
Enhancement of ORR catalytic activity by multiple heteroatom-doped carbon materials†
Dae-wook
Kim
a,
Oi Lun
Li
ab and
Nagahiro
Saito
*abc
aDepartment of Materials, Physics and Energy Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan. E-mail: hiro@rd.numse.nagoya-u.ac.jp; Fax: +81 52 789 3259; Tel: +81 52 789 3259
bGreen Mobility Collaborative Research Center, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan
cEcoTopia Science Institute, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, Japan
First published on 9th October 2014
Abstract
Heteroatom-doped carbon matrices have been attracting significant attention due to their superior electrochemical stability, light weight and low cost. Hence, in this study, various types of heteroatom, including single dopants of N, B and P and multiple dopants of B–N and P–N with a carbon matrix were synthesized by an innovative method named the solution plasma process. The heteroatom was doped into the carbon matrix during the discharge process by continuous dissociation and recombination of precursors. The chemical bonding structure, ORR activity and electrochemical performance were compared in detail for each single dopant and multiple dopants. According to the Raman spectra, the carbon structures were deformed by the doped heteroatoms in the carbon matrix. In comparison with N-doped structures (NCNS), the ORR potential of PN-doped structures (PNCNS) was positively shifted from −0.27 V to −0.24 V. It was observed that doping with N decreased the bonding between P and C in the matrix. The multiple doping induced additional active sites for ORR which further enhanced ORR activity and stability. Therefore, PNCNS is a promising metal-free catalyst for ORR at the cathode in a fuel cell.
1. Introduction
In energy conversion devices, in particular, fuel cells and metal–air batteries, the oxygen reduction reaction (ORR) occurs at the cathode and is crucial reaction for high performance.1,2 In order to improve cell potential, it is necessary to efficiently reduce oxygen at the cathode. Platinum (Pt) based materials are acknowledged as the best catalysts in terms of catalytic activity for ORR, but they have some problems in overall performance in operation such as high cost, durability and methanol tolerance, and are easily polluted by carbon monoxide (CO).3 Therefore, alternative materials are essential, and much research is focused on replacing Pt-based materials with others having similar ORR activity.Various types of metal alloys and metal oxides have been researched as alternative catalysts.4–6 However, there still remain difficulties in terms of durability. Decomposition, agglomeration and sintering of the metal particles are often encountered on exposure to acid or alkaline environments in fuel cell operation. This leads to a significant decrease in electrochemical performance.7 Therefore, for long-term operation, non-metal based catalysts, i.e. heteroatom-doped carbon materials, are worth considering as an alternative. Such materials have shown promising potential as effective catalysts for ORR.8,9 Other beneficial properties, such as light weight and low capital cost, further encourage commercial development of these materials.
Among many dopants attempted within the carbon matrix, nitrogen has been attracting significant attention due to its electron accepting ability and superior electrochemical stability.10,11 Several different types of nitrogen doped materials have been reported, including N-doped carbon nanotubes, graphene, ordered mesoporous carbon, carbon black, and activated carbon. The results demonstrated that N-doped carbon is a suitable catalyst for long-term operation in a cathode.11–14
Theoretically, doped nitrogen within the carbon matrix can induce defects due to the difference of atomic radius, bond length and electronegativity. The conjugated heteroatom and the surrounding site become uneven in the distribution of electric charge state.15,16 As a consequence, the carbon atoms are positively charged by the charge delocalization. The positively charged carbon atoms efficiently reduce oxygen molecules to water.17,18 Doped nitrogen results in a positive charge on carbon and the neighboring carbon can be an active site for ORR. Boron and phosphorus, on the other hand, have electronegativities lower than that of carbon. In comparison to nitrogen, B and P exhibit a different ORR mechanism. The doped B or P dopants become active sites instead of the carbon. Due to the different electronic structure and chemical properties, the simultaneous doping of N with B or P might further change the electronic structure of the carbon matrix. Therefore, the multiple heteroatom-doped carbon matrices were expected to possess new active sites corresponding to the ratio and position of various dopants.
In our previous work, we reported an innovative synthetic procedure, named the solution plasma process (SPP), for carbon-related materials. SPP is defined as non-equilibrium or cold plasma occurring in a liquid.19–21 The process has been used to successfully synthesize carbon materials with nitrogen-doped carbon.22 In this study, we focused on the evaluation of ORR activity and electrochemical performance of single and multiple heteroatom-doped carbon in the application to an alkaline anion exchange membrane fuel cell (AAEMFC). The chemical bonding structure of the prepared samples was investigated to explain the corresponding dominant active site and the relationship between dopants and ORR catalytic activity.
2. Experimental procedure
2.1 Preparation of single and multiple doped carbon nanoparticles by SPP
Carbon nanoparticles were prepared by the solution plasma process, with benzene (C6H6, Kanto Chemical) serving as the basic carbon precursor. In order to dope nitrogen, boron or phosphorous atoms into the carbon matrix, pyridine (C5H6N, Tokyo chemical industry), phenylboronic acid (C6H7BO2, Tokyo Chemical Industry) and phenylphosphonic acid (C6H7PO3, Tokyo Chemical Industry) were chosen as the corresponding precursors. The synthesis of N-doped carbon nanoparticles was performed by mixing benzene and pyridine solutions in different ratios. The boron and phosphorus were doped within the carbon matrix with and without nitrogen. For synthesizing B–N and P–N doped carbon matrices, phenylboronic and phenylphosphonic acid were added respectively into the mixture of benzene and pyridine. The obtained samples with different heteroatom doping were denoted as CNS, NCNS (N-doped), BCNS (B-doped), PCNS (P-doped), BNCNS (BN-doped), and PNCNS (PN-doped).The generation of plasma in solution was performed by using a pair of tungsten electrodes and a bipolar pulsed power generator (Kurita, Japan). The duration time of the plasma process is 30 minutes. The schematic of SPP is shown in Fig. 1. After the discharge process, the obtained solution was filtered and the residual carbon particles were dried in a vacuum oven at 80 °C. In order to increase the electronic conductivity of the carbon matrix, it was annealed at 700 °C for 30 minutes under an argon atmosphere.
 | ||
| Fig. 1 Schematic of the solution plasma process. | ||
2.2 Characterization
Transmission electron microscopy (TEM, JEOL-JEM2500SE at 200 kV) observations were conducted to study the microstructure, shape and size of the synthesized heteroatom-doped carbon nanoparticles. Raman spectra were obtained by Raman spectroscopy (NRS-1000, JASCO Corp.) operated at an excitation wavelength of 514 nm. X-ray diffraction patterns (XRD, Rigaku Smart lab) were obtained using Cu Kα (1.54 Å) as a target over a scan range of 15–90° with 0.02° step size and 2° min−1 scan speed. The chemical bonding structures were examined by X-ray photoelectron spectroscopy (XPS, VG Scientific, ESCALAB250 at 15 kV, 15 mA). All measurements were performed using a monochromatic Al Kα X-ray (1486 eV) source.2.3 Electrode preparation and electrochemical characterization
The electrochemical properties of the heteroatom-doped carbon were evaluated. In order to examine the electrochemical properties, cyclic voltammetry (CV) and linear sweep voltammetry (LSV) were performed using three electrode cells with a potentiostat (Hokuto Denko Inc. HZ5000). The three electrode cells consisted of a glassy carbon (GC) disk (3 mm in diameter) working electrode, platinum counter electrode and Ag/AgCl reference electrode. The working electrode was prepared by mixing 5 mg of sample, 0.5 ml of ethanol, and 50 μl of Nafion solution (Aldrich, 5 wt% Nafion) by sonication until a homogeneous suspension was formed. 15 μl of the obtained suspension was spread on the GC electrode and dried at room temperature prior to the measurement. The ORR catalytic activity of all samples will be evaluated based on future application in AAEMFC and the direct methanol fuel cell (DMFC). The CV method was conducted with a 0.1 M KOH electrolyte and saturated O2 concentration with a 20 mV s−1 scan rate from 0.3 V to −1 V (V vs. Ag/AgCl). The LSV method was performed with the same solution and a 10 mV s−1 scan rate from 0.1 V to −0.9 V (V vs. Ag/AgCl) at various rotation speeds from 900 to 3600 rpm using a rotating disk electrode (RDE). A commercial 20 wt% Pt/C catalyst was selected as a performance benchmark. The electrochemical durability was measured by chronoamperometric responses in 0.1 M KOH with saturated oxygen concentration for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s at −0.4 V. The methanol crossover effect was demonstrated to show the effect of methanol poisoning.
000 s at −0.4 V. The methanol crossover effect was demonstrated to show the effect of methanol poisoning.
3. Results and discussion
3.1 Synthesis mechanism and structural characterization
In our previous report, we successfully applied SPP to the synthesis of carbon particles in benzene with an extremely rapid reaction.22–25 The morphology of the obtained carbon exhibited a mesoporous structure with a high surface area and high pore volume.24,26 In this study, we focused on a different route to introduce heteroatoms as ORR active sites into the carbon material. We synthesized heteroatom-doped carbon based on the dissociation and recombination reactions within liquid plasma. The proposed synthesis mechanism of heteroatom-doped carbon is shown in Fig. 2. The proposed specific carbon structure was further analysed in terms of chemical bonding and structural deformation.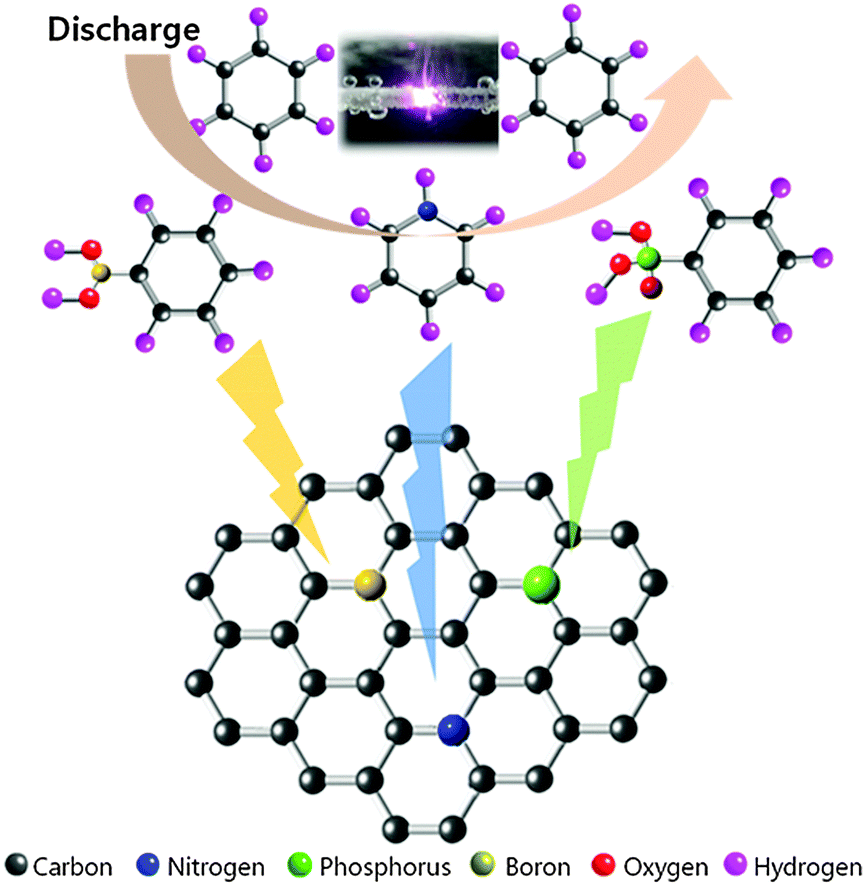 | ||
| Fig. 2 Schematic of SPP-synthesized carbon structure and synthesis mechanism. | ||
Fig. 3(a) shows the Raman spectra of various heteroatom-doped carbon materials. As the baseline, the pure carbon matrix, CNS, exhibited the lowest ID/IG values of the prepared samples. The ID/IG value increased when heteroatoms were doped into the carbon matrix. Among all prepared samples, PNCNS indicated the highest ID/IG values. The D peak corresponds to the distortion in the graphitic lattice. Generally, the ratio of D and G peaks has been used to determine the degree of carbon disorder.27,28 Introducing P dopants can result in defects due to the large atomic radius. Therefore, doping with P may cause deformation of the carbon matrix. The carbon structure was further examined by XRD pattern. Fig. 3(b) presents the XRD patterns of the prepared samples. As shown in the spectra, a broad peak of the carbon (002) plane was observed around 25°. However, the peak was slightly shifted to a lower angle and became broader with doped carbon matrices. These results confirmed that the heteroatom doping induced defects in the carbon structure and multiple doping might further deform the carbon structure. The results were in agreement with those of the Raman spectra, thus confirming that the heteroatoms were successfully doped into the carbon matrix and led to deformation.
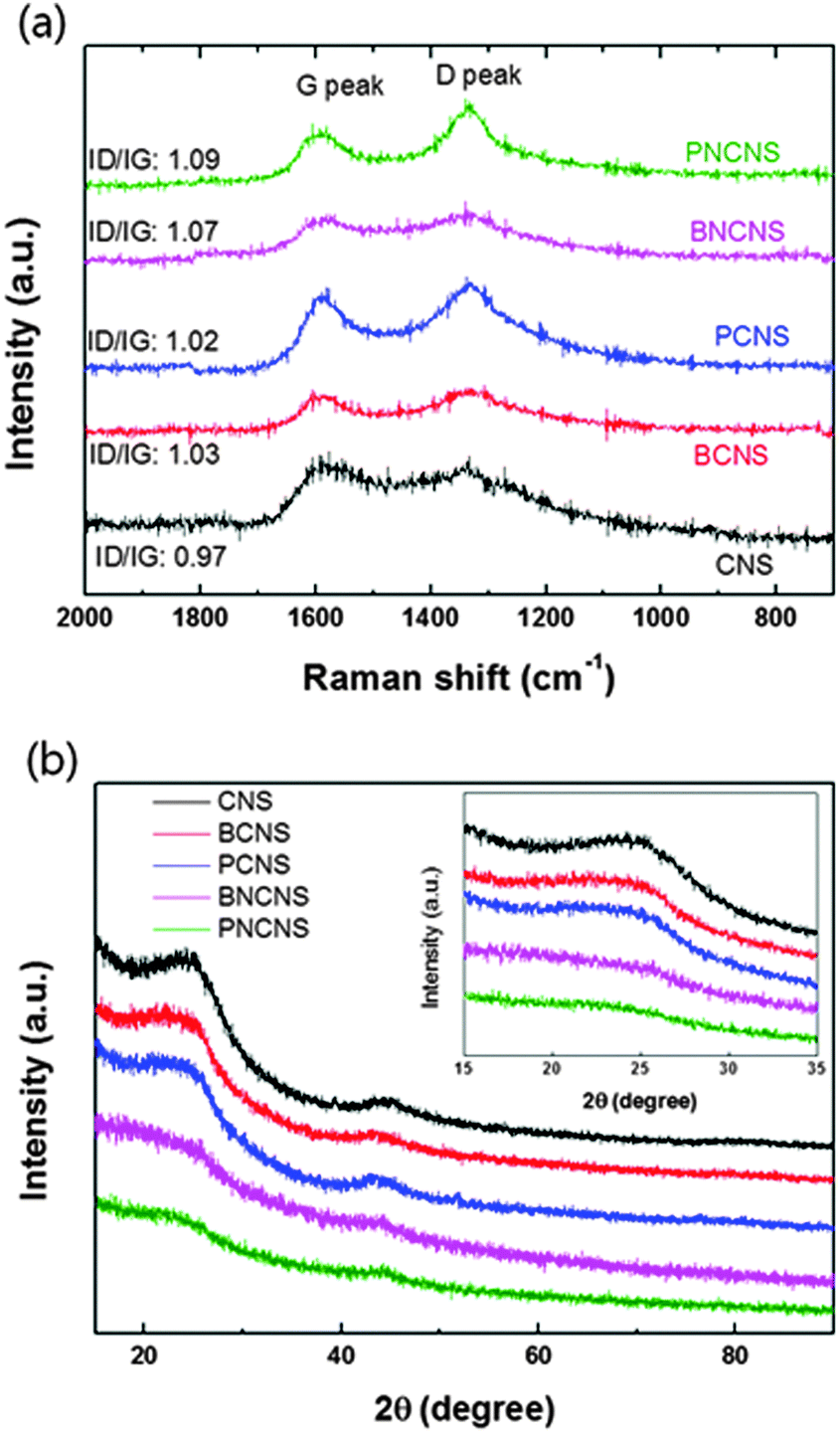 | ||
| Fig. 3 Raman spectra (a) and XRD patterns (b) of heteroatom-doped carbon nanoparticles. | ||
3.2 Chemical bonding state of heteroatom-doped carbons
The chemical bonding structure has been examined by XPS analysis. In Fig. 4(a), four typical peaks of nitrogen bonding (pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N) were observed for all prepared samples. The NCNS and PNCNS did not show obvious differences, while BNCNS showed an increase of the pyridinic peak ratio and the peak was shifted to a lower binding energy. This result might correspond to B–N bonding. As the pyridinic peak in nitrogen bonding has been reported as the most favourable structure as an ORR active site,29,30 the additional bonding with B might present a positive effect on the ORR catalytic activity.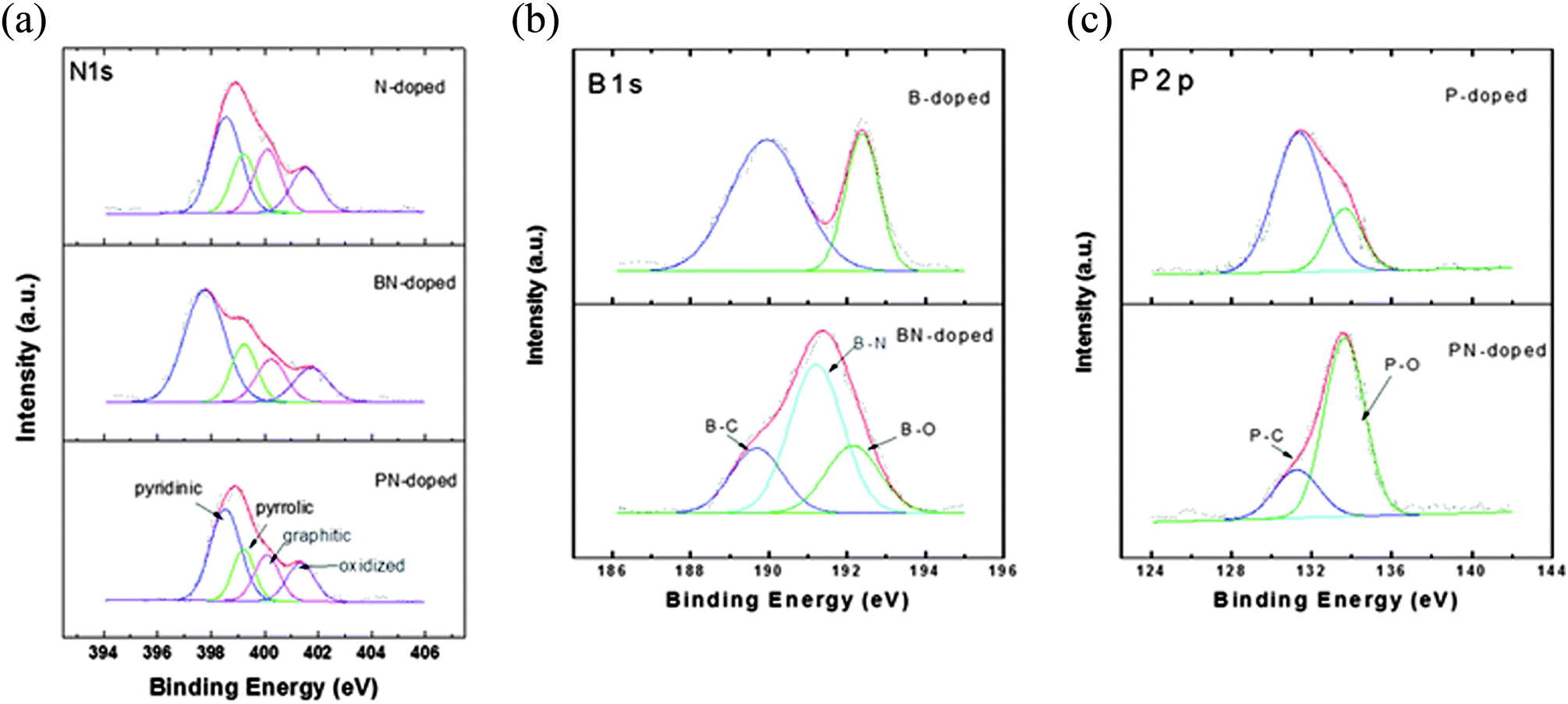 | ||
| Fig. 4 XPS spectra of heteroatom-doped carbon nanoparticles: (a) N1s, (b) B1s and (c) P2p. | ||
In Fig. 4(b), the BCNS presents two peaks in the B1s spectrum. The B–C peak shows that B–C bonding is dominant, followed by B–O bonding. In the case of BNCNS, an additional B–N peak appeared, whereas the intensity of B–C and B–O bonding decreased. The obtained result revealed that B and N bonded within the carbon matrix. In Fig. 4(c), the P2p spectra show contrary results for the PCNS and the PNCNS. The PCNS showed dominant P–C bonding, whereas PNCNS demonstrated P–O bonding as the dominant site. Therefore, the dominant sites shifted from B–C and P–C to B–N and P–O, respectively, when the solution was altered from benzene to a mixture of benzene and pyridine. This finding indicates that multiple doping with N might result in a reduction in the amount of P–C or B–C bonding.
The total dopant concentration of each sample was measured and is shown in the ESI,† Table S1. The total dopant concentration increased when the carbon matrices were doped by multiple atoms (e.g. BNCNS and PBCNS). Comparing single and multiple doping, the oxygen and phosphorus concentrations in multi-doped PNCNS were higher than those of single-doped PCNS. A similar trend was also observed between BCNS and BNCNS. In fact, BNCNS and PNCNS have a very similar ORR onset potential. The results indicated that the increase in B–N, P–O and C–N active sites can further enhance ORR activity. PNCNS, which showed the highest total doping concentration and active site ratio, demonstrated superior ORR activity among all the doped carbon matrices. In addition, the dominant active site changed from P–C to P–O bonding when the sample was dual doped in PNCNS. P–O bonding might be important for further enhancement of ORR activities. The catalytic activity of each sample will be discussed in detail in Section 3.3.
3.3 Electrochemical properties of heteroatom-doped carbons
The electrochemical performance of each doped carbon material was evaluated and discussed. Fig. 5 presents the cyclic voltammetry (CV) results of all samples in oxygen-saturated 0.1 M KOH electrolyte. In single-doped carbon, the cathodic peaks corresponding to ORR were observed in the range between −0.31 and −0.27 V. The effect of each dopant on ORR was as follows: N > P > B. However, multiple-doped carbon materials including BNCNS and PNCNS exhibited a further enhancement in ORR activity. Compared to single doping, the cathodic peak potential was positively shifted by about 0.043 V and the current density also increased by about 40%. In particular, PNCNS showed the highest ORR activity among all samples. In the case of single dopants such as B or P, the heteroatoms were positively charged and acted as the dominant site. Meanwhile, N dopants induced a positive charge on the carbon atom, so that the neighboring carbon became the active site for ORR instead. Thus, carbon materials doped with B–N and P–N might have the possibility to offset ORR activity due to the opposite characteristics of B–C or P–C sites and C–N sites. The bonding ratio of each doped carbon material is shown in Fig. 6(a). Multiple dopants induced a decrease in B–C and P–C bonding, while B–N and P–O bonding became the dominant sites. According to Table S1 (ESI†), the total doping concentration increased with multiple dopants. Comparing single and multiple doping, the oxygen and phosphorus concentrations were increased for PNCNS. Also, the nitrogen and boron concentrations were increased for BNCNS. Thus, an increase in the number of B–N, P–O and C–N active sites can further enhance ORR activity. In the case of PNCNS, the total doping concentration was the highest and the active site ratio was higher than that of BNCNS. In addition, the large atomic radius of phosphorus created more defects and distortions, which disturbed the delocalized electrons. In Fig. S3 (ESI†), the C1s peak of carbon shows C–C bonding at 284.4 eV. The binding energy of the C–C bonding was positively shifted by heteroatom doping, and phosphorus doping induced the highest binding energy. The increase in the binding energy implies that more charge transfer was occurred from the carbon atoms to the nearby heteroatoms.31,32 Weakening of oxygen molecule bond and reducing oxygen molecule efficiently induced by charge delocalization which could serve as an active.15 The PNCNS showed a higher binding energy than that of BNCNS and NCNS. This implies that the electrons were transferred to a greater degree, and consequently, the adsorbed oxygen molecule could be reduced easily by simultaneous doping of N with P. Therefore, we hypothesized that the multiple-doped carbon materials were expected to contain various active sites originating from B–N or P–N dopants.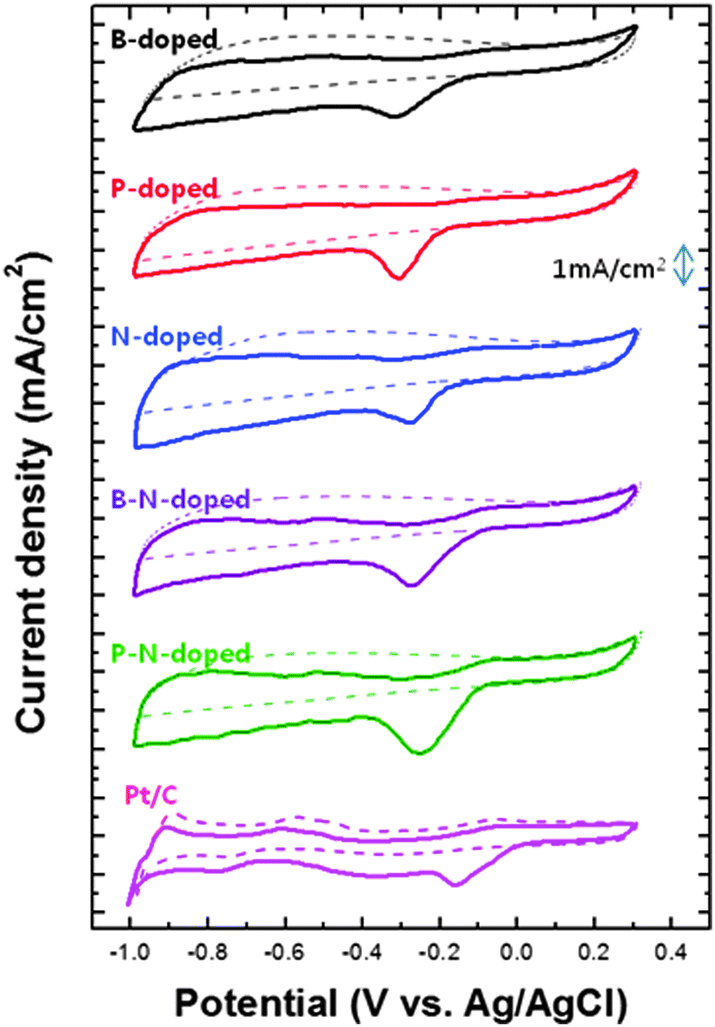 | ||
| Fig. 5 Cyclic voltammetry measurements of heteroatom-doped carbon nanoparticles compared with Pt/C catalyst in 0.1 M KOH saturated with N2 (dotted line) or O2 (dashed line). | ||
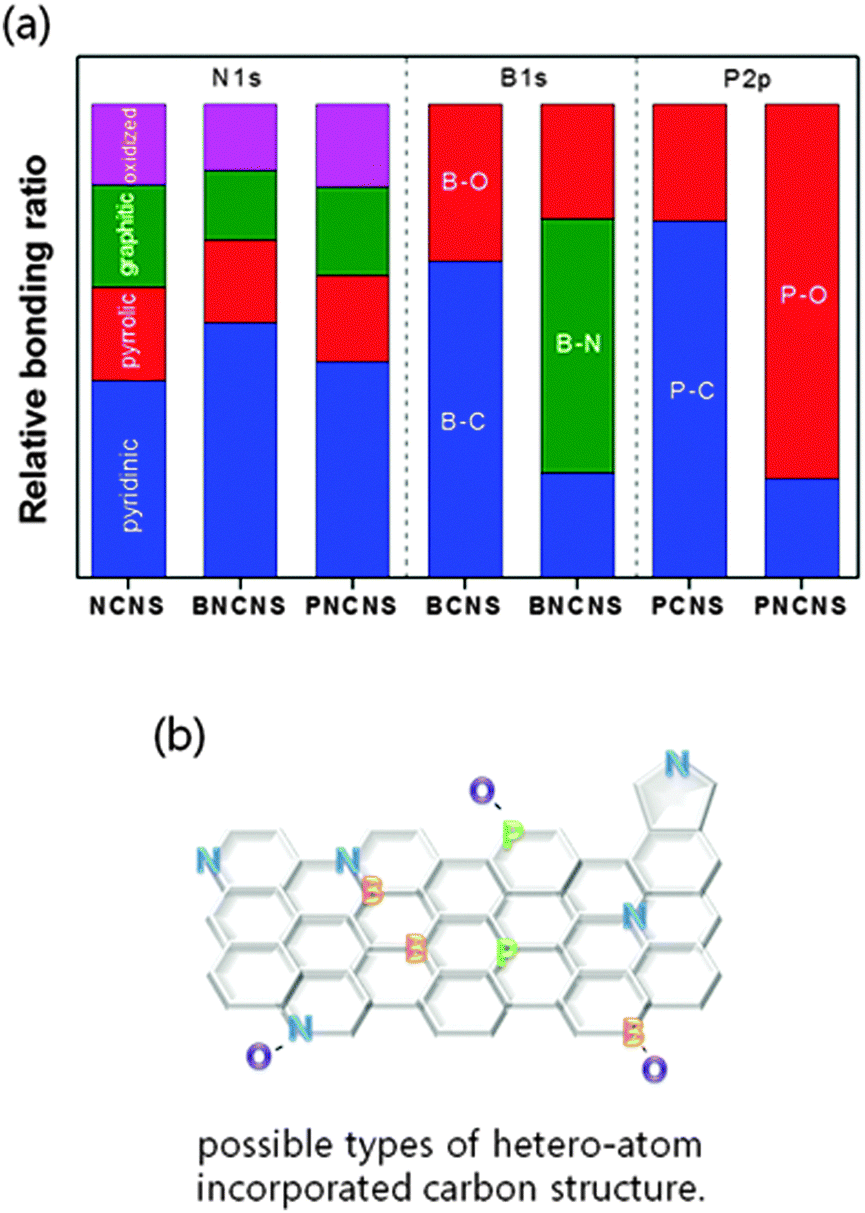 | ||
| Fig. 6 (a) Relative chemical bonding ratio and (b) possible types of structure derived from XPS measurements. | ||
In order to understand the electron transfer kinetics, we performed linear sweep voltammetry (LSV) measurements at different rotation speeds ranging from 900 to 3600 rpm using a rotating disk electrode (RDE). The kinetic and diffusion mechanisms of reaction can be addressed by the Koutecky–Levich equations using the obtained parameters:33
| 1/j = 1/jL + 1/jk | (1) |
| 1/j = 1/Bω1/2 + 1/jk | (2) |
| B = 0.62nFCo*Do2/3ν−1/6 | (3) |
485 C), n is the number of electrons transferred per oxygen molecule, Co* and Do are the oxygen bulk concentration (1.2 × 10−3 mol cm−3) and diffusion coefficient of O2 (1.9 × 10−5 cm2 s−1), respectively, and ν is the kinematic viscosity of the electrolyte (1.1 × 10−2 cm2 s−1). The obtained currents with respect to the different rotation speeds are plotted in Fig. S4 (ESI†). The electron transfer numbers were calculated from the plot in Fig. 7(c) using eqn (3). Fig. 7(b) presents the calculated electron transfer numbers for ORR. Single-doped carbon materials exhibited electron transfer numbers ranging from 2.8 to 3.3, while the multiple-doped carbon materials exhibited electron numbers of 3.5 and 3.7. In particular, PNCNS exhibited the highest value. These results clearly demonstrated that multiple doping induced interactions between dopants and enhanced the ORR activity.
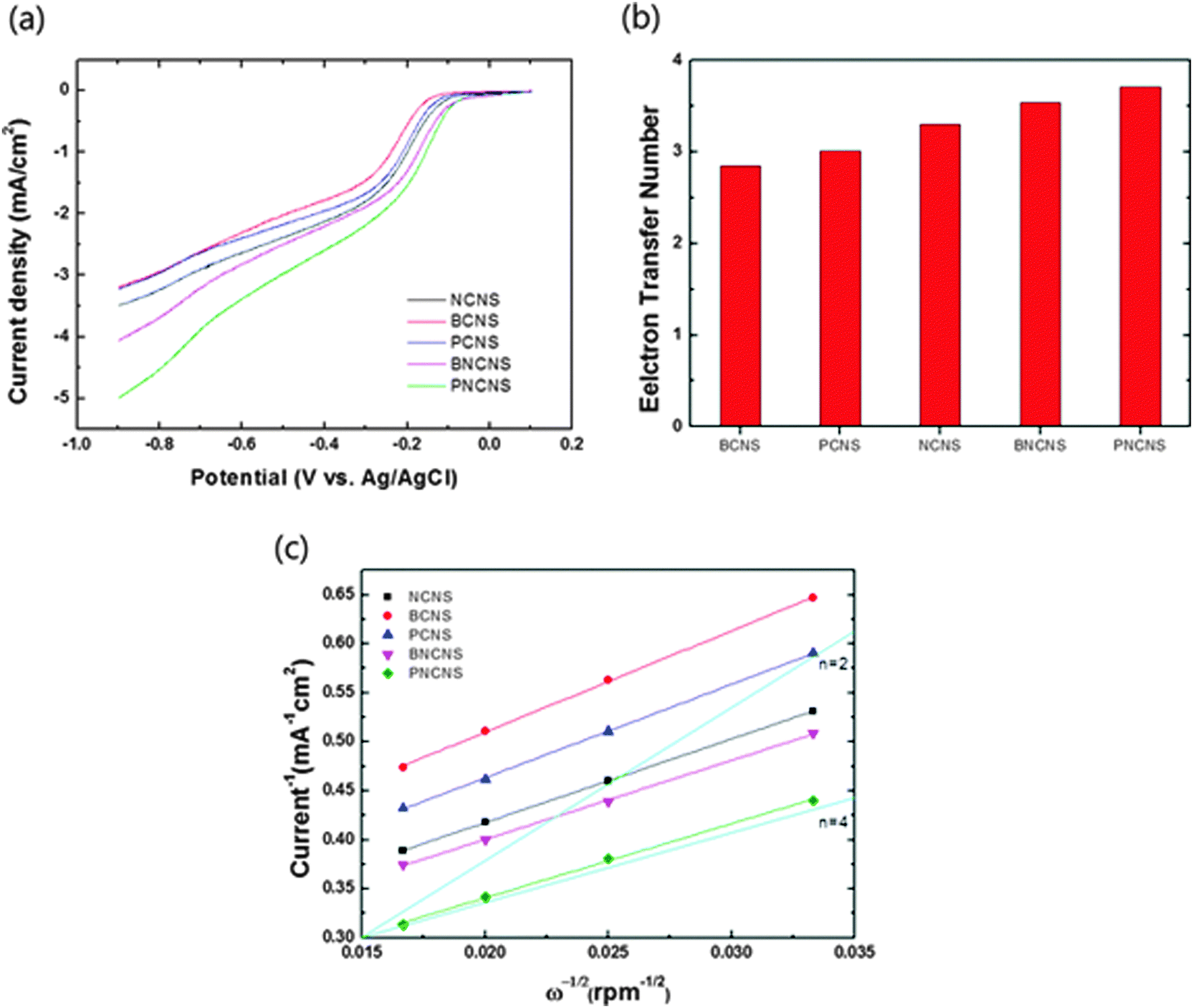 | ||
| Fig. 7 (a) Linear sweep voltammetry measurements at a rotation speed of 1600 rpm, (b) transferred electron numbers of heteroatom-doped carbon nanoparticles and (c) Koutecky–Levich plot. | ||
Another important issue in fuel cell applications concerns the low durability of Pt-based catalysts in long-term operation. In particular, methanol tolerance is essential for application in direct methanol fuel cells (DMFC). Carbon monoxide (CO) can be adsorbed on the surface of catalysts during the reaction with methanol. As a result, this depletes the catalytic activity for ORR. The effect of methanol crossover and long-term durability of various catalysts including NCNS, BNCNS, PNCNS and commercial Pt/C catalysts were measured in oxygen saturated solution with and without methanol. In Fig. 8, commercial Pt/C showed an obvious current drop corresponding to the methanol oxidation reaction (MOR). Besides, the oxygen reduction current could not recover to its initial current value, while NCNS, BNCNS and PNCNS did not show an evident change with methanol addition. The electrocatalytic durability of the prepared samples and Pt/C was also compared. PNCNS retained the highest relative current (96% of initial current) after 20000 s. NCNS and BNCNS also showed a high durability for long-term operation, with the relative current remaining at over 90% of the initial current. On the other hand, the ORR activity decreased to 53% of the initial current for Pt/C.
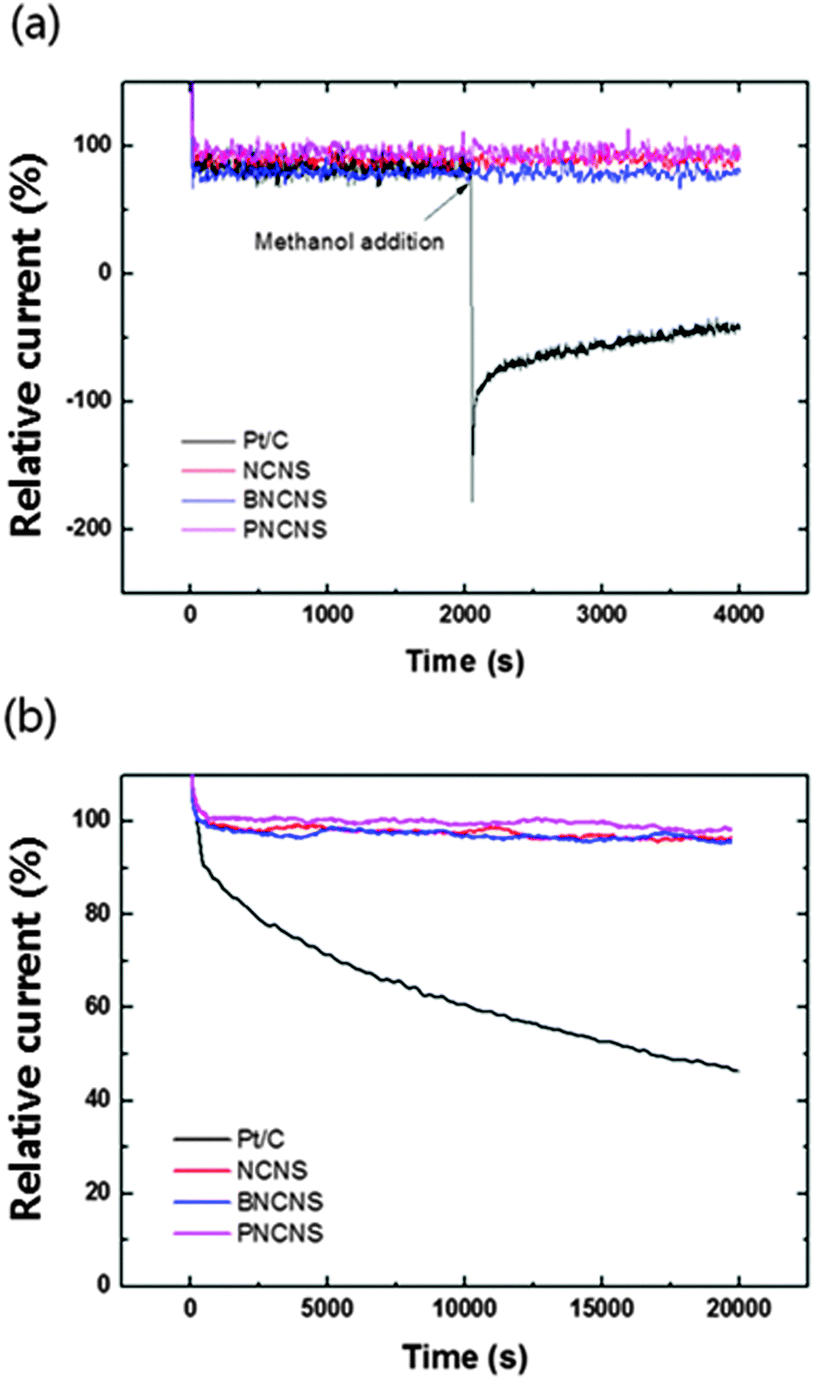 | ||
| Fig. 8 The current–time chronoamperometric responses of heteroatom-doped carbon nanoparticles compared with Pt/C catalyst in O2-saturated 0.1 M KOH (a) in the presence of methanol and (b) in the absence of methanol. | ||
Conclusions
Carbon matrices doped with various types of heteroatom, including N, B and P, were successfully synthesized by SPP. The obtained Raman spectra and XRD patterns confirmed that heteroatoms were successfully doped within the carbon matrix and the carbon structure was deformed by the doped heteroatoms. The XPS results determined that single dopants were bonded to the carbon as the dominant site, while multiple dopants mainly bonded with nitrogen or oxygen. In fact, the ORR activity was highly related to the type of chemical bonding. Among all samples, the PNCNS exhibited the best ORR activity and durability. In comparison with Pt/C catalysts, PNCNS presented comparable ORR activity and superior durability. Also, the presence of methanol has a negligible effect on its catalytic performance. Therefore, the SPP-synthesized multiple heteroatom-doped carbon matrix could be a promising alternative ORR catalyst for application to alkaline fuel cell devices.Notes and references
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- Y. Liu, A. Ishihara, S. Mitsushima, N. Kamiya and K. I. Ota, J. Electrochem. Soc., 2007, 154, 664 CrossRef PubMed.
- J. Kim, T. Oh, Y. Shin, J. Bonnett and K. Weil, Int. J. Hydrogen Energy, 2011, 36, 4557 CrossRef CAS PubMed.
- S. Doi, A. Ishihara, S. Mitsushima, N. Kamiya and K. I. Ota, J. Electrochem. Soc., 2007, 154, 362 CrossRef PubMed.
- Y. J. Wang, D. P. Wilkinson and J. J. Zhang, Chem. Rev., 2011, 111, 7625 CrossRef CAS PubMed.
- P. H. Matter, L. Zhang and U. S. Ozkan, J. Catal., 2006, 239, 83 CrossRef CAS PubMed.
- B. W. Jensen, O. W. Jensen, M. Forsyth and D. R. MacFarlane, Science, 2008, 321, 671 CrossRef PubMed.
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182 CrossRef CAS PubMed.
- D. Yu, Q. Zhang and L. Dai, J. Am. Chem. Soc., 2010, 132, 15127 CrossRef CAS PubMed.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS PubMed.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170 CAS.
- J. Chen, X. Wang, X. Cui, G. Yang and W. Zheng, Chem. Commun., 2014, 50, 557 RSC.
- K. P. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- Y. F. Tang, B. L. Allen, D. R. Kauffman and A. J. Star, J. Am. Chem. Soc., 2009, 131, 13200 CrossRef CAS PubMed.
- K. Parvez, S. B. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. L. Feng and K. Müllen, ACS Nano, 2012, 6, 9541 CrossRef CAS PubMed.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X. F. Zhou, H. T. Wang, J. J. Xu and E. G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155454 CrossRef.
- O. Takai, Pure Appl. Chem., 2008, 80, 2003 CrossRef CAS.
- P. Pootawang, N. Saito and O. Takai, Mater. Lett., 2011, 65, 1037 CrossRef CAS PubMed.
- M. A. Bratescu, S. P. Cho, N. Saito and O. Takai, J. Phys. Chem. C, 2011, 115, 24569 CAS.
- D. Kim, O. L. Li and N. Saito, RSC Adv., 2014, 4, 16813 RSC.
- J. Kang, O. L. Li and N. Saito, Carbon, 2013, 60, 292 CrossRef CAS PubMed.
- D. Kim, O. L. Li and N. Saito, Phys. Chem. Chem. Phys., 2014, 16, 14905 RSC.
- J. Kang, O. L. Li and N. Saito, J. Power Sources, 2014, 261, 156 CrossRef CAS PubMed.
- J. Kang, O. L. Li and N. Saito, Nanoscale, 2013, 5, 6874 RSC.
- K. Ghosh, M. Kumar, T. Maruyama and Y. Ando, Carbon, 2009, 47, 1565 CrossRef CAS PubMed.
- C. Choi, S. Park and S. Woo, J. Mater. Chem., 2012, 22, 12107 RSC.
- S. van Dommele, K. P. de Jong and J. H. Bitter, Chem. Commun., 2006, 4859 RSC.
- S. Van Dommele, A. R. Izquirdo, R. Brydson, K. P. de Jong and J. H. Bitter, Carbon, 2008, 46, 138 CrossRef CAS PubMed.
- S. Lim, H. Elim, X. Gao, A. wee, W. Ji, J. Lee and J. Lin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 1 CrossRef.
- G. Liu, X. Li, P. Ganesan and B. Porov, Appl. Catal., B, 2009, 93, 156 CrossRef CAS PubMed.
- R. Liu, C. Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2011, 133, 10372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp03868a |
| This journal is © the Owner Societies 2015 |