
A mechanistic investigation of morphology evolution in P3HT–PCBM films induced by liquid crystalline molecules under external electric field†
Weihua
Zhou
ab,
Jiangman
Shi
a,
Lingjian
Lv
a,
Lie
Chen
ac and
Yiwang
Chen
*ac
aInstitute of Polymers/Department of Chemistry, Nanchang University, 999 Xuefu Avenue, Nanchang 330031, China. E-mail: ywchen@ncu.edu.cn; Fax: +86 791 83969561; Tel: +86 791 83969562
bState Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
cJiangxi Provincial Key Laboratory of New Energy Chemistry, Nanchang University, 999 Xuefu Avenue, Nanchang 330031, China
First published on 5th November 2014
Abstract
We demonstrate that the morphology of poly(3-hexyl thiophene) and [6,6]-phenyl-C61-butyric acid methyl ester (P3HT–PCBM) bulk heterojunctions (BHJ) could be tuned by the 4-cyano-4′-pentylterphenyl (5CT) liquid crystalline molecules under electric field assisted treatment for enhanced solar cell performance. The miscibility and interactions between the components were carefully studied, showing that 5CT could induce the crystallization of P3HT to form edge-on structures in ternary blends after electric field assisted treatment as revealed by grazing-incidence wide-angle X-ray diffraction (GIXRD). The PCBM and 5CT are supposed to form the rod-like complexes, and the nanorods could orient to the direction of electric field, accompanied by the homogeneous distribution of nanorods in diameters of about 30 nm at an electric field of 600 V mm−1. The sizes of PCBM clusters and complexes are dependent on the 5CT doping ratios and intensity of electric field according to grazing-incidence small-angle X-ray scattering (GISAXS) analysis. When the active layers were processed under the atmospheric environment, the power conversion efficiency (PCE) could reach 3.5% at 5CT weight fraction of 6 wt% after treatment by an electric field of 600 V mm−1, in contrast to the PCE value of 2.4% for a pristine P3HT–PCBM blend. This work provides an attractive strategy for manipulating the nanostructure of BHJ layers and also increases insight into morphology evolution when liquid crystalline molecules are incorporated into BHJs.
1. Introduction
Polymeric semiconductor-based solar cells (PSCs) have emerged as promising low-cost and flexible large area devices in both academic and industrial fields.1–3 Present research showed that the nanoscale morphology of the bulk heterogeneous junction (BHJ) solar cells played a critical role in the high power conversion efficiency (PCE) devices, which has reached 10% for lab-scale devices.4–6 It is suggested that a conceptually optimal BHJ film morphology should consist of a bicontinuous interdigitated donor–acceptor network with a donor and acceptor nanodomain within the sizes of the exciton diffusion lengths (10 nm) for efficient charge generation and transport.7,8 However, the actual morphologies of most BHJ films have a large disparity due to the uncontrollability of the aggregation and phase separation by solution processing. Thermal annealing and solvent vapor annealing are frequently-used strategies to manipulate crystallization and control the morphology of polymer–fullerene BHJs.9–12 These post-processing methods are supposed to not only induce the crystallization of poly(3-hexyl thiophene) (P3HT), but also control the phase separation between P3HT and [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) aggregates in an optimized size. But a major concern attracts people's attention that annealing at a higher temperature made PCBM molecules diffuse into huge aggregates which create damage to the exciton dissociation and charge carrier transport. Meanwhile, solvent vapor annealing is not suitable for the real roll-to-roll process due to the dangers of solvent vapor.13,14Incorporation of an additive forming a ternary solar cell is another efficient method to provide a simple and promising approach to increase PCE.15–25 Peet and co-workers showed that incorporation of high-boiling-point liquid additive octanedithiol (ODT) or diiodooctane (DIO) into the poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]-dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (PCPDTBT)-based device active solution resulted in a near doubly increased PCE, from 2.8% to 5.5%.18 An increased ordering or crystallinity of the conjugated polymer and optimized structural organization of the BHJ blend was achieved by adding solvent additives. Besides, additives containing thiophene rings usually have a strong chemical affinity toward polymers, which can act as nucleating agents to improve the conformation of polymer chains and enhance the intra- and interplane stacking of P3HT chains with a high crystallinity.19,20 Using metal complex additives21,22 is another valid approach in which solar light is absorbed at longer wavelengths so as to enhance the charge mobility of the active layer. Amphiphilic molecules such as thiophene–C60 derivatives23 could increase the compatibility between the donor and the acceptor to obtain a micro-phase separation, leading to a thermodynamically stable nanoscale morphology. Besides, P3HT diblock copolymers24,25 as interfacial compatibilizers could improve the PCE for inducing favorable active layer morphology with interpenetrating nanoscale domains, and the enhanced P3HT crystallinity and orientation facilitated the hole transport within the active layer.
Recently, liquid crystalline molecules (LCs) seemed to be especially interesting additives in PSCs because of the unique properties.26–30 In our previous studies, we have intramolecularly incorporated the mesogenic cyano-biphenyl to the fluorene unit in the D–A copolymer poly[fluorene-alt-5,5-(4′,7′-di-2-thienyl-2′,1,3′-benzothiadiazole)] (PFDTBT) that enhanced the orientation degree of the polymer and also finetuned the energy levels.31 Jeong and co-workers27 found that the devices fabricated using the P3HT–PCBM layer blended with 3 wt% of 2,3,6,7,10,11-hexaacetoxytriphenylene discotic liquid crystals (DLCs) achieved an average PCE of 3.97%, compared to the reference cells with a PCE of 3.03%. It's attributed to the improved crystallization and ordering of P3HT chains with an aid of DLCs after thermal annealing. Meanwhile, the nematic liquid crystals (NLCs) as nucleating agents induced polymer crystallization or tuned phase separation in the active layer.29 However, the role of polymer rearrangement is at least partly understood in the ternary blend devices, yet there are many conflicting explanations for the effect that additives have on PCBM crystallites and aggregation, which are also critical to the device performance. It is of great significance to experimentally examine the effect of LCs on the crystalline structure of P3HT chains, and the aggregation behavior of PCBM in the P3HT–PCBM–LCs ternary blends by using multi-scale characterization techniques to spatially correlate device performance with morphology.
Due to the anisotropic property, the liquid crystalline molecules could be aligned under electric field and magnetic field treatment. The oriented molecules or crystals are believed to induce polymer aggregates with short range order in the casting solution, and then act as nuclei for the polymer crystallization during the film formation.32,33 The electric field is believed to be able to induce the orientation of the liquid crystalline molecules, serving as the template to induce the crystallization of P3HT and aggregation of PCBM. Similar research work has not been reported previously based on our knowledge. It's of great importance to study the mechanism behind these morphological changes induced by LCs in ternary blend systems under external electric fields, such as the structural and dynamical processes involving polymer, fullerene and LC molecules (crystallization, molecular motions, phase-separation, etc.), to gain deeper insight into the working principles of ternary organic solar cells.
In this contribution, a small percentage 4-cyano-4′-pentylterphenyl (5CT) was incorporated into the P3HT–PCBM system via the solution blending method. The ternary blend solution was then spin-coated onto an indium tin oxides (ITO) glass, followed by treatment with an electric field during the solvent evaporation process in an atmospheric environment. The absorption behavior and morphology of the ternary blend films, as well as the interactions and miscibility between the components were studied in detail. The grazing-incidence X-ray diffraction (GIXRD) and grazing-incidence small-angle X-ray scattering (GISAXS) techniques were applied to explore the crystalline structure of P3HT crystallites, and the aggregation behavior of PCBM clusters.34–38 The performance of the solar cell device and the hole and electron mobilities39–41 were found to be determined by the 5CT content and electric field intensity. The results obtained in this article are very helpful for designing a potential annealing method and characterizing the morphology of ternary blend films at different length scales.
2. Results and discussion
2.1 Interactions of 5CT with P3HT and PCBM
The structure of the electric field equipment, and the schematic GISAXS configuration are illustrated in Fig. 1. As revealed by many researchers,27–29 the liquid crystalline molecules were able to improve the crystallization of P3HT chains, even though the ordering of P3HT chains was hampered in the presence of PCBM aggregates. However, the effect of LCs on the crystalline structure of P3HT and the aggregation behavior of PCBM has not been deeply investigated. To explore the influence of 5CT on the crystallization of P3HT and PCBM, the corresponding P3HT–5CT and PCBM–5CT blends were prepared via the solution blending method. After the evaporation of chloroform solvents and drying in a vacuum oven at 40 °C for 6 h, the specimens were used for the differential scanning calorimetry (DSC) analysis. Although the specimens obtained by the solution blending method are different from the ultra-thin films used for the solar cell devices, the corresponding DSC heating and cooling curves could still give some information about the interactions such as miscibility between the different components. | ||
| Fig. 1 (a) Schematic of the apparatus used for applying electric field across the polymer layer during the solvent drying process. The gap between the two ITO glasses was about 0.5 mm. A film of the active layer solution on the substrate was kept at different intensities of electric field of 300, 600 and 1200 V mm−1. (b) Schematic of GISAXS configuration illustrating the in-plane scattering (Qx) direction, the out-of-plane scattering (Qz) direction, and the 2D scattering pattern with incident angle αi, in-plane scattering angle θ, and out-of-plane scattering angle αf of X-rays, respectively. In the center of the CCD detector, the X-ray signal was shielded with a rod-like beamstop. | ||
As shown in Fig. 2a, it is observed that the 5CT molecules exhibit three phase transitions on the heating curve below 190 °C. The 5CT molecules transform from the solid state to the liquid crystalline state at 79.6 °C. The other two exothermic peaks at 102.8 °C and 127.3 °C should be related to the transitions in smectic phases.42,43 For the pristine P3HT, only one melting peak at 231.3 °C is noticed. It is clear that the distinct crystalline P3HT-rich and 5CT-rich phases are present upon the incorporation of 5CT. In addition, with the increase of the 5CT weight fraction, the melting and crystallization point depression is associated with the P3HT phase. For example, the melting behavior of P3HT is not seriously affected upon incorporation of 3 wt% 5CT. Upon further increasing the 5CT doping amount to 10 wt%, the melting peak of P3HT becomes broader and shifts to a lower temperature of 220.3 °C. It is generally accepted that the melting temperature of a crystalline polymer is mainly related to the lamellar thickness of the crystals, the existence of impurities in the lamellar regions may obviously reduce the melting temperature.44 Melting point depression of the semi-crystalline P3HT phase is expected and can result from reduced P3HT crystallite sizes upon increasing the weight fraction of 5CT or from changes in molecular interaction due to the 5CT molecules. Thus, it is believed that some of the 5CT molecules may be trapped in the lamellar regions of P3HT crystallites. The phase transitions of 5CT in the blends are similar to those of pristine 5CT even at 5CT weight fraction of 3 wt%, indicating that the crystallization of 5CT is not disturbed in the presence of a large amount of P3HT. The 5CT molecules are believed to be immiscible with P3HT and could segregate to form separated regions. Based on the transmission electron microscopy (TEM) images shown in Fig. 2c and d, the phase-separated morphology could be discerned in the P3HT–5CT blend. The P3HT chains tended to form the nano-fibrils across the whole image where the bright regions contribute to the liquid crystalline phases. The existence of LCs seems to induce crystallization of P3HT to form more obvious nano-fibrils. The microphase separated LC regions are supposed to show liquid crystalline transitions during the heating process as revealed by DSC analysis.
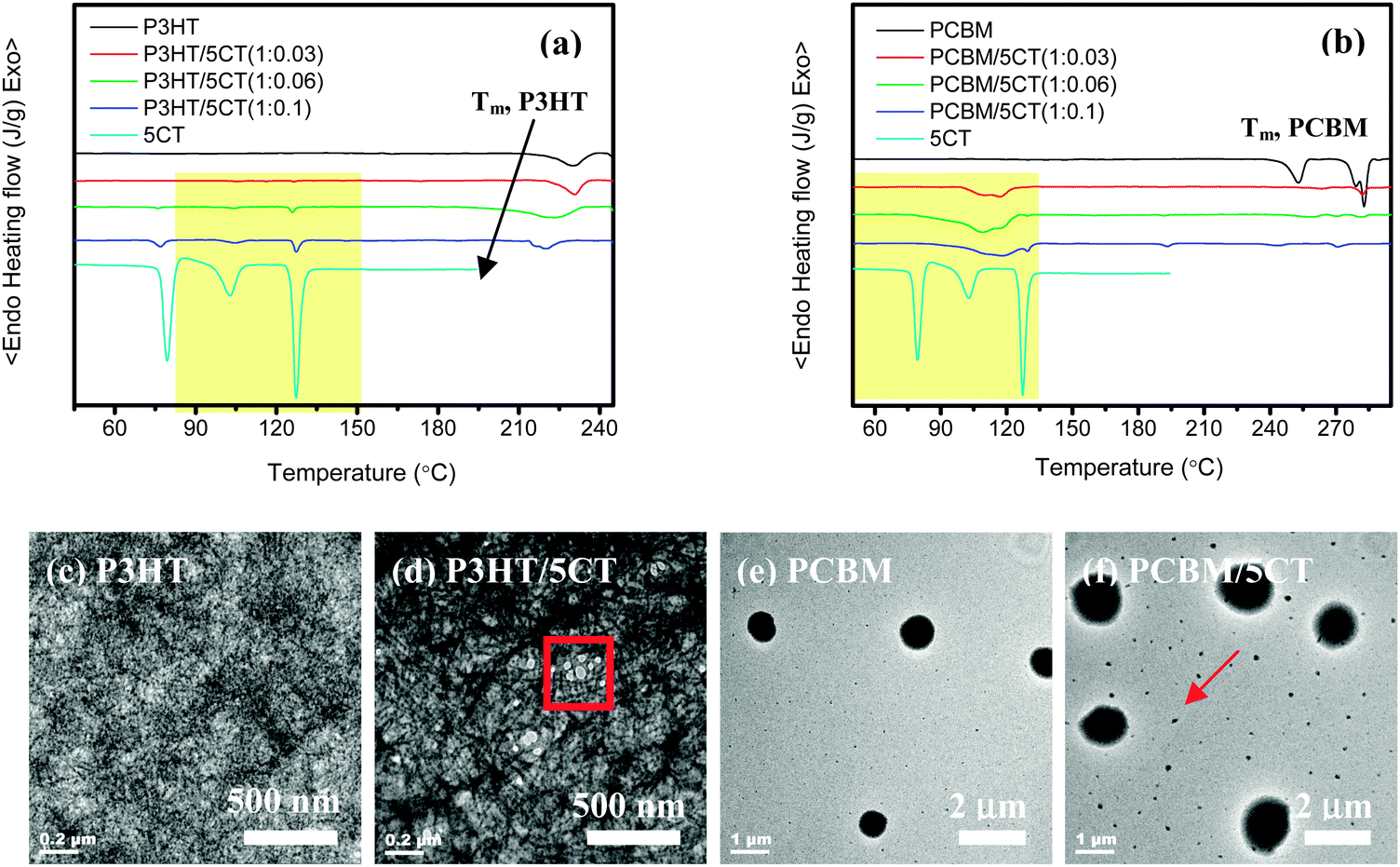 | ||
| Fig. 2 DSC heating curves of (a) P3HT–5CT and (b) PCBM–5CT blends at different 5CT weight fractions, and TEM images of (c) pristine P3HT, (d) P3HT–5CT blend at 5CT weight fraction of 6 wt%, (e) pristine PCBM and (f) PCBM–5CT blend at 5CT weight fraction of 6 wt%. | ||
In contrast to the P3HT–5CT system, the PCBM–5CT blends exhibit different phase transition behaviors and morphology. Based on the DSC heating curves (Fig. 2b), three obvious melting peaks at 253.0 °C, 279.0 °C and 283 °C could be observed for pristine PCBM. By the introduction of 3 wt% 5CT into the PCBM, only a tiny melting peak at 283.0 °C contributing to PCBM could be discerned. At 5CT content of 6 wt% and 10 wt%, the melting peak of PCBM becomes less obvious, showing that the crystallization of PCBM is significantly restricted in the presence of 5CT. Similarly, the liquid crystalline phase transitions of 5CT in the PCBM–5CT blends are also seriously affected by the PCBM, and no sharp phase transition peaks of 5CT could be discerned, exhibiting one broad exothermic peak. The shape and temperature of the exothermic peaks is found to be totally different from that of pristine 5CT. The enthalpy corresponding to 5CT exothermic peaks is calculated to be 487.0, 242.0 and 154.3 J g−1 for the blends containing 3 wt%, 6 wt% and 10 wt% 5CT, respectively. The enthalpy of 5CT exothermic peaks in blends is very high, even at 5CT content of 3 wt%, showing strong interactions between 5CT and PCBM. Due to the existence of a phenyl ring in both 5CT and PCBM, the similarity in chemical structure between the two molecules may eventually lead to the formation of some ordered complexes composed of 5CT and PCBM via the π–π interactions. Consequently, the strong π–π interactions between the two components restrict the crystallization of both PCBM and 5CT. The morphology of PCBM and the PCBM/5CT (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.06) blend was further analyzed by TEM. The pristine PCBM shows some black spheres with a diameter of about 500 nm, attributed to the PCBM clusters (Fig. 2e). Upon the incorporation of 5CT molecules, the diameter of the spheres grows bigger. In addition, the other dots with diameters less than 100 nm could also be discerned in the image (Fig. 2f). The existence of 5CT molecules is believed to induce the PCBM molecules to aggregate and form bigger clusters. Based on the above analysis, it is concluded that the 5CT is immiscible with P3HT and could segregate to form separated regions. However, the strong interactions between 5CT and PCBM molecules led to the formation of complexes, restricting the crystallization of each other.
:0.06) blend was further analyzed by TEM. The pristine PCBM shows some black spheres with a diameter of about 500 nm, attributed to the PCBM clusters (Fig. 2e). Upon the incorporation of 5CT molecules, the diameter of the spheres grows bigger. In addition, the other dots with diameters less than 100 nm could also be discerned in the image (Fig. 2f). The existence of 5CT molecules is believed to induce the PCBM molecules to aggregate and form bigger clusters. Based on the above analysis, it is concluded that the 5CT is immiscible with P3HT and could segregate to form separated regions. However, the strong interactions between 5CT and PCBM molecules led to the formation of complexes, restricting the crystallization of each other.
Furthermore, the effect of 5CT on the crystallization behaviors of P3HT and PCBM upon cooling from the melting state was also investigated, as shown in Fig. S1 (ESI†). The crystallization temperature (Tc) of P3HT shifts to lower temperatures as the 5CT content increases, showing that the presence of 5CT restricts the crystallization of P3HT from the melting state. Similarly, the crystallization of PCBM is also seriously restricted in the presence of 5CT. In P3HT–PCBM–5CT ternary blends (Fig. S1, ESI†), only a tiny melting peak ascribed to P3HT is observed on the DSC heating curves. Additionally, no obvious crystallization peak could be discerned based on the DSC cooling curves. It is believed that the crystallization of P3HT and PCBM from the melting state was restricted by 5CT.
2.2 Absorption and photovoltaic properties of P3HT–PCBM–5CT blends
Fig. 3a displays the UV-vis absorption spectrum of P3HT upon the incorporation of 5CT. The pristine P3HT film shows a peak at 511 nm associated with the π–π* transition, while two additional absorption peaks at 555 and 605 nm are attributed to the π–π stacking of P3HT chains.45 Upon increasing the 5CT weight fraction from 3 wt% to 6 wt%, the intensity of low energy absorption bands at 555 and 605 nm increases, indicating that 5CT could induce the crystallization of P3HT chains. However, the intensity of absorption bands for the specimen containing 10 wt% 5CT is even lower than that of pristine P3HT, which could be due to the relatively low content of P3HT in the blend. In the P3HT–PCBM–5CT ternary solid films, the intensity of absorption bands is also found to be dependent on the 5CT weight fraction (Fig. 3b). The intensity of absorption bands reaches the highest for the specimen containing 6 wt% 5CT, illustrating the ordered state of P3HT due to interchain interaction involved in aggregation and crystallization in the presence of 5CT. It is therefore suggested that an appropriate amount of 5CT facilitates the crystallization of P3HT in both binary and ternary blending systems.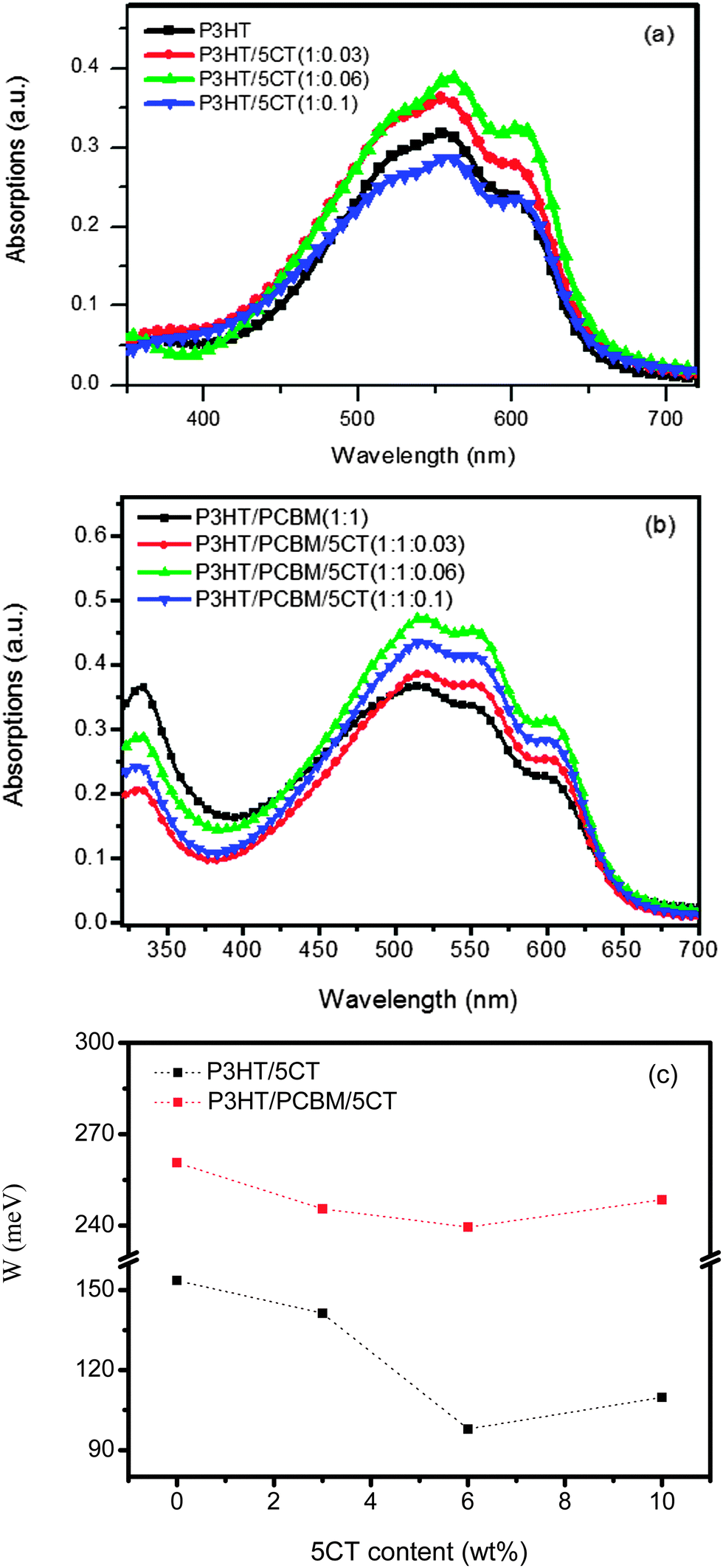 | ||
| Fig. 3 UV-vis spectra of the (a) P3HT–5CT binary blend films and (b) P3HT–PCBM–5CT ternary blend films at different weight fractions of 5CT, and (c) the evolution of exciton bandwidth W of ordered aggregates in the P3HT–5CT and P3HT–PCBM–5CT blend films as a function of 5CT content. | ||
Detailed analysis of UV-vis absorption spectra provides further insights relating to polymer conjugation length (intermolecular ordering). Examination of the P3HT absorption spectrum reveals bands associated with two phases, comprising of the crystalline and amorphous regions.46 According to Spano's model,47,48 the crystalline regions are supposed to be composed of weakly interacting H-aggregates, and the vibronic bands in the absorption spectrum are originated from the interchain coupling. Furthermore, the vibronic bands can be related to the free exciton bandwidth (W) which correlates with the conjugation length or intrachain ordering of an individual polymer chain. The decrease of W value indicates an increase in both average conjugation length and chain order.49 The W values could be calculated using the following equation:
 | (1) |
Then, the performance of the solar cell based on the P3HT–PCBM–5CT system was measured, and the corresponding current–voltage (J–V) characteristics of the devices are illustrated in Fig. 4a, and Table 1 summarizes the parameters of devices. The device fabricated using the P3HT–PCBM–5CT layer containing 6 wt% 5CT achieves the highest PCE value of 2.8%, in contrast to that of 2.4% for the pristine P3HT–PCBM device. The improvement of the device performance is mainly ascribed to the enhancement in both short-circuit current density (Jsc) and open-circuit voltage (Voc). It is well known that the Jsc strongly depends on the absorption intensity derived from the crystallinity of P3HT and the charge transport properties of networks in photovoltaic blend films.50 As demonstrated by the space-charge-limited-current (SCLC) hole and electron mobility measurement shown in Fig. S3 (ESI†) and Table 1, an appropriate amount of 5CT (6 wt%) facilitates the improvement of the carrier mobility. On the other hand, the maximal photovoltage is considered to be limited by the energy gap between the highest occupied molecular orbital (HOMO) level of a donor polymer and the lowest unoccupied molecular orbital (LUMO) level of an acceptor fullerene. The HOMO and LUMO energy levels of P3HT, PCBM and 5CT were estimated from the cyclic voltammogram (Fig. S4, ESI†) and are shown in Fig. 4b. Based on the electrochemical data, the HOMO and LUMO levels of 5CT are −5.50 and −3.36 eV, respectively. The 5CT possesses intermediate energy band edges between P3HT and PCBM, which is able to avoid the charge trapping in the ternary system. A cascade-energy alignment among P3HT–PCBM–5CT is established with the incorporation of 5CT, and 5CT exhibits a lower lying HOMO level as compared to P3HT as seen from Fig. 4b. The larger energy difference in EHOMO (D)–ELUMO (A) at the contact of 5CT to PCBM may contribute to the increased Voc. The LUMO level of the acceptor should be at least 0.3 eV lower than that of the donor to drive charge separation after exciton formation. Therefore, the excitons generated by P3HT will not be able to dissociate at the interface of the P3HT and 5CT junction while the excitons generated by 5CT could dissociate at the PCBM/5CT interface as revealed by the time-resolved photoluminescence spectra in Fig. S5 (ESI†). However, the fill factor (FF) of the solar cells is relatively low. The 5CT molecules lack the viscosity necessary for film casting, resulting in well mixed morphologies lacking the percolation pathways required to transport the photogenerated electrons and holes.
 | ||
| Fig. 4 (a) J–V characteristics of the P3HT–PCBM–5CT blend with different weight fractions of 5CT prepared by the solvent slow drying method without electric field assisted treatment. (b) The energy level diagram for P3HT, 5CT and PCBM. | ||
| P3HT/PCBM/5CT | Ea (V mm−1) | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | Hole mobilityb (cm2 V−1 s−1) | Electron mobilityc (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|
| a The intensity of electric field. b The ITO/PEDOT:PSS/P3HT:PCBM:5CT/MoO3/Ag device configuration using for the hole mobility measurement. c The ITO/ZnO/P3HT:PCBM:5CT/LiF/Al device configuration using for the electron mobility measurement. | |||||||
| 1:1:0 |
0 | 7.68 | 0.581 | 52.8 | 2.4 | 1.17 × 10−4 | 2.39 × 10−4 |
| 600 | 7.89 | 0.569 | 54.9 | 2.5 | — | — | |
| 1:1:0.03 |
0 | 8.26 | 0.578 | 55.5 | 2.7 | 1.66 × 10−4 | 4.02 × 10−4 |
| 600 | 8.47 | 0.596 | 62.2 | 3.1 | — | — | |
| 1:1:0.06 |
0 | 8.68 | 0.608 | 52.5 | 2.8 | 2.51 × 10−4 | 4.63 × 10−4 |
| 300 | 9.48 | 0.614 | 53.8 | 3.1 | 2.64 × 10−4 | 5.35 × 10−4 | |
| 600 | 9.87 | 0.621 | 56.6 | 3.5 | 2.91 × 10−4 | 7.61 × 10−4 | |
| 1200 | 7.98 | 0.604 | 52.4 | 2.5 | 1.66 × 10−4 | 1.54 × 10−4 | |
| 1:1:0.1 |
0 | 7.40 | 0.584 | 49.8 | 2.2 | 3.43 × 10−5 | 1.26 × 10−5 |
| 600 | 8.59 | 0.605 | 46.4 | 2.4 | — | — | |
2.3 Morphology under electric field assisted treatment
Based on POM images and PCE values, the incorporation of 6 wt% 5CT into the P3HT–PCBM blend is supposed to maintain the optimized morphology as well as the best photovoltaic performance. We thus choose the P3HT/PCBM/5CT (1:1:0.06) system for solvent drying in the presence of a constant electric field across active layer film solution, to induce the orientation of liquid crystalline molecules and improve the performance of the solar cell devices. In order to confirm the influence of electric field on the orientation of 5CT molecules, the POM images of 5CT films without or with electric field assisted treatment are shown in Fig. S6 (ESI†). The specimen without electric field assisted treatment tends to form needle-like crystals, showing strong anisotropy. Using the electric field assisted treatment, bright dots distributing homogeneously in the visual field could be observed. It is suggested that the 5CT molecules should orient to the electric field direction during the solvent evaporation process. In Fig. 5, the morphology of the P3HT–PCBM–5CT films displays strong dependence on the electric field strength. At E = 0 V mm−1, the anisotropic bundles comprising of 5CT and PCBM could be observed (Fig. 5a). The orientation of the bundles is random, indicative of bundles parallel and vertical to the surface of the films. At an electric field strength of 300 V mm−1, more dark dots could be observed, suggesting that more bundles orient vertically to the film surface (Fig. 5b). At an electric field of 600 V mm−1, smaller dark dots are observed in the whole visual field (Fig. 5c). Moreover, the bright dots with larger diameter are detected at an electric field strength of 1200 V mm−1 (Fig. 5d). The corresponding TEM images of the P3HT–PCBM–5CT films are presented on the right corner of the POM images. In Fig. 5a, the calamitic (rod-like) molecules parallel to the substrate demonstrate that the 5CT and PCBM molecules aggregate to form the rod-like structure. The length distribution of nanofibrils is centered at around 1.6 μm, while the width distribution is centered at around 0.15 μm. In addition, many dots with a diameter of about 0.17 μm could also be discerned in the TEM image, showing that the dots are nanorods vertical to the substrate. It is suggested that some of the nanorods tend to orient along the direction of solvent evaporation in contrast to those parallel to the substrates. After the electric field assisted treatment, most of the nanorods tend to align to the direction of electric field. At an electric field of 300 V mm−1 (Fig. 5b), the black dots with a diameter of 40–70 nm dominate the whole image. The nanofibrils with a length of about 1 μm and diameter of about 40 nm could also be noticed in the image, implying that most of the nanorods orient vertically to the substrate after the electric field assisted treatment. At an electric field of 600 V mm−1, the black dots attributing to 5CT and PCBM complexes with a diameter of about 30 nm distribute homogeneously in the matrix, in contrast to those at 300 V mm−1 (Fig. 5c). At an electric field of 1200 V mm−1, the dark dots with a larger diameter of about 100 nm could be observed (Fig. 5d). It is suggested that the appropriate intensity of electric field facilitates the aggregation of 5CT and PCBM molecules to form nanorods with smaller diameter. At lower or higher intensity of electric field, some of the 5CT and PCBM molecules tend to aggregate together to form nanofibrils with larger diameters. The schematic illustration of morphology of the PCBM–5CT complexes after electric field assisted treatment is also shown in Fig. 5. In the TEM observation of the conjugated polymer–fullerene blends, the domains of aggregated fullerene clusters were greater than 100 nm, due to the low electron density contrast between the conjugated polymer-rich and fullerene-rich domains.51 In this article, the 5CT and PCBM complexes are easier to be discerned, attributing to the large difference in the electron scattering densities between complexes and P3HT. The relatively homogeneous phase structure should benefit the exciton separation and charge transportation.
 | ||
| Fig. 5 Dark-field POM images and the corresponding TEM images of P3HT/PCBM/5CT (1:1:0.06) films spin-cast onto an ITO substrate covered by a PEDOT:PSS layer prepared under different electric field-assisted annealing conditions: (a) solvent only, (b) E = 300 V mm−1, (c) E = 600 V mm−1 and (d) E = 1200 V mm−1. The scheme below illustrates the morphology of the PCBM–5CT complexes after different intensities of electric field assisted treatment. | ||
The meso-scale film morphology in the lateral direction of P3HT–PCBM–5CT layers before and after electric field (600 V mm−1) assisted treatment was further analyzed using atomic force microscopy (AFM) in tapping mode as shown in Fig. 6. It is found that the surface topology of the electric field treated film is smoother than that of the un-annealed film. The root mean square (RMS) surface roughness of the active layer is 2.03 nm for the electric field treated film, and 4.95 nm for the un-annealed film. For the specimen without electric field assisted treatment, some aggregates with a diameter of about 400 nm could be observed, showing that the complexes based on 5CT and PCBM could remain on the surface of the films. By the electric field assisted treatment, a homogenous morphology could be obtained. The AFM images in a larger scale are shown in Fig. S7 (ESI†), which further confirms that the aggregates composed of 5CT and PCBM tend to migrate to the surface of films during the solvent evaporation process. After the electric field assisted treatment, the aggregates composed of 5CT and PCBM tend to align to the direction of electric field, showing smaller size in diameter. It is believed that the surface of the films is mainly composed of PCBM–5CT complexes, which is beneficial to the electron transport to the corresponding electrode. Thus, a vertically separated morphology could be achieved during the solvent evaporation process in the presence of electric field. The optimized morphology of the film after electric field assisted treatment may facilitate the interfacial contact between the electrode and the active layer, resulting in an improvement of the solar cell performance.
 | ||
| Fig. 6 AFM images of P3HT/PCBM/5CT (1:1:0.06) blend films on the ITO substrate covered by a PEDOT:PSS layer under different conditions: (a) solvent slow drying and (b) in the presence of electric field E = 600 V mm−1. The aggregates contribute to the complexes composed of PCBM and 5CT. | ||
It is revealed that an optimized morphology for the P3HT–PCBM–5CT film could be obtained at an electric field of 600 V mm−1. Whether the oriented nanorods composed of PCBM and 5CT could serve as the template to induce the orientation of P3HT chains? Fig. 7 shows the reflection spectra of P3HT–PCBM–5CT blend films in the out-of-plane and in-plane patterns after electric field assisted treatment at different intensities. Depending on the chain orientation of P3HT, diffraction peaks from the lamellar structure repeating along the alkyl chain direction and the π–π stacking should be visible in either the out-of-plane or in-plane measurements. In the out-of-plane pattern, the peaks at 5.6° and 10.8° are assigned to the (100) and (200) diffractions of P3HT crystallites. The position of the reflection peaks remains almost the same independent of the intensity of electric field, and the lamellar spacing of the P3HT crystalline domain is calculated to be about 1.58 nm. In the in-plane pattern, the minor peaks at 22.9° are attributed to the (010) diffraction of P3HT crystallites, and the distance of π–π stacking is calculated to be 0.39 nm. The diffraction patterns of the films can be attributed to the edge-on orientation, which is usually observed for P3AT thin films because of the hydrophobic interactions between the alkyl chains and the substrate.52 The lower surface energy of the alkyl side chains may also induce the edge-on orientation at the surface during spin-coating.53 As revealed by Su,54 the interfacial interactions between P3HT and the substrates determined the crystalline structure of P3HT. For example, the edge-on orientation was dominated by the electron-withdrawing ability of the surface functional groups while the face-on orientation was induced by the strong charge transfer interaction between the highly oriented pyrolytic graphite surface and the thiophene rings. In this article, the substrate is the PEDOT:PSS layer, and the interfacial interactions between P3HT and substrate should be stronger than those between P3HT and the oriented templates composed of 5CT and PCBM. Moreover, the 5CT and PCBM complexes are found to be immiscible and phase-separated with P3HT based on TEM and POM observation. Under the electric field assisted treatment, the oriented templates composed of 5CT and PCBM should have no significant effect on the orientation of P3HT chains. Thus, only an edge-on crystalline structure could be noticed in the P3HT–PCBM–5CT system, which is independent of the intensity of electric field and the oriented template. It is further found that the reflection peaks arising from 5CT or PCBM could not be discerned from the GIXRD spectra, revealing that the crystallization of 5CT and PCBM was restricted by each other in the P3HT matrix. Although the anisotropic structure composed of 5CT and PCBM could be observed in the POM images, 5CT and PCBM are unable to form the crystalline structure as detected by GIXRD analysis.
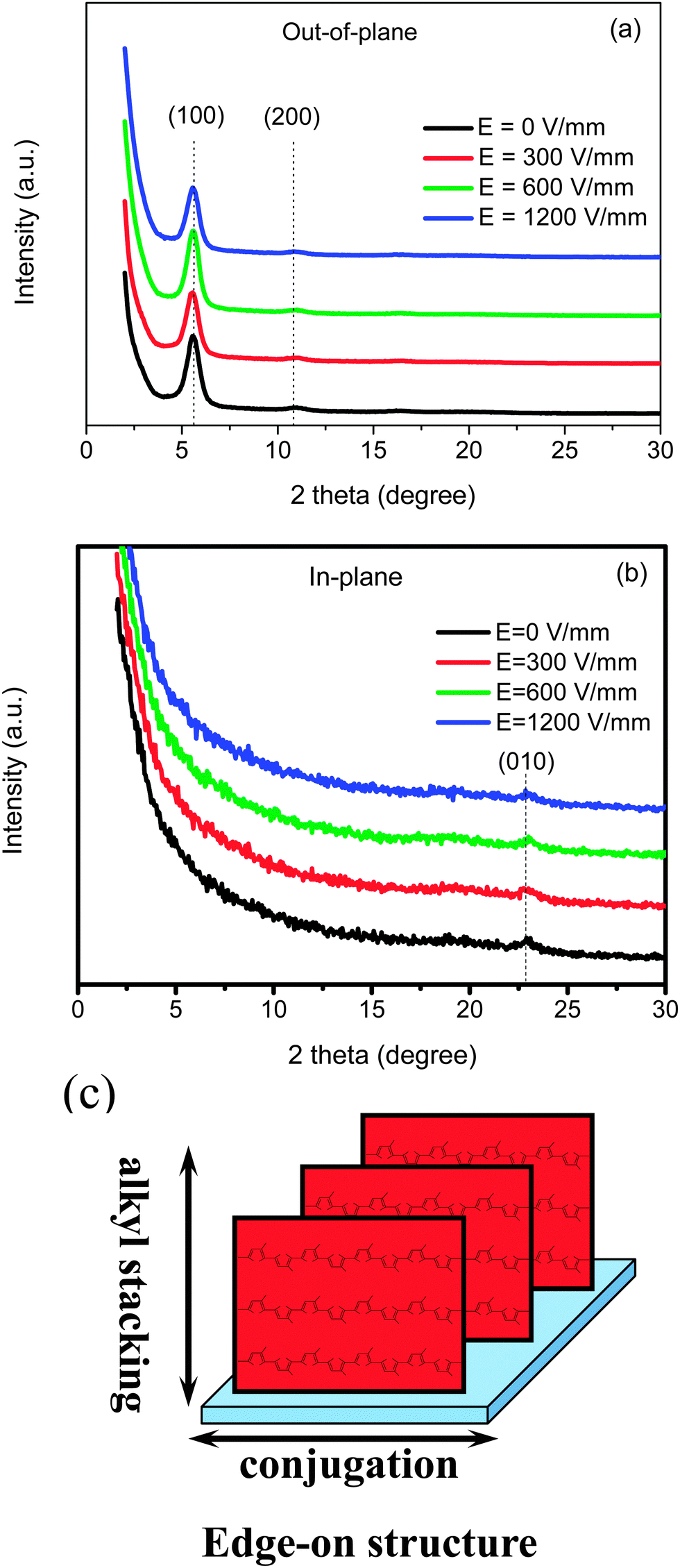 | ||
| Fig. 7 (a) Out-of-plane and (b) in-plane GIXRD profiles for P3HT/PCBM/5CT (1:1:0.06) films spin-cast onto an ITO substrate covered by a PEDOT:PSS layer prepared under electric field assisted treatment at different intensities of 0, 300, 600 and 1200 V mm−1. (c) Schematic representation of the P3HT chains adopting an edge-on orientation. | ||
Furthermore, GISAXS can give interpretations for structures across different lengths in the form of fractal sizes and provide more details on the hierarchical structures that cannot well be distinguished by TEM. We used the FitGISAXS and IGOR Pro software to obtain the 2D and 1D profiles from GISAXS data,55 as shown in Fig. 8 and 9 for the as-spun and electric field assisted treated P3HT–PCBM–5CT films. Extracting quantitative information from GISAXS profiles requires fitting experimental data to a model, however, modeling a BHJ OPV blend is extremely challenging given the irregularly shaped, polydisperse, interconnected and randomly distributed domains characteristic of the BHJ morphology. Due to the complexities, there exists no widely accepted GISAXS model for BHJ OPV blends. Su et al. have successfully used the Debye–Anderson–Brumberger (DAB) model and the polydisperse hard sphere model to describe the cluster size and distribution of PCBM domains.56 However, the combination of the DAB and polydisperse hard sphere model is unable to fit the profiles in the ternary system. We elect to interpret the data qualitatively based on the analysis of the intensity variation of the profiles. The as-cast P3HT/PCBM and P3HT/PCBM/5CT (1:1:0.03) thin films exhibit a power-law dependence of 2 and do not show obvious Guinier regime, both indicating that the domains are poorly phase segregated (i.e., highly intermixed) with no well-defined domain sizes within the length scales probed, which is also consistent with the result in POM observation. Upon increasing the 5CT content to 10 wt%, a discernible shoulder at about 0.25 nm−1 is noticed, and the power-law exponent increases to about 3. These features indicate that phase-segregation has taken place during the solvent evaporation process for the P3HT–PCBM blend at 5CT content above 6 wt%. The 5CT molecules are supposed to induce the aggregation of PCBM molecules into larger clusters. Upon increasing the intensity of the electric field, the shoulder at about 0.25 nm−1 becomes more obvious and seems to shift to lower q values. Therefore, it is believed that the 5CT and electric field could induce the aggregation of PCBM molecules into larger clusters.
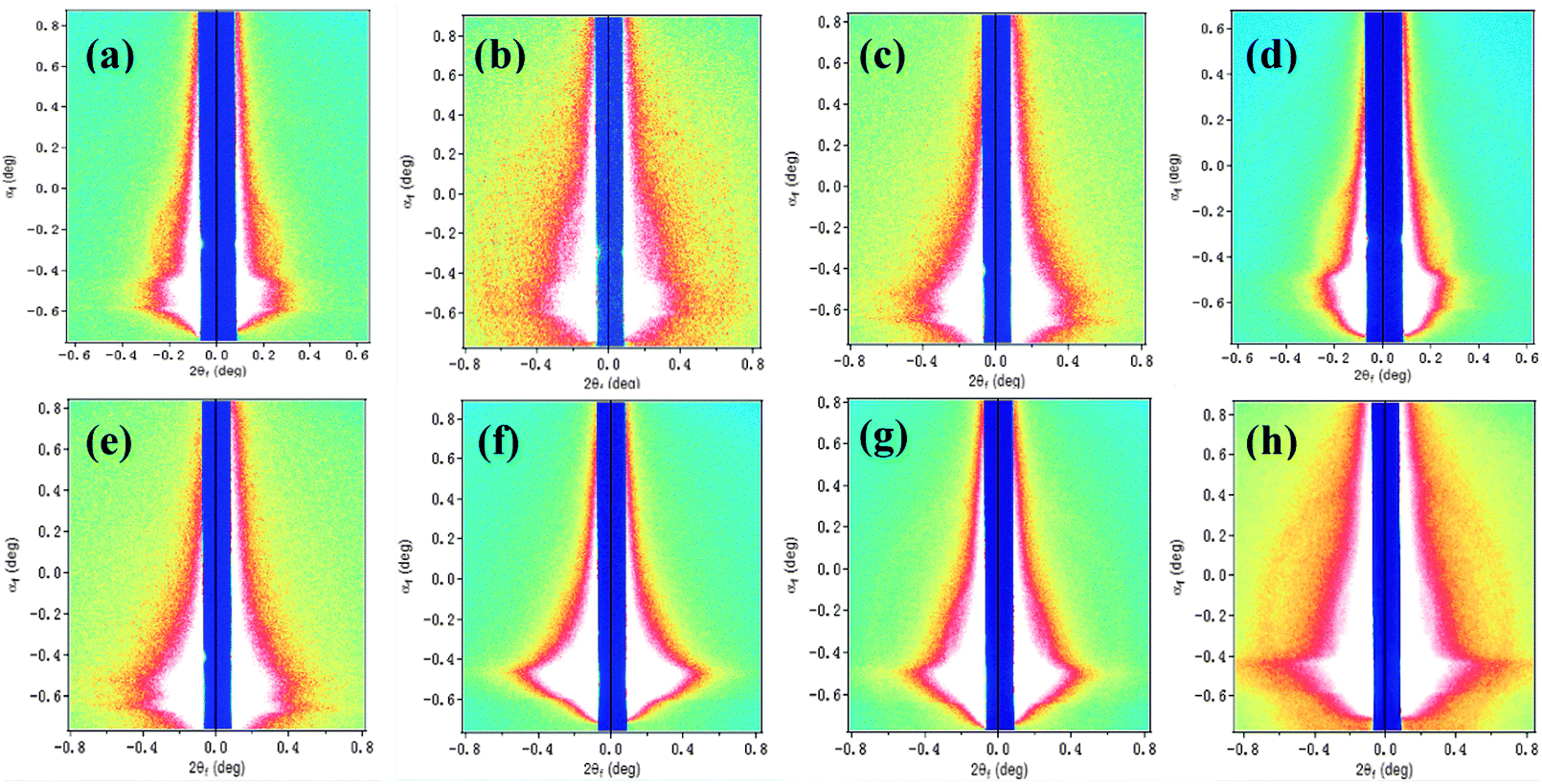 | ||
| Fig. 8 Two-dimensional GISAXS images of the P3HT–PCBM–5CT films at different weight fractions (a) 0 wt%, (b) 3 wt%, (c) 6 wt% and (d) 10 wt% of 5CT, and the P3HT–PCBM–5CT films containing 6 wt% 5CT after treatment by electric field of different intensities of (e) 0, (f) 300, (g) 600 and (h) 1200 V mm−1, respectively. | ||
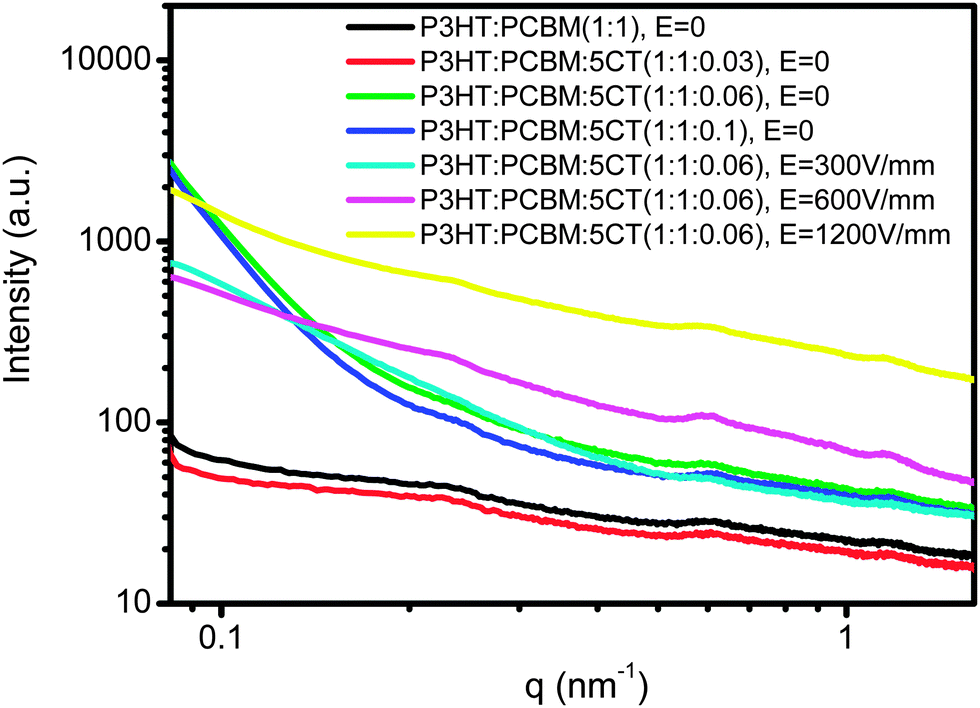 | ||
| Fig. 9 Selected GISAXS profiles measured for the P3HT–PCBM–5CT films at different weight fractions of 5CT and the films at 5CT content of 6 wt% after treatment by electric field of different intensities. | ||
2.4 Photovoltaic performance under electric field assisted treatment
The influence of electric field on the photovoltaic properties of the corresponding solar cell devices containing 6 wt% 5CT was also investigated. The J–V curves are shown in Fig. 10, and the corresponding photovoltaic performance parameters are summarized in Table 1. The device based on P3HT–PCBM–5CT exhibits an improved performance with a Jsc of 8.68 mA cm−2, a Voc of 0.608 V, and a FF of 52.5%, resulting in a PCE of 2.8%, which is comparable with the previous values in the literature. As the intensity of electric field increases to 600 V mm−1, a systematic enhancement in photovoltaic performance can be observed, giving the best power conversion efficiency of 3.5% with the Jsc, Voc and FF values of 9.87 mA cm−2, 0.621 V and 56.6%, respectively. The absorption bands of the films after electric field assisted treatment are similar to those without electric field treatment (Fig. S8, ESI†). Thus, the enhancement of the Jsc value mainly contributes to the increase of carrier mobility, especially for the electron mobility reaching the highest value of 7.61 × 10−4 cm2 V−1 s−1. The improvement of the electron mobility may be related to the formation of an ordered transport way by the rod-like PCBM–5CT complexes induced by the electric field. A further increase of the intensity of electric field results in a decline in the photovoltaic performance of the devices. The relationship between the parameters of Jsc, Voc, FF and PCE and the 5CT content at different electric field intensities is shown in Fig. S9 (ESI†). It should be noted that both the Jsc and Voc values increase after the electric field assisted treatment, showing a better interfacial contact between P3HT and PCBM. Based on the above result, the structure and morphology evolution in the pristine P3HT–PCBM blend and the P3HT–PCBM–5CT blend after electric field assisted treatment could be described as shown in Fig. 11. The PCBM and 5CT are believed to form the complexes which may orient to the direction of electric field. The oriented nanorods could provide more pathways for the charge transport to the electrodes. It should be noted that the electric field assisted treatment was performed in an air environment, providing a novel annealing method to fabricate the active layer. | ||
| Fig. 10
J–V characteristics of the P3HT/PCBM/5CT (1:1:0.06) blend after treatment by electric field of different intensities. | ||
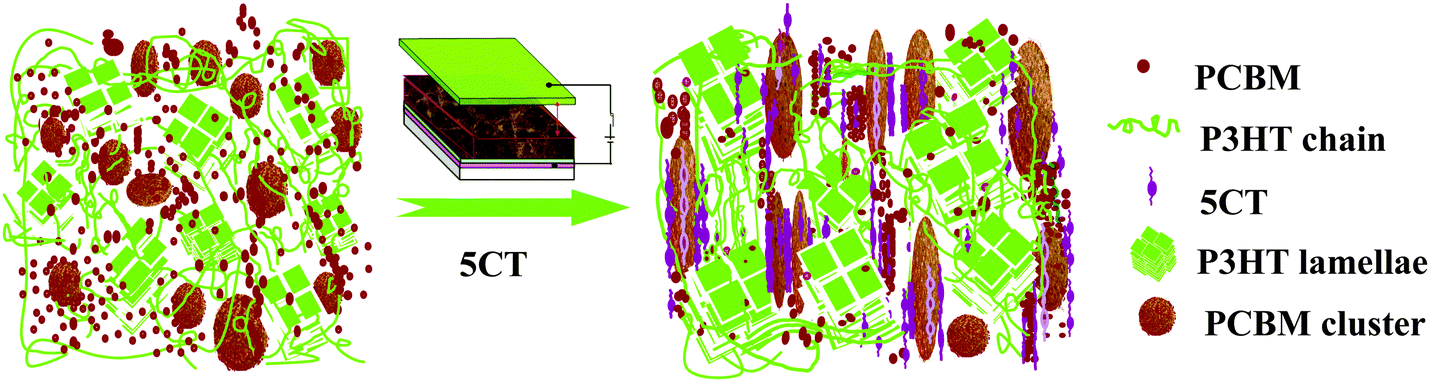 | ||
| Fig. 11 Schematic illustration of the morphology of P3HT–PCBM films before and after the incorporation of 5CT molecules under the electric field assisted treatment. | ||
3. Conclusions
The mechanism of morphology evolution in P3HT–PCBM films after blending with 5CT under electric field assisted treatment has been deeply investigated. The structural characteristics are well correlated to the device performance and photovoltaic properties. The 5CT was immiscible with P3HT and the crystallization of 5CT was not disturbed in the presence of large amounts of P3HT. Additionally, the crystallization, the conjugation length and chain order of P3HT could be improved by the 5CT molecules as revealed by TEM and UV-vis results. The crystallization of PCBM and 5CT was restricted by each other, showing a minor melting peak of PCBM and a broad liquid crystalline transition peak of 5CT as revealed by the DSC analysis. The existence of 5CT was found to induce the aggregation of PCBM molecules to form larger clusters in diameter. Furthermore, in the P3HT–PCBM–5CT ternary system, the PCBM and 5CT was able to form rod-like complexes, which could orient to the direction of electric field as shown in POM and TEM images. The oriented nanorods with diameters of about 30 nm distributed homogeneously in the matrix could be observed in TEM images at the 5CT weight fraction of 6 wt% and electric field of 600 V mm−1. In addition, only the edge-on structure could be discerned for P3HT as confirmed by the GIXRD, independent of the 5CT weight fraction and intensity of electric field. The 5CT and electric field could induce the aggregation of PCBM molecules into larger clusters as revealed by GISAXS. After processing in the atmospheric environment, the PCE of the solar cell devices reached the highest value of 3.5% at 5CT weight fraction of 6 wt% and electric field of 600 V mm−1, in contrast to the PCE value of 2.4% for the pristine P3HT–PCBM blend. The results provide new insights into the role of molecular dopants in affecting the morphology evolution in the active layer and provide a novel annealing method to fabricate BHJs.Acknowledgements
The financial support for this work is provided by the National Natural Science Foundation of China (51273088 and 51303077), National Basic Research Program of China (973 Program 2014CB260409), National Science Fund for Distinguished Young Scholars, PhD Programs Foundation for Young Teachers of Ministry of Education of China (Grants 20123601120010), and Fund by State Key Laboratory of Luminescent Materials and Devices, South China University of Technology (Grants 2013-skllmd-04). The experiments are partially carried out in Shanghai Synchrotron Radiation Facility (SSRF), and we also thank the team in SSRF for support during the GISAXS measurement. We thank Professor Qiu Dong in the Institute of Chemistry, Chinese Academy of Sciences, for his helpful discussion in the analysis of the experimental data.References
- G. Yu, J. Gao, J. C Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1790 CAS.
- G. Li, V. Shrotriya, J. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- N. C. Miller, E. Cho, M. J. Junk, R. Gysel, C. Risko, D. Kim and M. D. McGehee, Adv. Mater., 2012, 24, 6071–6079 CrossRef CAS PubMed.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446–1455 CrossRef PubMed.
- Y. Zheng and J. G. Xue, Polym. Rev., 2010, 50, 420–453 CrossRef CAS.
- L. G. Kaake, J. J. Jasieniak, R. C. Bakus, G. C. Welch, D. Moses, G. C. Bazan and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 19828–19838 CrossRef CAS PubMed.
- L. G. Kaake, D. Moses and A. J. Heeger, J. Phys. Chem. Lett., 2013, 4, 2264–2268 CrossRef CAS.
- A. J. Parnell, A. J. Cadby, O. O. Mykhaylyk, A. D. F. Dunbar, P. E. Hopkinson, A. M. Donald and R. A. L. Jones, Macromolecules, 2011, 44, 6503–6508 CrossRef CAS.
- L. H. Nguyen, H. Hoppe, T. Erb, S. Guenes, G. Gobsch and N. S. Sariciftci, Adv. Funct. Mater., 2007, 17, 1071–1078 CrossRef CAS.
- E. Verploegen, R. Mondal, C. J. Bettinger, S. Sok, M. F. Toney and Z. Bao, Adv. Funct. Mater., 2010, 20, 3519–3529 CrossRef CAS.
- T. Agostinelli, S. Lilliu, J. G. Labram, M. Campoy-Quiles, M. Hampton, E. Pires, J. Rawle, O. Bikondoa, D. D. C. Bradley, T. D. Anthopoulos, J. Nelson and J. E. Macdonald, Adv. Funct. Mater., 2011, 21, 1701–1708 CrossRef CAS.
- S. Berson, R. D. Bettignies, S. Bailly and S. Guillerez, Adv. Funct. Mater., 2007, 17, 1377–1384 CrossRef CAS.
- Y. M. Chang and L. Wang, J. Phys. Chem. C, 2008, 112, 17716–17720 CAS.
- J. T. Rogers, K. Schmidt, M. F. Toney, E. J. Kramer and G. C. Bazan, Adv. Mater., 2011, 23, 2284–2288 CrossRef CAS PubMed.
- E. Lim, S. Lee and K. K. Lee, Chem. Commun., 2011, 47, 914–916 RSC.
- Y. C. Chen, C. Y. Hsu, R. Y. Y. Lin, K. C. Ho and J. T. Lin, ChemSusChem, 2013, 6, 20–35 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- L. Q. Yang, H. X. Zhou, S. C. Price and W. You, J. Am. Chem. Soc., 2012, 134, 5432–5435 CrossRef CAS PubMed.
- S. S. Sharma, G. D. Sharma and J. A. Mikroyannidis, Sol. Energy Mater. Sol. Cells, 2011, 95, 1219–1223 CrossRef CAS PubMed.
- T. Hasobe, H. Imahori, P. V. Kamat, T. K. Ahn, S. K. Kim, D. Kim, A. Fujimoto, T. Hirakawa and S. Fukuzumi, J. Am. Chem. Soc., 2005, 127, 1216–1228 CrossRef CAS PubMed.
- T. Hasobe, A. S. D. Sandanayaka, T. Wada and Y. Araki, Chem. Commun., 2008, 3372–3374 RSC.
- I. C. Wu, C. H. Lai, D. Y. Chen, C. W. Shih, C. Y. Wei, B. T. Ko, C. Ting and P. T. Chou, J. Mater. Chem., 2008, 18, 4297–4303 RSC.
- S. Honda, T. Nogami, H. Ohkita, H. Benten and S. Ito, ACS Appl. Mater. Interfaces, 2009, 1, 804–810 CAS.
- J. B. Kim, K. Allen, S. J. Oh, S. Lee, M. F. Toney, Y. S. Kim, C. R. Kagan, C. Nuckolls and Y. L. Loo, Chem. Mater., 2010, 22, 5762–5773 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119–1122 CrossRef CAS PubMed.
- S. Jeong, Y. Kwon, B. D. Choi, H. Ade and Y. S. Han, Appl. Phys. Lett., 2010, 96, 183305 CrossRef PubMed.
- Y. Lou, Z. Wang, S. Naka and H. Okada, Appl. Phys. Lett., 2011, 99, 033305 CrossRef PubMed.
- S. Jeong, Y. Kwon, B. D. Choi, G. Kwak and Y. S. Han, Macromol. Chem. Phys., 2010, 211, 2474–2479 CrossRef CAS.
- Q. Zheng, G. J. Fang, W. B. Bai, N. H. Sun, P. L. Qin, X. Fan, F. Cheng, L. Y. Yuan and X. Z. Zhao, Sol. Energy Mater. Sol. Cells, 2011, 95, 2200–2205 CrossRef CAS PubMed.
- K. Yao, Y. W. Chen, L. Chen, F. Li, X. E. Li, X. Y. Ren, H. M. Wang and T. X. Liu, Macromolecules, 2011, 44, 2698–2706 CrossRef CAS.
- K. Schmidt, C. J. Tassone, J. R. Niskala, A. T. Yiu, O. P. Lee, T. M. Weiss, C. Wang, J. M. J. Frechet, P. M. Beaujuge and M. F. Toney, Adv. Mater., 2014, 26, 300–305 CrossRef CAS PubMed.
- L. Kaake, X. D. Dang, W. L. Leong, Y. Zhang, A. J. Heeger and T. Q. Nguyen, Adv. Mater., 2013, 25, 1706–1712 CrossRef CAS PubMed.
- M. Shin, H. Kim, J. Park, S. Nam, K. Heo, M. Ree, C. S. Ha and Y. Kim, Adv. Funct. Mater., 2010, 20, 748–754 CrossRef CAS.
- E. Verploegen, C. E. Miller, K. Schmidt, Z. N. Bao and M. F. Toney, Chem. Mater., 2012, 24, 3923–3931 CrossRef CAS.
- W. R. Wu, U. S. Jeng, C. J. Su, K. H. Wei, M. S. Su, M. Y. Chiu, C. Y. Chen, W. B. Su, C. H. Su and A. C. Su, ACS Nano, 2011, 5, 6233–6243 CrossRef CAS PubMed.
- H. C. Liao, C. S. Tsao, T. H. Lin, M. H. Jao, C. M. Chuang, S. Y. Chang, Y. C. Huang, Y. T. Shao, C. Y. Chen, C. J. Su, U. S. Jeng, Y. F. Chen and W. F. Su, ACS Nano, 2012, 6, 1657–1666 CrossRef CAS PubMed.
- A. Sharenko, M. Kuik, M. F. Toney and T. Q. Nguyen, Adv. Funct. Mater., 2014, 24, 3543–3550 CrossRef CAS.
- H. Q. Zhou, Y. Zhang, J. Seifter, S. D. Collins, C. Luo, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, Adv. Mater., 2013, 25, 1646–1652 CrossRef CAS PubMed.
- V. D. Mihailetchi, L. J. A. Koster, P. W. M. Blom, C. Melzer, B. Boer, J. K. J. Duren and R. A. J. Janssen, Adv. Funct. Mater., 2005, 15, 795–801 CrossRef CAS.
- Y. Zhang and P. W. M. Blom, Appl. Phys. Lett., 2011, 98, 143504 CrossRef PubMed.
- A. Itaya, T. Imamura, M. Hamaguchi, Y. Tsuboi, H. Miyasaka, T. Asahi and H. Masuhara, Thin Solid Films, 1997, 311, 277–285 CrossRef CAS.
- D. Zhou, Y. W. Chen, L. Chen, W. H. Zhou and X. H. He, Macromolecules, 2009, 42, 1454–1461 CrossRef CAS.
- H. D. Keith and F. J. Padden, J. Appl. Phys., 2004, 35, 1270–1285 CrossRef PubMed.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J. S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 064203 CrossRef.
- K. Zhao, L. Xue, J. Liu, X. Gao, S. Wu, Y. Han and Y. Geng, Langmuir, 2009, 26, 471–477 CrossRef PubMed.
- A. R. Aiyar, J. I. Hong, R. Nambiar, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2011, 21, 2652–2659 CrossRef CAS.
- J. Clark, J. F. Chang, F. C. Spano, R. H. Friend and C. Silva, Appl. Phys. Lett., 2009, 94, 163306 CrossRef PubMed.
- M. Chang, J. Lee, N. Kleinhenz, B. Fu and E. Reichmanis, Adv. Funct. Mater., 2014, 24, 4457–4465 CrossRef CAS.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642–6671 CrossRef CAS PubMed.
- C. M. Liu, M. S. Su, J. M. Jiang, Y. W. Su, C. J. Su, C. Y. Chen, C. T. Tsao and K. H. Wei, ACS Appl. Mater. Interfaces, 2013, 5, 5413–5422 CAS.
- D. H. Kim, Y. D. Park, Y. Jang, H. Yang, Y. H. Kim, J. I. Han, D. G. Moon, S. Park, T. Chang, C. Chang, M. Joo, C. Y. Ryu and K. Cho, Adv. Funct. Mater., 2005, 15, 77–82 CrossRef CAS.
- D. H. Kim, Y. Jang, Y. D. Park and K. Cho, Macromolecules, 2006, 39, 5843–5847 CrossRef CAS.
- Y. Guo, X. J. Ma and Z. H. Su, Macromolecules, 2013, 46, 2733–2739 CrossRef CAS.
- D. J. Babonneau, Appl. Crystallogr., 2010, 43, 929–936 CrossRef CAS.
- H. C. Liao, C. S. Tsao, T. H. Lin, C. M. Chuang, U. S. J. Chen, C. H. Su, Y. Y. Chen and W. F. Su, J. Am. Chem. Soc., 2011, 133, 13064–13073 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text giving the experimental details, instrumentation, and characterization. See DOI: 10.1039/c4cp04128c |
| This journal is © the Owner Societies 2015 |