
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromoting the “water-wire” mechanism of double proton transfer in [2,2′-bipyridyl]-3,3′-diol by porous gold nanoparticles
Tarak Nath
Nag
,
Tarasankar
Das
,
Somen
Mondal
,
Arnab
Maity
and
Pradipta
Purkayastha
*
Department of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, India. E-mail: pradiptp@gmail.com; Fax: +91 33 25873020; Tel: +91 33 2587 3019
First published on 23rd January 2015
Abstract
The effect of nanopores in porous gold nanoparticles (Au NPs) on excited-state double proton transfer (DPT) in [2,2′-bipyridyl]-3,3′-diol (BP(OH)2) in an aqueous environment is the main focus of the present work. DPT in BP(OH)2 is known to take place through two mechanisms. In a bulk environment, an open solvated molecule facilitates the process and emits at 460 nm whereas, in a confined situation, formation of a “water wire” between the prototropic centers leads to the transfer of protons. It has been shown spectroscopically in the present study that in the nanovessels provided by nanoporous Au NPs, the unconventional mechanism of DPT through the formation of a “water wire” is promoted due to the presence of a limited number of water molecules around the probe. Experiments in the presence of solid pure Au, Ag and Au/Ag NPs were performed to support our proposition. Time-resolved fluorescence spectral changes confirm our findings.
1. Introduction
It is now well known that proton transfer plays important roles in chemistry and biology. Ground- and excited-state proton transfers have become important from the mechanistic point of view.1–6 Single and double proton transfer probes are quite well known and serve as important and interesting means to discover many chemical and biological consequences. For a long time, double proton transfer (DPT) has drawn the attention of many in the scientific world.7–9 In this context, [2,2′-bipyridyl]-3,3′-diol (BP(OH)2) has been considered to be a classical probe. Bulska et al. were one of the first groups to report about single and double proton transfer in BP(OH)2.10,11 According to experimental findings, it was determined theoretically that only one single tautomeric form of BP(OH)2 exists in both the ground state (di-enol form) and the lowest excited singlet state (di-keto form), respectively.12 In another report on sub-picosecond proton transfer dynamics in BP(OH)2, it was mentioned that the emissive keto-enol tautomeric form of the compound develops in the excited state through a stepwise proton transfer mechanism.13,14 Studies on DPT in BP(OH)2 were recently carried out extensively by Abou-Zied in different solvents and confined nanocavities of cyclodextrins (CDs).15,16 In accordance with the previous studies, it was observed that a di-zwitterion (DZ) tautomer is produced in the excited state after intramolecular DPT. The results indicate that the tautomers are sensitive to solvent polarity.15,16 Further experiments were performed on the binding of BP(OH)2 with ionic and neutral surfactants,17 Nafion membranes,18 proteins,19,20 and lipid vesicles.21 DPT dynamics were also studied in nanocavities of different forms, such as in cyclodextrins (CDs),15 molecular sieves of AlPO4-5,22 and molecular containers of cucurbituril.23 BP(OH)2 is reported to be devoid of any aqueous solvation inside cucurbituril-7, hence promoting concerted DPT, whereas the surrounding water molecules inside β-CD lead to sequential DPT in BP(OH)2.23Abou-Zied has explained in one of his reports that a special photoinduced tautomerization mechanism takes place in water via a “water wire” that connects both of the two hydrogen-bonding centres of the molecule.16 Proton transfer in this manner is very similar to that explained by the Grotthuss mechanism.24,25 Theoretical studies on the mechanism and dynamics of DPT through hydrogen-bonded bridging water in a “water wire” show that all molecules hydrogen-bonded to the water wire reduce the tunnelling coefficients of DPT and transfer protons near the transition state.26 Proton transfer through a “water wire” is extremely important in biology and the phenomenon readily takes place in proteins.27,28
In the present report, we have developed a nanoporous gold nanoparticulate surface by dealloying Au/Ag alloy nanoparticles (NPs) and shown that the “water wire” mechanism is promoted for DPT in BP(OH)2 confined in the nanopores. The findings were compared with the effects of pure Au and Ag NPs and Au/Ag alloy NPs on DPT in BP(OH)2. The results confirmed the remarkable superiority of the porous Au NPs in promoting the “water wire” mechanism for DPT in BP(OH)2. The unique properties of porous materials develop due to their very high surface-to-mass ratio along with a high adsorption capacity.29,30
2. Experimental section
2.1 Materials
Silver nitrate (AgNO3), chloroauric acid trihydrate (HAuCl4·3H2O), sodium borohydride (NaBH4), sodium citrate (Na3C6H5O7) and (2,2-bipyridyl)-3,3-diol were purchased from SDFCL Mumbai, Sigma and Merck, Germany. Methanol and 70% nitric acid (HNO3) were purchased from Merck, India. Triple-distilled water was used throughout the experiment and all chemicals were used without further purification. A neutral pH of all experimental solutions was carefully maintained.2.2 Instruments
The absorption spectra were recorded on a Cary 300 Bio UV-vis spectrophotometer and fluorescence measurements were taken using a QuantaMaster 40 spectrofluorometer from Photon Technology International, Inc. The fluorescence lifetimes were measured by the method of time-correlated single photon counting (TCSPC) on a picosecond spectrofluorometer from Horiba Jobin Yvon IBH equipped with a FluoroHub single photon counting controller, Fluoro3PS precision photomultiplier power supply and FC-MCP-50SC MCP-PMT detection unit. The source of excitation was a 340 nm LED with <1 ns time resolution. Atomic force microscopy (AFM) was performed using an NT-MDT NTEGRA instrument procured from NT-MDT, CA, USA. TEM measurements were carried out in a CM 12 (Philips) TEM.2.3 Synthesis of silver, gold and Au–Ag alloy nanoparticles
Au/Ag NPs were synthesized following a procedure published elsewhere.31 In brief, Au/Ag alloy NPs were synthesized by varying the initial Au/Ag molar ratios (0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 0.5:0.5, and 1:0). The synthesis was performed by reducing HAuCl4 and AgNO3 with NaBH4 in the presence of sodium citrate. The concentration of the metal salts was chosen in such a way that the reaction quotient (Q) was 2.5 × 10−11, which is below the solubility product (Ksp) of AgCl(s), 1.77 × 10−10.
:1, 0.5:0.5, and 1:0). The synthesis was performed by reducing HAuCl4 and AgNO3 with NaBH4 in the presence of sodium citrate. The concentration of the metal salts was chosen in such a way that the reaction quotient (Q) was 2.5 × 10−11, which is below the solubility product (Ksp) of AgCl(s), 1.77 × 10−10.
In three clean 50 mL flasks, 20 mL H2O and 10 μL 0.01 M sodium citrate were taken. Varying mole fractions of 0.01 M HAuCl4 and 0.01 M AgNO3 were added to each of the solutions to achieve a final metal salt concentration of 5 μM. 10 μL freshly prepared 0.01 M NaBH4 was added to the flasks all at once with vigorous stirring. The solution was allowed to stir for an additional 30 s. A faint colour change occurred almost immediately. The formation of pure Ag, Au and Au/Ag alloy NPs was confirmed from UV-visible spectroscopic analysis. Pure Ag and Au NPs show characteristic absorption peaks at 406 nm and 525 nm, respectively, whereas Au/Ag alloy NPs absorb at 473 nm in an aqueous medium (Fig. 1A).31
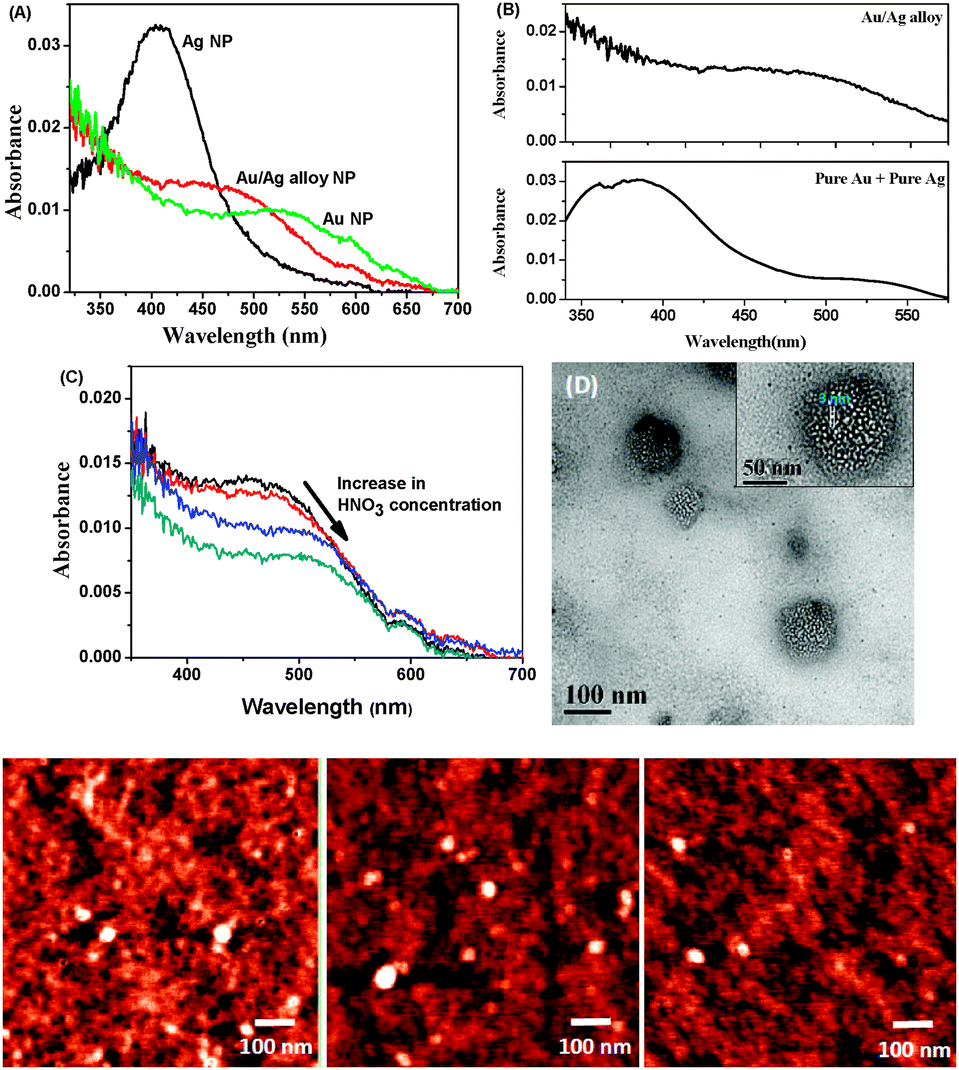 | ||
| Fig. 1 Formation and characterisation of porous Au NPs from Au/Ag alloy NPs: (A) comparison of absorption spectrum of Au/Ag alloy NP with those of pure Au and Ag NPs; (B) absorption spectrum of Au/Ag alloy NP (upper panel) and mixture of 50% (v/v) pure Au and Ag NPs; (C) demonstration of generation of pores due to addition of dilute HNO3; (D) HR-TEM image of porous NPs (inset shows pore size); and (lowest panel) (from left to right) AFM images of synthesised Au NPs, Ag NPs and porous Au NPs. | ||
We compared the absorption spectra of the alloy NPs with a mixture of 50% (v/v) pure Ag and Au NPs that produces two distinctly separated peaks at 390 and 523 nm corresponding to the absorbances of pure Ag and Au, respectively (Fig. 1B). To synthesize porous Au NPs, we dealloyed the Au/Ag alloy NPs with dilute HNO3.32 On addition of HNO3 to the alloy NPs, the plasmon peak shifted from 473 nm to 510 nm, clearly indicating that, during addition of the acid, Ag atoms in the alloy were oxidized to Ag+ ions that, in turn, came out of the alloy NPs, creating pores (Fig. 1C). Along with Ag+, some Au3+ may also have been eliminated from the alloy NPs. Since the standard reduction potential of Au3+/Au is 1.52 V, which is much higher than that of Ag+/Ag (0.799 V), Ag is more easily oxidized to Ag+ than is Au to Au3+. The formation of porous NPs was confirmed by absorption spectroscopy and high-resolution transmission electron microscopy (HR-TEM) (Fig. 1D). The diameter of the NPs was measured by atomic force microscopy (AFM) and found to be around 60 nm (Fig. 1, lowest panel). HR-TEM analysis of the porous NPs indicates well-distributed 3 nm-sized pores (Fig. 1D).
3. Results and discussion
3.1 Ground-state behaviour of BP(OH)2 in different nanoparticle environments
The absorption spectrum of BP(OH)2 in water shows a peak at around 344 nm due to transition to the lowest 1(ππ*) state of the DE tautomer and the two peaks at around 400 to 450 nm are due to intermolecular hydrogen bonds with the surrounding water molecules.15 These peaks do not appear in aprotic solvents, nor in hydrophobic pockets.15,16,23 No appreciable change in the absorption spectrum of BP(OH)2 was observed in either pure Au, Ag and Au/Ag alloys or the porous NPs (Fig. 2). This indicates that water molecules and the DE tautomer coexist under all circumstances.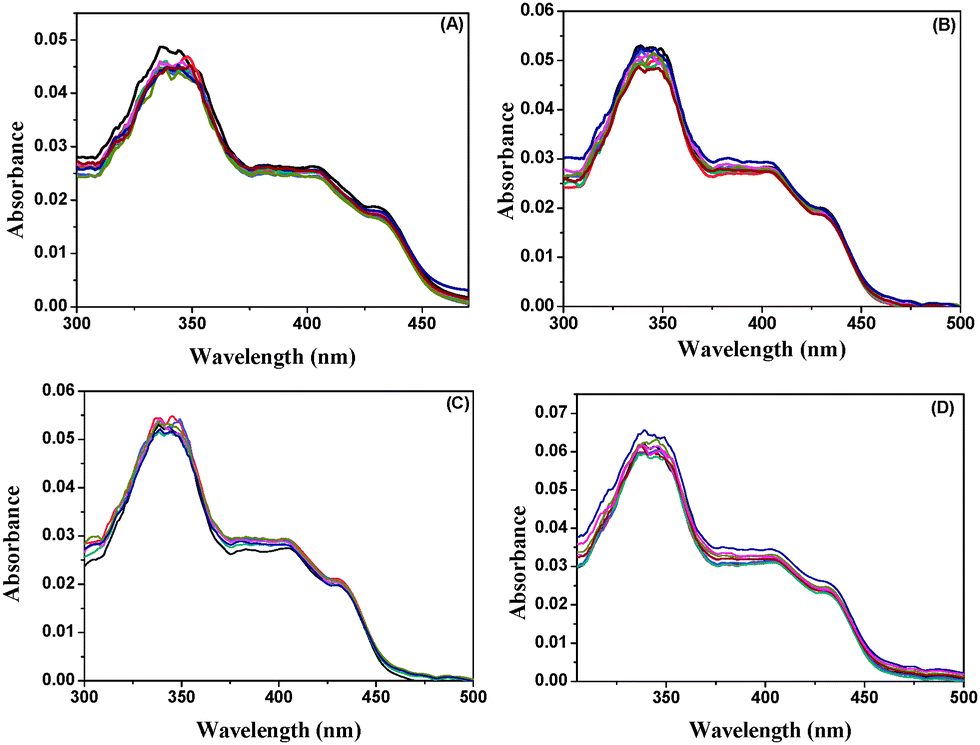 | ||
| Fig. 2 Absorption spectra of BP(OH)2 with increase in concentration of (A) Ag NPs, (B) Au NPs, (C) Au/Ag alloy NPs, and (D) porous Au NPs. The probe concentration was 4.5 μM. | ||
On explaining DPT in BP(OH)2 in solvents of different polarity and cyclodextrins (CDs), Abou-Zied proposed that the phenomenon may take place through two different mechanisms, as shown in Scheme 1.16 Mechanism I involves interaction of water molecules with each polar part in BP(OH)2, leading to enol deprotonation and imine protonation to produce the DZ tautomer. In a subsequent work, contradicting his previous report,15 Abou-Zied mentioned that DZ tautomer formation takes place in the ground state that absorbs in the 400–450 nm region.16 However, absorption of the hydrogen-bonded prototropic centres of BP(OH)2 with surrounding water molecules before tautomerization seems to be more realistic. Hence, we have changed the schematic (Scheme 1) slightly from that proposed by Abou-Zied.16 We propose that DPT takes place in the excited state in both mechanisms.
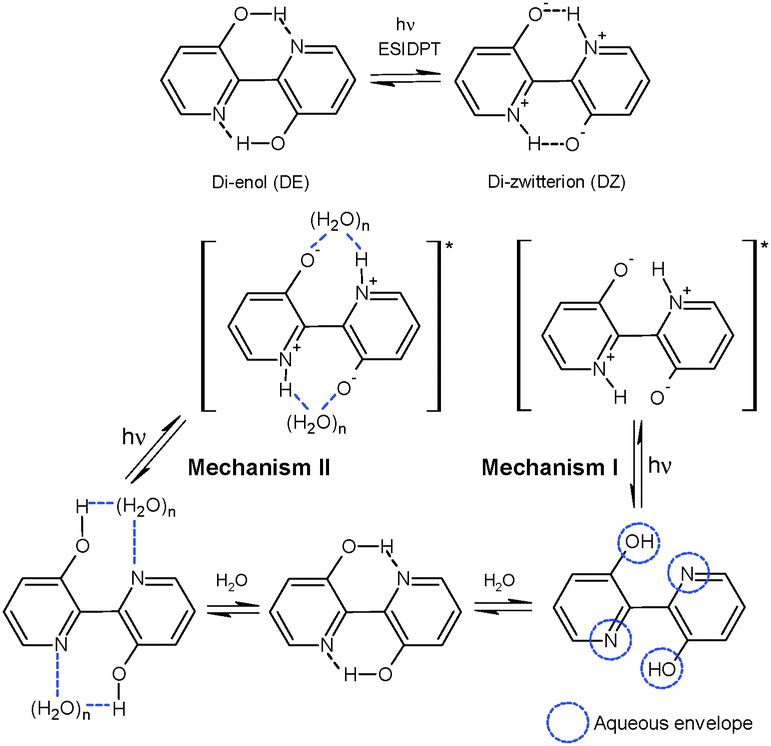 | ||
| Scheme 1 Tautomerisation of BP(OH)2 and the two most probable mechanistic pathways of tautomerisation of BP(OH)2 in water. | ||
Mechanism II in Scheme 1 shows a different DPT pathway through the formation of a “water wire” between the prototropic centres in the DE tautomer in the ground state. This solvation pattern produces the DZ tautomer in the excited state following a relay transfer of protons through the water wire. As described previously, due to strong solvation of the polar groups by the “water wire” between both hydrogen-bonding centres, the DZ tautomer may exhibit restricted rotation around the central bond between the two aromatic rings.16
3.2 Effect of different nanoparticle environments on the excited state of BP(OH)2
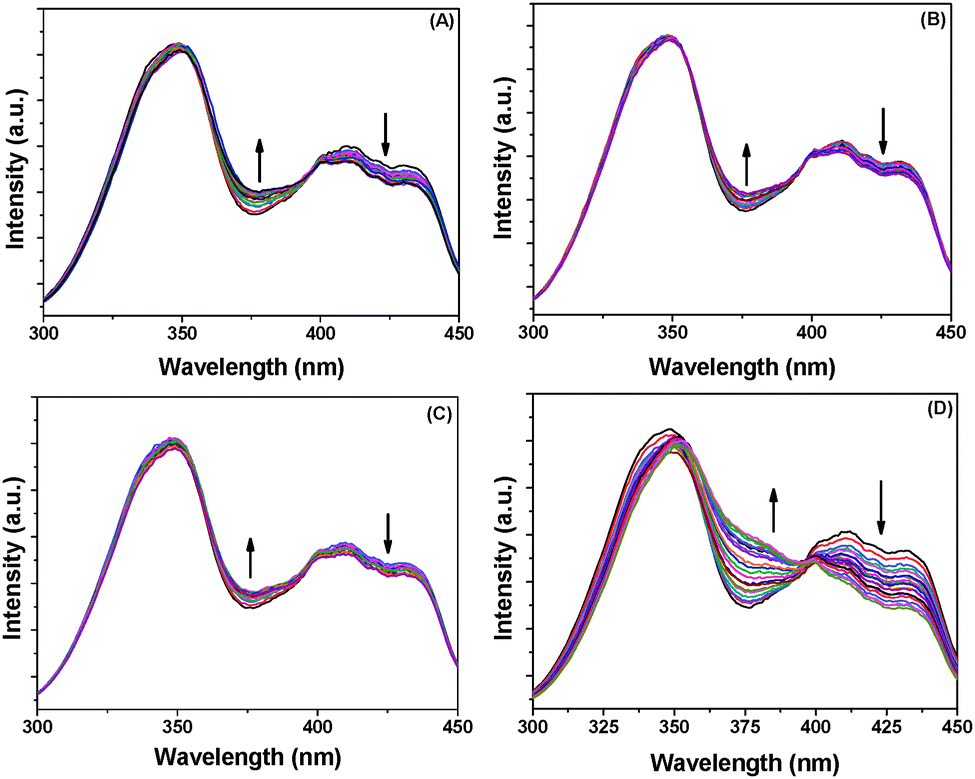
BP(OH)2 emits at 460 nm on excitation at 340 nm. This is the emission from the DZ tautomer that forms in the excited state.15–21 However, numerous works have been reported on DPT in BP(OH)2 and the main focus everywhere was on the behaviour of the 460 nm peak. Abou-Zied first pointed out the appearance of the low-wavelength weak band at 425 nm when he used a β-CD host to encapsulate BP(OH)2. This was explained as appearing due to DPT through the less favoured Mechanism II.16 This band is at a lower wavelength than that due to Mechanism I because of the restricted rotation of the water-bridged form of BP(OH)2.16Fig. 3 presents the emission spectra of BP(OH)2 with an increase in the concentration of the different solid and porous NPs. It is clearly observed that both mechanisms of DPT in BP(OH)2 are followed in all cases. It is interesting to note that for the solid pure and alloy NPs we observed a slight promotion of Mechanism II at the expense of Mechanism I. The number of water molecules is reduced at the interface of the NPs, which somewhat supports Mechanism II of DPT in BP(OH)2. The effect is overwhelming in the case of porous NPs, where we see a remarkably high promotion of Mechanism II. The relative effects of the different NPs on DPT in BP(OH)2 are provided in Fig. 4 for better understanding. It is observed that there is nearly a fivefold increase in the possibility of Mechanism II through the formation of a “water wire”. The results clearly indicate that tautomerization in BP(OH)2 through Mechanism II may be promoted in the presence of a limited number of water molecules around the probe. To consolidate our view, we focused our study on the development of the band at 425 nm on addition of porous Au NPs to the aqueous medium, which appears due to a confined aqueous environment.16 The nanopores on the surface of the NPs provide numerous nanovessels that can trap guest molecules, along with some water molecules. An increase in the concentration of the porous NPs in solution results in a noticeable enhancement in the intensity of the 425 nm band at the expense of that at 460 nm (Fig. 3D). Hence, it can be presumed that Mechanism II is reinforced inside the Au nanopores.
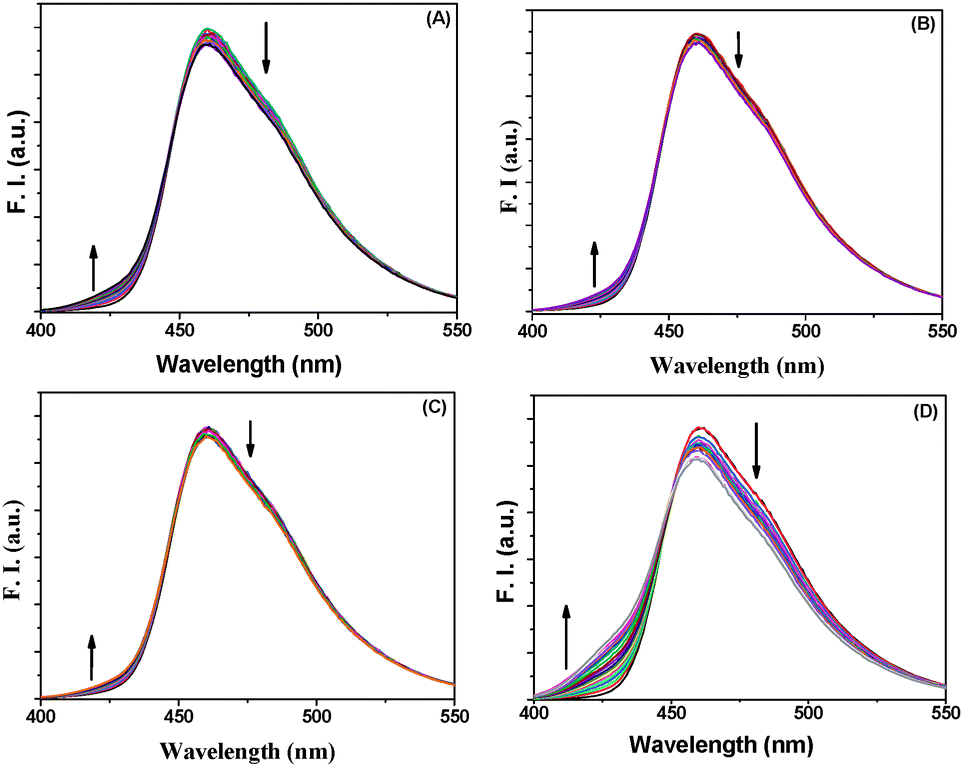 | ||
| Fig. 3 Emission spectra of BP(OH)2 with increase in concentration of (A) Ag NPs, (B) Au NPs, (C) Au/Ag alloy NPs, and (D) porous Au NPs. The probe concentration was 4.5 μM and the excitation wavelength was 340 nm. | ||
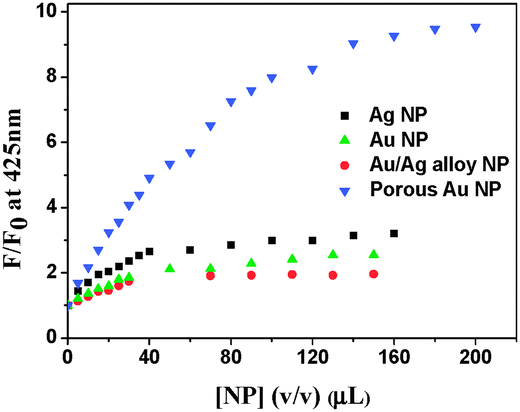 | ||
| Fig. 4 Relative increase in emission intensity of BP(OH)2 at 425 nm in the presence of the different nanoparticles. The data were taken from the emission spectra in Fig. 3. | ||
To confirm that DPT in BP(OH)2 takes place in the excited state in water, we collected the excitation spectra with an increase in the concentration of the porous Au NPs (Fig. 5). The excitation spectrum of BP(OH)2 showed the emergence of a new band at 375 nm at the expense of those between 400 and 450 nm when the 460 nm emission was monitored. The emergence of the new band at 375 nm is also observed in the presence of the solid NPs. This corroborates the description in the previous section. The effect of the porous NPs is overwhelmingly high compared to the other cases. Hence, the new band at 375 nm can be assigned to DPT in BP(OH)2 due to Mechanism II. Such changes were not observed in the absorption spectra. So this also confirms that the DPT phenomenon in the present probe is an excited-state process.
 | ||
| Fig. 5 Excitation spectra of BP(OH)2 with increase in concentration of (A) Ag, (B) Au, (C) Au/Ag alloy, and (D) porous Au NPs. The 460 nm emission was monitored. | ||
3.3 Time-resolved spectral analysis
Time-resolved fluorescence studies were performed to obtain the decay times and contributions of the responsible species in porous NPs. Both 425 nm and 460 nm emissions were monitored. The data could be fitted with a two-exponential function in each case, indicating the coexistence of two emitting species. The decay profiles and parameters are given in Fig. 6 and Table 1, respectively. The shorter-lifetime component (τ1) is due to the species obtained through Mechanism I and the longer one (τ2) is because of that produced through Mechanism II as described previously by Abou-Zied.16 The contribution of the water-wired BP(OH)2 is observed to be more than that of its open counterpart when monitored at 425 nm. Similar circumstances occur on monitoring the 460 nm emission. This clearly indicates the existence of two distinct species in solution. In the porous NPs, monitoring the 425 nm emission shows that the contribution of the shorter-lifetime component decreases significantly, along with an even faster decay, indicating that Mechanism II is favoured in this case. This is also reflected when the 460 nm emission is monitored, where we find that the contribution from the longer-lifetime species increases substantially.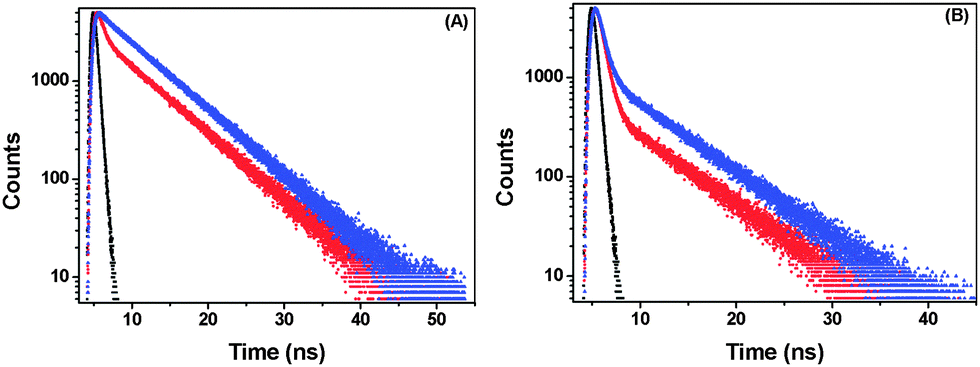 | ||
| Fig. 6 Time-resolved emission of BP(OH)2 in the absence (red) and presence (blue) of porous Au NPs; the excitation wavelength is 340 nm and the monitored emission wavelengths are (A) 425 nm and (B) 460 nm. The black data are for the prompt. | ||
| λ ex (nm) | λ em (nm) | τ 1 (ps) | τ 2 (ns) | χ 2 | |
|---|---|---|---|---|---|
| 340 | 425 | Blank | 540 (17%) | 6.40 (83%) | 1.03 |
| Porous NPs | 440 (3%) | 6.40 (97%) | 1.06 | ||
| 340 | 460 | Blank | 560 (66%) | 6.30 (34%) | 1.00 |
| Porous NPs | 560 (44%) | 6.37 (56%) | 1.00 |
4. Conclusions
DPT in BP(OH)2 was studied in an aqueous medium in the presence of porous Au NPs. It is known that the process takes place in the excited state in water through two different mechanisms. These mechanisms stem from the different solvation characteristics of the probe by water molecules. In one category of solvation, water molecules solvate both of the prototropic centres in the compound and an open structural form develops. In the other type, a “water wire” is created between both of the two centers responsible for proton transfer in BP(OH)2. The second mechanism becomes predominant with lower availability of water molecules around the probe. We find from our study that the nanopores of porous NPs provide nanovessels, where the limited number of water molecules promotes DPT in BP(OH)2 through formation of a “water wire” between the prototropic centers. The suppositions have been clarified by comparing the phenomenon of DPT in the presence of solid Au, Ag and Au/Ag alloy NPs.Acknowledgements
Financial support from the Department of Science and Technology (SR/S1/PC-35/2011) is gratefully acknowledged. TN thanks IISER Kolkata for support; TD, SM and AM acknowledge the Council of Scientific and Industrial Research and University Grants Commission for their fellowships. The authors are grateful to IIT Kharagpur for the TEM facility.Notes and references
- S. Y. Park, O. H. Kwon, T. G. Kim and D. J. Jang, J. Phys. Chem. C, 2009, 113, 16110–16115 CAS.
- Y. Arasaki, K. Yamazaki, M. T. N. Varella and K. Takatsuka, Chem. Phys., 2005, 311, 255–268 CrossRef CAS PubMed.
- S. Y. Park, Y. S. Lee and D. J. Jang, Phys. Chem. Chem. Phys., 2011, 13, 3730–3736 RSC.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 1–20 CrossRef CAS.
- L. G. Arnaut and S. J. Formosinho, J. Photochem. Photobiol., A, 1993, 75, 21–48 CrossRef.
- S. K. Mondal, S. Ghosh, K. Sahu, P. Sen and K. Bhattacharyya, J. Chem. Sci., 2007, 119, 71–76 CrossRef CAS PubMed.
- J. C. Hargis, E. Vöhringer-Martinez, H. L. Woodcock, A. Toro-Labbé and H. F. Schaefer, J. Phys. Chem. A, 2011, 115, 2650–2657 CrossRef CAS PubMed.
- H. Lim, S. Y. Park and D. J. Jang, Photochem. Photobiol., 2011, 87, 766–771 CrossRef CAS PubMed.
- O. H. Kwon and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8703–8708 CrossRef CAS PubMed.
- H. Bulska, Chem. Phys. Lett., 1983, 98, 398–402 CrossRef CAS.
- H. Bulska, A. Grabowska and Z. R. Grabowski, J. Lumin., 1986, 35, 189–197 CrossRef CAS.
- A. L. Sobolewski and L. Adamowicz, Chem. Phys. Lett., 1996, 252, 33–41 CrossRef CAS.
- H. Zhang, P. van der Meulen and M. Glasbeek, Chem. Phys. Lett., 1996, 253, 97–102 CrossRef CAS.
- F. Plasser, M. Barbatti, A. J. A. Aquino and H. Lischka, J. Phys. Chem. A, 2009, 113, 8490–8499 CrossRef CAS PubMed.
- O. K. Abou-Zied, J. Photochem. Photobiol., A, 2006, 182, 192–201 CrossRef CAS PubMed.
- O. K. Abou-Zied, J. Phys. Chem. B, 2010, 114, 1069–1076 CrossRef CAS PubMed.
- D. De and A. Datta, J. Phys. Chem. B, 2011, 115, 1032–1037 CrossRef CAS PubMed.
- E. S. S. Iyer and A. Datta, J. Phys. Chem. B, 2012, 116, 5302–5307 CrossRef CAS PubMed.
- O. K. Abu-Zied, J. Phys. Chem. B, 2007, 111, 9879–9885 CrossRef PubMed.
- D. De, K. Santra and A. Datta, J. Phys. Chem. B, 2012, 116, 11466–11472 CrossRef CAS PubMed.
- P. Ghosh, A. Maity, T. Das, S. Mondal and P. Purkayastha, Soft Matter, 2013, 9, 8512–8518 RSC.
- K. Rurack, K. Hoffmann, W. Al-Soufi and U. Resch-Genger, J. Phys. Chem. B, 2002, 106, 9744–9752 CrossRef CAS.
- K. Gavvala, A. Sengupta, R. K. Koninti and P. Hazra, Phys. Chem. Chem. Phys., 2014, 16, 933–939 RSC.
- X. L. Hu, J. Klimeš and A. Michaelides, Phys. Chem. Chem. Phys., 2010, 12, 3953–3956 RSC.
- A. Hassanali, F. Giberti, J. Cuny, T. D. Kühne and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 13723–13728 CrossRef CAS PubMed.
- B. K. Mai, K. Park, M. P. T. Duong and Y. Kim, J. Phys. Chem. B, 2013, 117, 307–315 CrossRef CAS PubMed.
- S. Narayan, D. L. Wyatt, D. S. Crumrine and S. Cukierman, Biophys. J., 2007, 93, 1571–1579 CrossRef CAS PubMed.
- E. Freier, S. Wolf and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11435–11439 CrossRef CAS PubMed.
- T. Limnell, J. Riikonen, J. Salonen, A. M. Kaukonen, L. Laitinen, J. Hirvonen and V. P. Lehto, Int. J. Pharm., 2007, 343, 141–147 CrossRef CAS PubMed.
- H. A. Santos, J. Riikonen, J. Salonen, E. Mäkilä, T. Heikkilä, T. Laaksonen, L. Peltonen, V. P. Lehto and J. Hirvonen, Acta Biomater., 2010, 6, 2721–2731 CrossRef CAS PubMed.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235–1237 CrossRef CAS.
- J. Cheng, R. Bordes, E. Olsson and K. Holmberg, Colloids Surf., A, 2013, 436, 823–829 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |