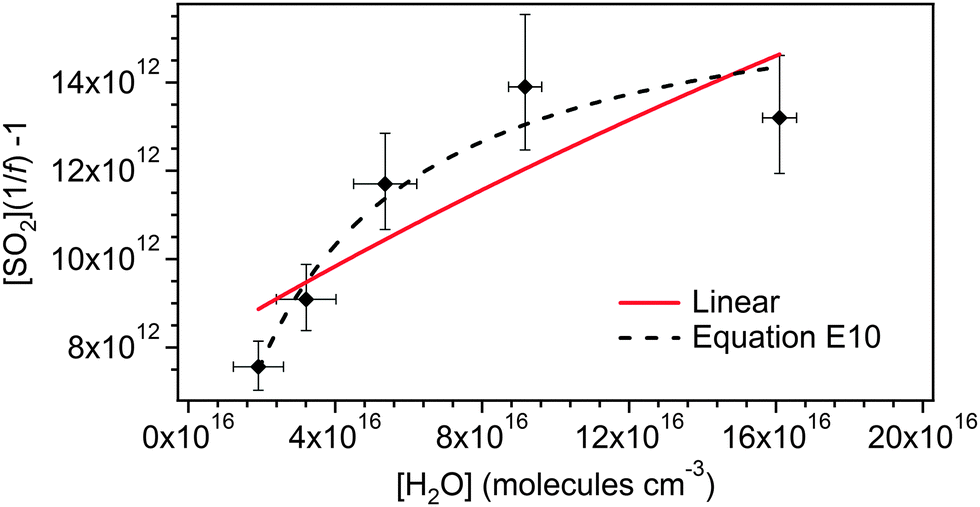 | ||
| Fig. 7 Data for TME ozonolysis (as shown in Fig. 3), with fit to eqn (E10), and linear fit to the full dataset. | ||
Fig. 7 also includes a linear fit (i.e.eqn (E3)) to the full (CH3)2COO dataset (including the highest RH experiment). While it seems unlikely that the curvature observed in the data is a result of measurement error (as described in Section 3.1), this must be considered as a possibility for (CH3)2COO in light of the two conformer explanation not being applicable. The linear fit in Fig. 7 gives a k3/k2 value of 3.8 (±3.2) × 10−5 cm3 s−1, a factor of two smaller than the k3/k2 value presented in Table 2.
The most obvious candidate for an additional oxidant present to consume SO2 is OH. OH radicals are produced in the chamber, primarily (in the absence of sunlight and NOx) through the alkene + ozone reaction.11 However cyclohexane was added in excess at the beginning of each experiment to act as an OH scavenger, such that SO2 reaction with OH was calculated to be ≤1% of the total chemical SO2 removal in all experiments. Other potential candidates for this oxidant species include the (stabilised) vinyl hydroperoxide (VHP) intermediate, a secondary ozonide (formed through an SO2–SCI cyclic adduct), and dioxirane (Scheme 1).
Drozd et al.10 have presented evidence for substantial VHP stabilisation (derived from the (CH3)2COO CI) at pressures of a few hundred Torr, with a lifetime of the order of a few hundred milliseconds with respect to decomposition to form OH, providing scope for bimolecular reactions of this species to occur. This would be consistent with the kinetic observations presented here: a small yield in the systems with syn-SCIs, while for ethene, no VHP intermediate is available, and the standard chemistry ((R1)–(R4)) can be used to satisfactorily reproduce the observations. However, no significant SO2 reactivity is known for the peroxide or alkene functionalities present in the closed shell VHP in isolation, hence it may be surprising if this species reacted rapidly with SO2, although theoretical studies have suggested that the VHP may react with H2O.23
Secondary ozonide species, formed as an adduct from the SCI + SO2 reaction, have been suggested to account for the observed isotopic exchange in alkene–ozone–SO2 systems,25 and are suggested to have lifetimes of seconds or longer.19 Such a secondary ozonide could react with a further SO2 molecule (i.e. a two-component secondary ozonide catalysed oxidation route for SO2 to SO3 conversion), however a substantial humidity dependence to the overall process may still be anticipated (on the basis of SCI removal through the SCI + H2O reaction), which is not observed here.
The ‘hot acid/ester channel’ (rearrangement and decomposition via a dioxirane intermediate) is the dominant isomerisation route available for anti-SCIs. Although the hydroperoxide channel is the principal isomerisation route for syn-SCIs, the ester mechanism is also available,25 and it is likely that a small proportion of the syn-CI will isomerise through this channel to form a dioxirane. Dioxiranes are known to be highly reactive and selective oxidising agents,51,52 and have a particular affinity for sulphur compounds.53 Additional oxidation of SO2 by the dioxirane, over and above that arising directly from reaction with the SCI, could then explain the observed behaviour of SO2 in the TME experiments (and contribute to the behaviour observed for the but-2-ene systems). For the small CI CH2OO, formed in the ethene system, it has been predicted that the dioxirane formed is considerably less stable than the methyl substituted dioxiranes formed from but-2-ene and TME ozonolysis, and furthermore will decompose promptly due to chemical activation.
The possibility of non-CI products of the ozonolysis system being responsible for some of the observed SO2 oxidation has been suggested previously as an alternative (or additional) explanation for the observed behaviour of SO2 in the atmosphere.1,54,55 In their laboratory study Berndt et al.19 note that their data were not perfectly described by a model in which a single (SCI related) SO2 oxidation process was assumed, and commented that the SO2 oxidation in ozonolysis systems may in fact be more complex. Taatjes et al.56 have recently suggested that these data19 are consistent with a two-component oxidation system, either two processes removing SO2 in parallel (as described above), or through a sequential two-step SO2 removal mechanism, such as the secondary ozonide route outlined above. However, this latter mechanism cannot (in isolation) account for the observations presented here.
The presence of an additional oxidant would have implications for the role of alkene ozonolysis in oxidation of trace gases in the atmosphere. If an oxidant is being formed that reacts slowly with H2O then this, perhaps in addition to SCI, may contribute to the additional (non-OH) SO2 oxidation observed in recent field experiments.1 Further investigation of this possibility is needed.
As for CH3CHOO, the analysis performed in this section has implications for the determination of the SCI ((CH3)2COO) yield. Using the value of kd derived from the linear fit to all the data shown in Fig. 7 would indicate an SCI yield of 0.33. Using the value of kd derived from the ‘additional oxidant fit in Fig. 7, and taking into account that ∼10 ppb of the SO2 loss in the high SO2 experiment would be attributed to reaction with the additional oxidant rather than the SCI, determines a slightly lower φmin of 0.23, and a corrected yield of 0.32, very similar to that shown in Table 1. Both of these possible alternative SCI yields lie within the uncertainties in the SCI yield from TME ozonolysis presented in Table 1.
As for CH3CHOO, it is not possible to quantitatively consider the contribution of the water dimer to the SCI loss given the limited data, but theory53 predicts k(H2O)2/k(H2O) to be three orders of magnitude smaller than that for CH2OO suggesting that reaction with the water dimer would be unimportant for (CH3)2COO at typical atmospheric boundary layer [H2O].
4. Atmospheric implications
The derived values for k3 reported in Table 3 correspond to loss rates for reaction of SCI with H2O in the atmosphere of 650 s−1 for CH2OO, ∼3500 s−1 for CH3CHOO and ∼1050 s−1 for (CH3)2COO (assuming [H2O] = 5 × 1017 molecules per cm−3, equivalent to an RH of 65% at 298 K). Comparing this to the derived kd values it is seen that reaction with H2O is predicted to be the main sink for SCI in the atmosphere, but also that loss through decomposition cannot be neglected for some SCIs – contributing on the order of 0–1% for CH3CHOO and 13% for (CH3)2COO.An estimate of a mean steady state SCI concentration in the background atmospheric boundary layer can then be calculated using eqn (E11).
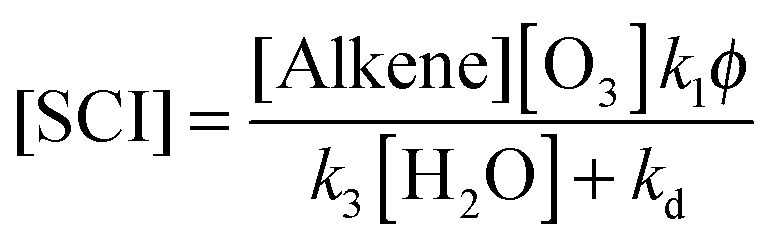 | (E11) |
Using the data given below, a steady state SCI concentration of 1.7 × 103 molecules per cm−3 is estimated for an ozonolysis source (noting that other potential atmospheric sources of SCI exist, e.g. photolysis of alkyl-iodides in the marine boundary layer, and sinks, e.g. reaction with NO and NO2). This assumes an ozone mixing ratio of 40 ppbv, an alkene mixing ratio of 2 ppbv, φ of 0.35, and mean reaction rate constants k1 (alkene–ozone) of 1 × 10−16 cm3 s−1; k2 (SCI + SO2) of 3.5 × 10−11 cm3 s−1, k3 (SCI + H2O) of 2 × 10−15 cm3 s−1, kd of 30 s−1 with [H2O] of 5 × 1017 cm−3 (RH ∼ 65%).
However, in the case of CH3CHOO the data shown in Fig. 3, and the discussion above, indicate contributions from multiple species – syn- and anti-conformers with contrasting behaviour. It is clear that the burden of CH3CHOO in the atmosphere would be better described by considering these two fractions of SO2 loss separately. Eqn (E12) expands eqn (E11) to treat the two conformers separately, where [SCI] = [anti-SCI] + [syn-SCI], making the same assumptions as for the analysis of CH3CHOO in eqn (E5) and (E6). kanti3 is estimated to be 1 × 10−14 cm3 s−1 (taking into account that CH2OO is considered as an anti-SCI in this analysis and that the derived k3(CH2OO) is more than an order of magnitude smaller than the derived k3(CH3CHOO) of 2.3 × 10−14 cm3 s−1), ksynd is assumed to be 200 s−1 and φanti = φsyn = 0.175. Additionally the anti-SCI + water dimer reaction is also considered, using a value of 5.6 × 10−13 cm3 s−1 as derived for CH2OO in this work.
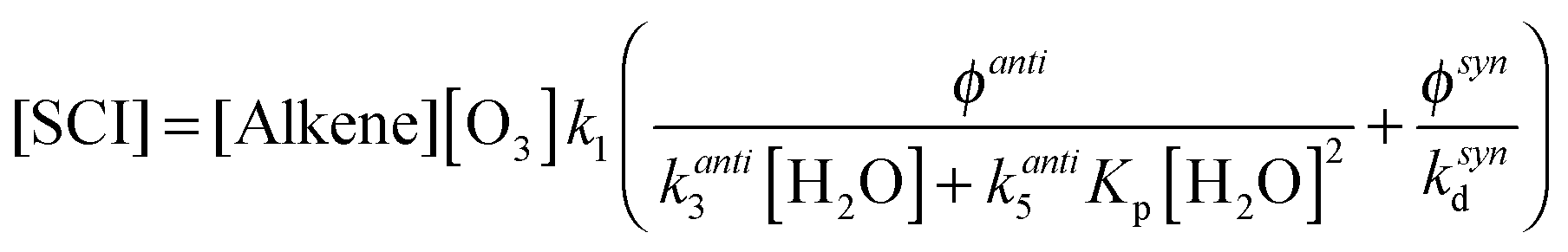 | (E12) |
Using these values in eqn (E12) determines [anti-SCI] = 164 molecules per cm−3 and [syn-SCI] = 4.4 × 103 molecules per cm−3. The formation of an additional oxidant during alkene ozonolysis would be expected to have a similar effect to the two component contribution presented in eqn (E12) based on the apparent yields from the experiments presented here. From this analysis the atmospheric SCI burden is seen to likely be dominated by syn-SCI since this term is at least an order of magnitude greater than the anti-SCI term.
A typical diurnal loss rate of SO2 to OH (kOH × [OH]) is 9 × 10−7 s−1,15 while the SO2 loss rate due to reaction with SCI, using the values derived from eqn (E12), would be 1.2 × 10−7 s−1. This suggests, for the conditions and assumptions given above, the loss of SO2 to SCI to be about 13% of loss to OH. This analysis neglects additional chemical sinks for SCI, which would reduce SCI abundance but are unlikely to be competitive with the two main SCI loss processes identified herein. SCI concentrations are expected to vary greatly depending on the local environment, e.g. alkene abundance may be considerably higher (and with a different reactive mix of alkenes) in a forested environment, compared to a rural background environment. The majority of the SCI burden, particularly in forested regions, is likely to be dominated by larger SCI derived from (C5) isoprene and (C10) monoterpenes. The chemistry of these species could differ greatly from the small SCI reported here (which we have found to be structure specific, even for small alkene systems), especially for tethered SCI derived from ozonolysis of internal double bonds within (for example) some monoterpenes. It is clear that the total SCI loss rate is dependent upon SCI identity and configuration, and that further work is required to quantify speciated SCI in the atmosphere, and to accurately calculate SCI concentrations for use in atmospheric modelling.
5. Conclusions
It has been shown that at relatively low [H2O] (<1 × 1017 cm−3) the loss of SO2 in the presence of four ozone–alkene systems: ethene, cis-but-2-ene, trans-but-2-ene and 2,3-dimethyl-but-2-ene significantly decreases with increasing water vapour. This is consistent with production of a stabilised Criegee intermediate from the ozonolysis reaction and subsequent reaction of this species with SO2 and H2O. Competition between H2O and SO2 for reaction with the SCI leads to the observed relationship which is sensitive to water vapour abundance over a relatively narrow range of RH. Derived kinetic data for these ozonolysis systems shows that the reaction rates are dependent on the structure of the SCI. At [H2O] > 1 × 1017 cm−3 the SO2 loss in the presence of cis- and trans-but-2-ene, and 2,3-dimethyl-but-2-ene appears to show a reduced dependence upon H2O. The results suggest that there is an H2O dependent and an H2O independent fraction to the observed SO2 loss in these systems. These two fractions may be attributable to differing kinetics of the two conformers produced in but-2-ene ozonolysis or to other oxidant products of the alkene ozonolysis reaction. This observation means that SCI structure must be considered in atmospheric modelling of SCI production from alkene ozonolysis, and suggests that the atmospheric SCI burden (and hence the oxidation of trace gases) will be dominated by syn-SCI.This work provides constraints on the behaviour of SCI formed through alkene ozonolysis under conditions relevant to the atmospheric boundary layer, but also highlights the complex nature and incomplete current understanding of the ozonolysis system. Further research is needed to definitively quantify the impact of this chemistry upon atmospheric oxidation.
Acknowledgements
The assistance of the EUPHORE staff is gratefully acknowledged. Marie Camredon, Stephanie La and Mat Evans are thanked for helpful discussions. This work was funded by EU FP7 EUROCHAMP 2 Transnational Access activity (E2-2012-05-28-0077), the UK NERC (NE/K005448/1) and Fundacion CEAM. Fundación CEAM is partly supported by Generalitat Valenciana, and the projects GRACCIE (Consolider-Ingenio 2010) and FEEDBACKS (Prometeo – Generalitat Valenciana). EUPHORE instrumentation is partly funded by the Spanish Ministry of Science and Innovation, through INNPLANTA project: PCT-440000-2010-003. LV is supported by the Max Planck Graduate Center with the Johannes Gutenberg-Universität Mainz (MPGC).Notes and references
- R. L. Mauldin III, T. Berndt, M. Sipilä, P. Paasonen, T. Petäjä, S. Kim, T. Kurtén, F. Stratmann, V.-M. Kerminen and M. Kulmala, Nature, 2012, 488, 193 CrossRef PubMed.
- G. Sarwar, K. Fahey, R. Kwok, R. C. Gilliam, S. J. Roselle, R. Mathur, J. Xue, J. Yu and W. P. L. Carter, Atmos. Environ., 2013, 68, 186 CrossRef CAS PubMed.
- P. Forster and V. Ramaswamy, et al., IPCC 4, 2007, ch. 2, vol. 160 Search PubMed.
- R. A. Cox and S. A. Penkett, Nature, 1971, 230, 321 CrossRef CAS.
- R. Criegee and G. Wenner, Liebigs Ann. Chem., 1949, 564, 9 CrossRef CAS.
- D. Johnson and G. Marston, Chem. Soc. Rev., 2008, 37, 699 RSC.
- H. Niki, P. D. Maker, C. M. Savage, L. P. Breitenbach and M. D. Hurley, J. Phys. Chem., 1987, 91, 941 CrossRef CAS.
- R. I. Martinez and J. T. Herron, J. Phys. Chem., 1987, 91, 946 CrossRef CAS.
- G. T. Drozd, J. Kroll and N. M Donahue, J. Phys. Chem. A, 2011, 115, 161 CrossRef CAS PubMed.
- G. T. Drozd and N. M Donahue, J. Phys. Chem. A, 2011, 115, 4381 CrossRef CAS PubMed.
- M. S. Alam, A. R. Rickard, M. Camredon, K. P. Wyche, T. Carr, K. E. Hornsby, P. S. Monks and W. J. Bloss, J. Phys. Chem. A, 2013, 117, 12468 CrossRef CAS PubMed.
- K. Izumi, M. Mizuochi, K. Murano and T. Fukuyama, Atmos. Environ., 1987, 21, 1541 CrossRef CAS.
- J. G. Calvert, R. Atkinson, J. A. Kerr, S. Madronich, G. K. Moortgat, T. J. Wallington and G. Yarwood, The Mechanisms of Atmospheric Oxidation of the Alkenes, Oxford University Press, New York, 2000 Search PubMed.
- C. A. Taatjes, G. Meloni, T. M. Selby, A. J. Trevitt, D. L. Osborn, C. J. Percival and D. E. Shallcross, J. Am. Chem. Soc., 2008, 130, 11883 CrossRef CAS PubMed.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177 CrossRef CAS PubMed.
- D. Stone, M. Blitz, L. Daubney, N. U. M. Howes and P. Seakins, Phys. Chem. Chem. Phys., 2014, 16, 1139 RSC.
- B. Ouyang, M. W. McLeod, R. L. Jones and W. J. Bloss, Phys. Chem. Chem. Phys., 2013, 15, 17070 RSC.
- T. Berndt, T. Jokinen, R. L. Mauldin III, T. Petaja, H. Herrmann, H. Junninen, P. Paasonen, D. R. Worsnop and M. Sipila, J. Phys. Chem. Lett., 2012, 3, 2892 CrossRef CAS.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2012, 14, 14682 RSC.
- J. R. Pierce, M. J. Evans, C. E. Scott, S. D. D’Andrea, D. K. Farmer, E. Swietlicki and D. V. Spracklen, Atmos. Chem. Phys., 2013, 13, 3163 CrossRef.
- M. Olzmann, E. Kraka, D. Cremer, R. Gutbrod and S. Andersson, J. Phys. Chem. A, 1997, 101, 9421 CrossRef CAS.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Phys. Chem. Chem. Phys., 2014, 16, 19941 RSC.
- K. T. Kuwata, M. R. Hermes, M. J. Carlson and C. K. Zogg, J. Phys. Chem. A, 2010, 114, 9192 CrossRef CAS PubMed.
- J. T. Herron, R. I. Martinez and R. E. Huie, Int. J. Chem. Kinet., 1982, 14, 201 CrossRef CAS.
- J. D. Fenske, A. S. Hasson, A. W. Ho and S. E. Paulson, J. Phys. Chem. A, 2000, 104, 9921 CrossRef CAS.
- J. H. Kroll, S. R. Sahay, J. G. Anderson, K. L. Demerjian and N. M. Donahue, J. Phys. Chem. A, 2001, 105, 4446 CrossRef CAS.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Am. Chem. Soc., 2012, 134, 19104 CrossRef PubMed.
- J. M. Beames, F. Liu, L. Lu and M. I. Lester, J. Chem. Phys., 2013, 138, 244307 CrossRef PubMed.
- K. H. Becker, EUPHORE: Final Report to the European Commission, Contract EV5V-CT92-0059, Bergische Universität, Wuppertal, Germany, 1996 Search PubMed.
- M. S. Alam, M. Camredon, A. R. Rickard, T. Carr, K. P. Wyche, K. E. Hornsby, P. S. Monks and W. J. Bloss, Phys. Chem. Chem. Phys., 2011, 13, 11002 RSC.
- T. Berndt, T. Jokinen, M. Sipilä, R. L. Mauldin, H. Herrmann, F. Stratmann, H. Junninen and M. Kulmala, Atoms. Environ., 2014, 89, 603 CrossRef CAS PubMed.
- S. M. Saunders, M. E. Jenkin, R. G. Derwent and M. J. Pilling, Atmos. Chem. Phys., 2003, 3, 161 CrossRef CAS.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, J. Phys. Chem., 1981, 85, 1024 CrossRef CAS.
- S. Hatakeyama, H. Kobayashi and H. Akimoto, J. Phys. Chem., 1984, 88, 4736 CrossRef CAS.
- O. Horie and G. K. Moortgat, Atmos. Environ., Part A, 1991, 24, 1881 CrossRef.
- P. Neeb, O. Horie and G. K. Moortgat, Int. J. Chem. Kinet., 1996, 28, 721 CrossRef CAS.
- A. S. Hasson, G. Orzechowska and S. E. Paulson, J. Geophys. Res., 2001, 106, 34131 CrossRef CAS.
- A. R. Rickard, D. Johnson, C. D. McGill and G. Marston, J. Phys. Chem. A, 1999, 103, 7656 CrossRef CAS.
- H. Niki, P. D. Maker, C. M. Savage and L. P. Breitenbach, Chem. Phys. Lett., 1977, 46, 327 CrossRef CAS.
- R. A. Cox and S. A. Penkett, J. Chem. Soc., Faraday. Trans., 1972, 68, 1735 RSC.
- H. G. Kjaergaard, T. Kurtén, L. B. Nielsen, S. Jørgensen and P. O. Wennberg, J. Phys. Chem. Lett., 2013, 4, 2525 CrossRef CAS.
- L. Vereecken, H. Harder and A. Novelli, Phys. Chem. Chem. Phys., 2014, 16, 4039 RSC.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, E. P. F. Lee, J. M. Dyke, D. W. K. Mok, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2012, 14, 10391 RSC.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, M. Anwar H. Kahn, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Angew. Chem., Int. Ed., 2014, 18, 4547 CrossRef PubMed.
- T. Berndt, J. Voigtländer, F. Stratmann, H. Junninen, R. L. Mauldin III, M. Sipilä, M. Kulmala and H. Herrmann, Phys. Chem. Chem. Phys., 2014, 16, 19130 RSC.
- A. B. Ryzhkov and P. A. Ariya, Phys. Chem. Chem. Phys., 2004, 6, 5042 RSC.
- Y. Scribano, N. Goldman, R. J. Saykally and C. Leforestier, J. Phys. Chem. A, 2006, 110, 5411 CrossRef CAS PubMed.
- L. Sheps, A. M. Scully and K. Au, Phys. Chem. Chem. Phys., 2014, 16, 26701 RSC.
- J. M. Anglada, J. Gonzalez and M. Torrent-Sucarrat, Phys. Chem. Chem. Phys., 2011, 13, 13034 RSC.
- W. Adam, R. Curci and J. O. Edwards, Acc. Chem. Res., 1989, 22, 205 CrossRef CAS.
- R. W. Murray, Chem. Rev., 1989, 89, 1187 CrossRef CAS.
- R. Curci, A. Dinoi and M. F. Rubino, Pure Appl. Chem., 1995, 67, 811 CAS.
- D. Heard, Nature, 2012, 488, 164 CrossRef CAS PubMed.
- J. Prousek, Chem. Listy, 2009, 103, 271 CAS.
- C. A. Taatjes, D. E. Shallcross and C. J. Percival, Phys. Chem. Chem. Phys., 2013, 16, 1704 RSC.
| This journal is © the Owner Societies 2015 |