
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An experimental and theoretical study of the gas phase kinetics of atomic chlorine reactions with CH3NH2, (CH3)2NH, and (CH3)3N†
J. M.
Nicovich
a,
S.
Mazumder
a,
P. L.
Laine‡
b,
P. H.
Wine
*ab,
Y.
Tang
c,
A. J. C.
Bunkan
d and
C. J.
Nielsen
*d
aSchool of Chemistry & Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332-0400, USA. E-mail: paul.wine@chemistry.gatech.edu
bSchool of Earth & Atmospheric Sciences, Georgia Institute of Technology, Atlanta, GA 30332-0340, USA
cSchool of Environmental and Municipal Engineering, Qingdao Technological University, Fushun Road 11, 266033 Qingdao, Shandong, P. R. China
dCentre for Theoretical Computational Chemistry, Department of Chemistry, University of Oslo, P.O. Box 1033 Blindern, 0316 Oslo, Norway. E-mail: c.j.nielsen@kjemi.uio.no
First published on 11th November 2014
Abstract
The rate coefficients for the reactions of Cl(2PJ) with methylamine (R1), dimethylamine (R2) and trimethylamine (R3) have been measured using the laser flash photolysis – resonance fluorescence technique as a function of temperature (274–435 K) and pressure (25–400 Torr N2). The experimental data are well-represented by the following temperature- and pressure-independent rate coefficients (1010 × k/cm3 molecule−1 s−1): kR1 = 2.90 ± 0.44, kR2 = 3.89 ± 0.58, kR3 = 3.68 ± 0.55; the uncertainties are estimates of accuracy at the 95% confidence level. Potential energy surfaces (PES) for the reactions have been characterized at the MP2/cc-pVTZ level and improved single point energies of stationary points obtained in CCSD(T)-F12a calculations. The PES for all reactions are characterized by the formation of pre and post reaction complexes and submerged barriers. Rate coefficients for the reactions were calculated as a function of temperature and pressure using a master equation model based on the coupled cluster theory results. The calculated rate coefficients are in good agreement with experiment; the overall rate coefficients are relatively insensitive to variations of the barrier heights within typical chemical accuracy, but the predicted branching ratios vary significantly. The inclusion of tunnelling has no effect.
Introduction
Several reviews of the atmospheric occurrence, thermodynamic properties and chemistry of amines have recently appeared.1–4 In spite of around 150 different amines having been identified in the atmosphere,1 they were almost left out of atmospheric and environmental sciences due to their low ppbV-range mixing ratios and their short lifetimes.5 It has now been demonstrated from modeling of field observations,6 and from controlled experiments in the CLOUD chamber at CERN,7 that amines are important in new particle formation through their gas phase acid–base reaction with sulphuric acid. A recent matrix isolation study shows that trimethylamine and sulfuric acid may even form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of ionic character, in which a proton is nearly completely transferred: (CH3)3NH+⋯−OSO3H.8
:1 complex of ionic character, in which a proton is nearly completely transferred: (CH3)3NH+⋯−OSO3H.8
The primary tropospheric sink for amines is generally accepted to be reaction with the OH radical. It has been reported that levels of Cl atoms in the marine boundary layer can be 1–10 percent of OH levels,9 and findings suggest a significant Cl production rate even in the middle of the continental United States.10 Laboratory and theoretical research demonstrates that heterogeneous reaction of N2O5 with HCl(aq) may represent a significant source of tropospheric ClNOx species that can rapidly photolyze under daytime conditions to generate Cl atoms.11 Hence, it appears that reaction with Cl could be a significant tropospheric sink for any trace gas that reacts with Cl significantly more rapidly than with OH. There are no kinetic data for Cl + amine reactions reported in the literature, although one reaction dynamics study of Cl + CH3NH2 has been published showing yields of the two hydrogen abstraction products to be 48% ˙CH2NH2 and 52% ˙NHCH3 at a collision energy of ∼2000 cm−1.12 Since it is a reasonable expectation that Cl + amine reactions are very fast, laboratory studies to quantify the kinetics of these reactions are needed.
In this paper, we report a combined experimental and theoretical study of the reactions of Cl atoms with mono-, di- and tri-methyl amine.
| Cl + CH3NH2 → products | (R1) |
| Cl + (CH3)2NH → products | (R2) |
| Cl + (CH3)3N → products | (R3) |
Experimental determinations of temperature- and pressure-dependent rate coefficients for (R1)–(R3) are reported for the first time, as are theoretical analyses of reaction potential energy surfaces and kinetics. The potential influence of (R1)–(R3) on the atmospheric chemistry of the studied amines is qualitatively assessed.
Experimental approach
The kinetics of Cl reactions with CH3NH2, (CH3)2NH, and (CH3)3N have been studied under pseudo-first order conditions with the amine as the excess reagent using the laser flash photolysis (LFP) – resonance fluorescence (RF) technique. In the LFP-RF approach, Cl atoms are produced on a nanosecond time scale via LFP of a suitable Cl-containing precursor. A chlorine resonance lamp, which consists of an electrodeless microwave discharge through a flowing gas mixture containing a trace of Cl2 in helium, continuously excites vacuum-UV fluorescence in the photolytically produced Cl atoms. The fluorescence signal is monitored by a solar blind photomultiplier and signals are processed using photon-counting techniques in conjunction with multichannel scaling. As long as the Cl atom concentration is relatively low (less than 1012 atoms per cm3 under typical operating conditions), the fluorescence signal is proportional to the Cl atom concentration.A schematic diagram of the LFP-RF apparatus is published elsewhere.13 The apparatus is similar in configuration to those employed in a number of earlier studies of chlorine atom kinetics carried out at Georgia Tech.14–19 Details of the experimental approach that are specific to this study are provided below.
A jacketed Pyrex® reaction cell with an internal volume of 150 cm3 was used in all experiments. The cell was maintained at a constant temperature by circulating ethylene glycol from a thermostated bath through the outer jacket. A copper-constantan thermocouple was inserted into the reaction zone through a vacuum seal, thus allowing measurement of the gas temperature under the precise pressure and flow rate conditions of the experiment. The temperature variation in the reaction volume, i.e., the volume from which fluorescence could be detected, was less than 2 K at the highest temperature employed in the study (435 K) and less than 1 K at the lowest temperature employed (274 K).
Atomic chlorine was produced by 248 nm laser flash photolysis of phosgene, Cl2CO.
| Cl2CO + hν (248 nm) → 2 Cl + CO | (R4) |
A GAM EX50 KrF excimer laser served as the 248 nm light source; its pulse width is ∼20 ns and fluences employed in the study ranged from 3 to 67 mJ cm−2 pulse−1.
All details concerning the operation of the resonance lamp and signal processing electronics are published elsewhere.14–19 For each chlorine atom decay rate measured, signals from a large number of laser shots (100–20000) were averaged to obtain a well-defined pseudo-first order temporal profile over (typically) three e-folding times of chlorine atom decay.
Both excited spin–orbit state chlorine atoms (2P1/2) and ground state chlorine atoms (2P3/2) can be produced by the ultraviolet photo-dissociation of phosgene; the fraction of excited Cl(2P1/2) has been reported to be <10% at 248 nm.20 The RF detection scheme is sensitive to both spin orbit states. To ensure rapid deactivation of Cl(2P1/2) atoms, approximately 0.5 Torr CO2 was added to each Cl2CO–amine–N2 reaction mixture. Since the rate coefficient for deactivation of Cl(2P1/2) by CO2 is (1.2 ± 0.3) × 10−11 cm3 molecule−1 s−1,21–23 the time scale for spin–orbit state equilibration was always very rapid compared to the time scale for chemical reaction of Cl atoms.
In order to avoid accumulation of photochemically generated reactive species, all experiments were carried out under “slow flow” conditions. The linear flow rate through the reactor was typically 3 cm s−1 (1.4–9 cm s−1 was the complete range), while the laser repetition rate was typically 6 Hz (3–7 Hz was the complete range). Since the direction of flow was perpendicular to the photolysis laser beam, no volume element of the reaction mixture was subjected to more than a few laser shots. As expected, observed kinetics were independent of linear flow rate and laser repetition rate over the ranges investigated. Phosgene (Cl2CO) and amines were introduced into the reaction cell from 12-liter Pyrex® bulbs containing dilute mixtures in N2, while CO2 and N2 were flowed directly from their high pressure storage cylinders. All gas flows were controlled by needle valves and measured using calibrated mass flow meters. The amine–N2 gas mixture, CO2, and additional N2 were premixed before entering the reaction cell whereas the Cl2CO–N2 mixture was injected into reaction mixture flow (typically) 2 cm upstream from the reaction zone; this approach minimized interferences from hydrolysis of Cl2CO on reactor walls and dark reaction of amines with the HCl product of Cl2CO hydrolysis. At 298 K, kinetics results were found to be independent of injector position over the range 2–10 cm upstream from the reaction zone, and also independent of the fraction of total flow attributable to the Cl2CO–N2 mixture over the ranges 2–18% for R1, 0.5–13% for R2, and 0.5–12% for R3. These observations demonstrate that mixing of Cl2CO into the overall flow was complete by the time the flow reached the reaction zone.
Concentrations of each component in the reaction mixture were determined from the corresponding bulb concentrations, the mass flow rates and the total pressures. The bulb concentrations of each amine were measured frequently by UV photometry at 213.86 nm using a zinc penray lamp as the light source. The absorption cross sections employed to convert measured absorbances to concentrations were determined as part of this study and are, in unit of 10−18 cm2 molecule−1, 2.35 ± 0.12 for CH3NH2, 1.27 ± 0.06 for (CH3)2NH and 4.39 ± 0.22 for (CH3)3N. In excellent agreement with our results, Tannenbaum et al.24 report the following cross sections at 213.86 nm (0.1 nm resolution): 2.34 for CH3NH2, 1.34 for (CH3)2NH, and 4.33 for (CH3)3N. One other reported cross section for CH3NH2 at 213.86 nm (0.05 nm resolution) is 1.80,25i.e., about 25% smaller than the cross section we report.25
The gases used in this study had the following stated minimum purities: CO2, 99.99%; Cl2CO, 99.9%; CH3NH2, 98%; (CH3)2NH, 99%; and (CH3)3N, 99%. The above purities all refer to the liquid phase in the high-pressure storage cylinders. The N2 used in this study was the gas obtained as seep-off from a high-pressure liquid nitrogen cylinder. Nitrogen and CO2 were used as supplied while the other gases were degassed repeatedly at 77 K before being used to prepare mixtures with N2.
Computational methods and details
Electronic structure calculations
Geometries and frequencies of the stationary points on the amine + chlorine atom potential energy surfaces were calculated using second order Møller–Plesset perturbation theory (MP2) with Dunning's correlation consistent cc-pVTZ basis set.26 The pre- and post-reaction adducts were localized by calculating the reaction path in mass weighted coordinates (IRC).Improved single point energies of the stationary points were calculated using explicitly correlated Coupled Cluster Singles and Doubles with perturbative triples, CCSD(T)-F12a,27,28 with Dunnings triple-zeta basis set augmented with diffuse functions, aug-cc-pVTZ.29 The MP2 calculations were performed using Gaussian09,30 while the explicitly correlated coupled cluster calculations were performed in Molpro 2012.1.31,32
Calculation of rate coefficients
The kinetics of the different methyl amine + chlorine atom reactions may in principle be governed by the formation of a pre-reaction adduct, one or more tight transition states and possible stabilization of the pre-reaction complex. A master equation model was therefore used to simulate the kinetics of the reactions. Rate coefficients for the inner transition states were calculated using RRKM theory with energies and rovibrational data from the electronic structure calculations, while rate coefficients for the outer transition states were calculated using long-range transition state theory with a dispersion force potential.33 Experimental values for the employed polarizabilities, αi, and the first ionisation potentials, Ii, are summarized in Table S1 (ESI†) and stem from the NIST database.34 All master equation calculations were performed in MESMER 3.0.35The two spin–orbit states 2P3/2 (lowest) and 2P1/2 of the chlorine atom, having degeneracies of 4 and 2, respectively, and separated by 882 cm−1 were included in the calculation of the electronic partition function. Since spin–orbit coupling present in the Cl atom becomes smaller during the reaction it will contribute to the potential energy surface by effectively lowering the non-relativistic energy of the reactants by 1/3 of the SO coupling constant of Cl (3.5 kJ mol−1) assuming negligible SO coupling in the transition state.
Results
Kinetic experiments
As mentioned above, all experiments were carried out under pseudo-first order conditions with [amine] ≫ [Cl]0. Hence, in the absence of side reactions that remove or produce Cl(2PJ) atoms, the Cl(2PJ) temporal profile following the laser flash is described by the following relationship:| ln{[Cl]0/[Cl]t} = ln{S0/St} = (kRi[amine] + kR5)t = k′t | (E1) |
In eqn (E1), S0 is the RF signal at a time immediately after the laser fires, St is the RF signal at a later time t; ki (i = 1, 2 or 3) is the total bimolecular rate coefficient for all Cl(2PJ) + amine reaction channels that are irreversible on the experimental time scale; k′ is the pseudo-first order Cl(2PJ) fluorescence signal decay rate coefficient; and kR5 is the first-order rate coefficient for background Cl(2PJ) atom loss:
| Cl(2PJ) → loss by diffusion from the detector field of view and/or reaction with background impurities. | (R5) |
The bimolecular rate coefficients of interest, kRi(P,T), i = 1–3, are obtained from the slopes of plots of k′ versus [amine] for data obtained at constant temperature and total pressure. Although numerous possible impurities in the methyl amine samples can react rapidly with atomic chlorine, we can assume impurity reactions are of negligible importance because the rate coefficients for reactions (R1)–(R3) are measured to be very fast (see below) and, as reported above, the amine purities were ≥98%.
Overall, the observed kinetics are consistent with the behavior predicted by eqn (E1), i.e., observed Cl(2PJ) temporal profiles are exponential and observed k′ are found to increase linearly with increasing [amine]. Furthermore, observed kinetics were found to be independent of significant variations in laser fluence, confirming the expectation that radical concentrations were low enough to render radical–radical side reactions too slow to be a significant kinetic interference on the time scale of Cl(2PJ) decay. Typical Cl(2PJ) temporal profiles are shown in Fig. 1 and typical plots of k versus [amine] are shown in Fig. 2.
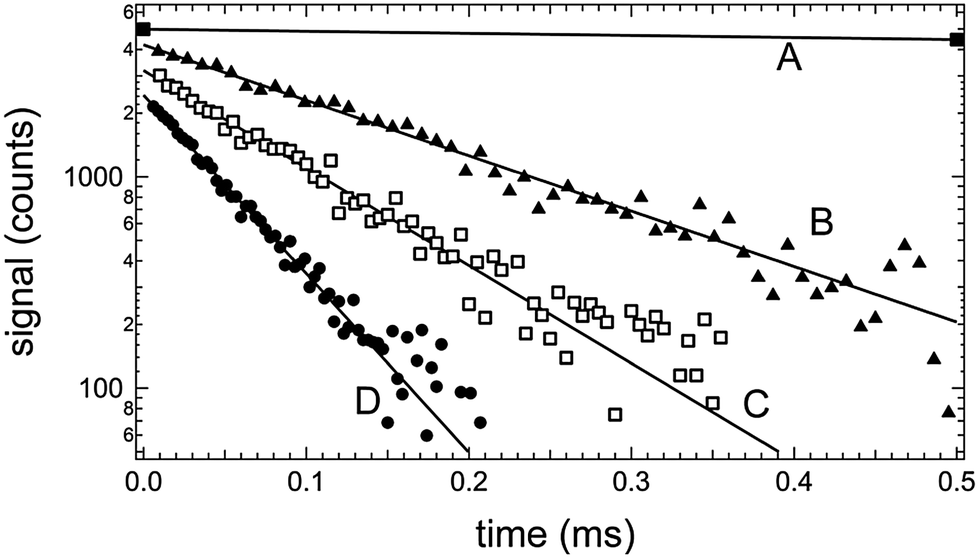 | ||
| Fig. 1 Typical resonance fluorescence temporal profiles observed in kinetic studies of (R1)–(R3). Experimental conditions: T = 296 K, P = 25 Torr, linear flow rate through reactor = 3.0 cm s−1. Concentrations (1011 cm−3): [Cl2CO] = (A) 481, (B) 391, (C) 378 and (D) 481; [CO2] = 210000; [Cl]0 = (A) 3, (B) 0.8, (C) 0.8, (D) 2; [CH3NH2] = (A) 0, (B) 212, (C) 364 and (D) 678. Number of laser shots averaged = (A) 20, (B) 6000, (C) 9000 and (D) 11000. Solid lines are obtained from nonlinear least-squares analyses of the Cl(2PJ) fluorescence signal versus time data and give the following pseudo-first-order decay rates (k′) in units of s−1: (A) 76, (B) 6050, (C) 10600 and (D) 19500. For clarity, traces (A), (B) and (C) are scaled upwards by factors of 3.0, 2.4 and 2.0, respectively. Most of the data used to determine the decay rate for trace (A) were obtained at longer times than those shown in the figure. | ||
 | ||
| Fig. 2 Plots of k′ versus [CH3NH2] for data obtained at different temperatures and pressures. Solid lines are obtained from linear least-squares analyses and lead to the bimolecular rate coefficients reported in Table S2 (ESI†). Blue: 296 K, 25 Torr. Green: 296 K, 200 Torr. Red: 419 K, 25 Torr. | ||
For all three Cl + amine reactions studied, bimolecular rate coefficients were, within experimental uncertainties, found to be independent of pressure over the range 25–400 Torr N2. Such observational evidence supports the idea that the dominant pathway for Cl(2PJ) + amine reactions over the full range of temperature and pressure investigated is H-transfer. Measured bimolecular rate coefficients for reactions (R1)–(R3) are summarized in Tables S2–S4 (ESI†).
Because the precisions of tabulated kRi values are quite good (2σ < 5% at 298 K and 2σ < 11% at other temperatures), we estimate that the absolute uncertainty of reported values for kRi is ±10% at 298 K and ±16% at other temperatures. Since interfering side reactions appear to be of negligible importance (see above), the primary source of systematic error appears to be associated with amine concentration determinations.
Arrhenius plots for reactions (R1)–(R3) are shown in Fig. 3. The following best fit Arrhenius expressions are derived from linear least-squares analyses of the lnkRiversus T−1 data (units are 10−10 cm3 molecule−1 s−1):
| kR1(T) = (2.63 ± 0.30) exp{(+33 ± 38)/T} |
| kR2(T) = (4.46 ± 1.45) exp{(−49 ± 113)/T} |
| kR3(T) = (3.47 ± 0.46) exp{(+18 ± 78)/T} |
Uncertainties in the above expressions are 2σ and represent the precision of the Arrhenius parameters. Given that the statistical uncertainties in measured activation energies are larger than the activation energies themselves, the following temperature independent rate coefficients (obtained from computing unweighted averages of experimental kRi values) are also considered adequate representations of the experimental data (units are 10−10 cm3 molecule−1 s−1): kR1 = 2.90 ± 0.13, kR2 = 3.89 ± 0.46, and kR3 = 3.68 ± 0.35, where the uncertainties are two standard deviations of the average. Absolute uncertainties in these rate coefficients are estimated to be ±15% at the 95% confidence level.
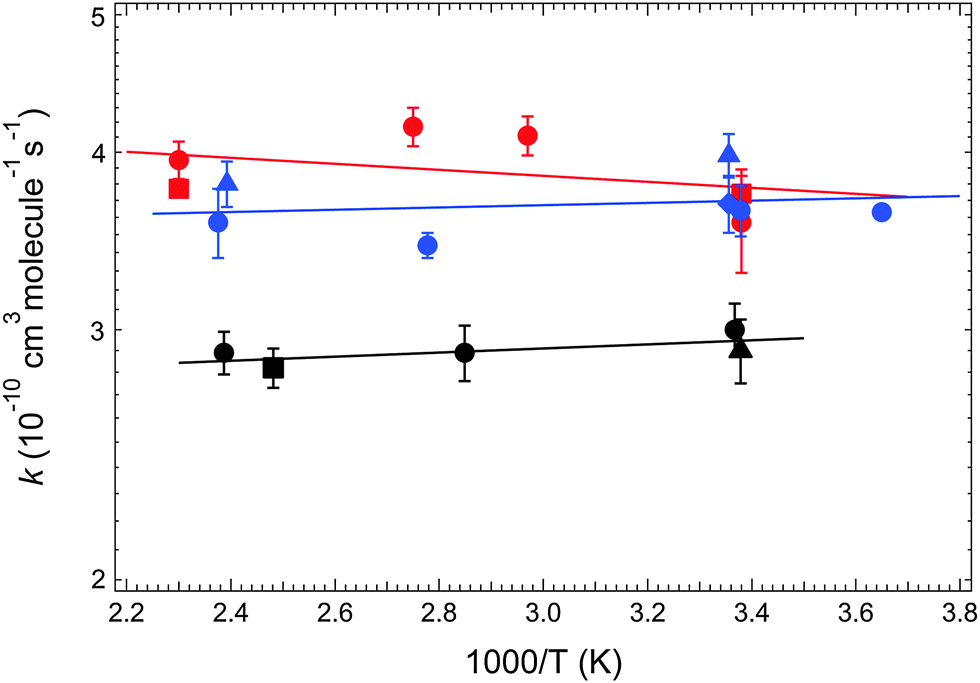 | ||
| Fig. 3 Arrhenius plots for Cl(2PJ) reactions with CH3NH2 (R1, black), (CH3)2NH (R2, red), and (CH3)3N (R3, blue). Solid lines are obtained from linear least squares analyses of the unweighted ln kRiversus T−1 data; the best fit Arrhenius expressions in units of 10−10 cm3 molecule−1 s−1 are kR1 = 2.63 exp(+33/T), kR2 = 4.46 exp(−49/T), and kR3 = 3.47 exp(+18/T). Error bars are 2σ, precision only. | ||
Structures and energies of stationary points
The stationary points on the potential energy surfaces (PES) of the Cl reactions with MA, DMA and TMA were located in MP2/cc-pVTZ calculations. Improved energies were obtained in CCSD(T)-F12a/aug-cc-pVTZ calculations; the results are summarized in Tables S5–S7 (ESI†) and illustrated in Fig. 4. Cartesian coordinates of reactants, products and stationary points on the PES obtained in MP2/cc-pVTZ calculations are given in Table S8 (ESI†), which also includes illustrations of the stationary point structures. The vibrational wave numbers of the saddle points are collected in Table S9 (ESI†). The minimum energy path (MEP) connecting reactants and products of the Cl reactions with MA, DMA and TMA were computed using the intrinsic reaction coordinate (IRC) method36 at the MP2/cc-pVTZ level of theory. In addition to the saddle points of the hydrogen abstraction reactions we have located a pre-reaction adduct on the MEP for all reactions. On the product side of the MEP there is a post-reaction van der Waals adduct between amino radicals and HCl. In summary, the general and prominent features of the amine + Cl reaction PES are strong pre-reaction complexes and saddle points with energies below that of the corresponding reactants. | ||
| Fig. 4 Relative energies of stationary points on the potential energy surfaces of the CH3NH2 + Cl, (CH3)2NH + Cl and (CH3)3N + Cl reactions. Results from CCSD(T)-F12a/aug-cc-pVTZ//MP2/cc-pVTZ calculations. | ||
Calculated rate coefficients
Results from the kinetics calculations are summarized in Table S10 (ESI†); for the temperature range 200–600 K the overall theoretical rate coefficients can conveniently be parameterized (units are 10−10 cm3 molecule−1 s−1):| kR1(T) = 4.20 exp{+3.6/T} or 4.15 (T/298 K)0.01 |
| kR2(T) = 5.43 exp{−48/T} or 4.64 (T/298 K)0.13 |
| kR3(T) = 5.61 exp{−45/T} or 4.74 (T/298 K)0.14 |
The calculations confirm that the reaction rates are independent of pressure; the energy transfer parameter for the pre-reaction complex, 〈ΔEdown〉, was initially set to 250 cm−1, but since the reaction rates do not show any pressure dependence, the calculations are independent the value of 〈ΔEdown〉. The calculated rate coefficients are in very good agreement with the experimental values with the largest deviations being less than a factor of two. This good agreement is to a large extent caused by the fact that the reactions are very close to being collision controlled, with overall rate coefficients being only slightly less than the LRTST capture rate coefficients. The calculated branching ratios are 9C:91N and 0C:100N at 298 K for MA and DMA, respectively.
The sensitivity of the rate coefficients and branching ratios to the calculated energy barriers was tested by shifting the calculated barriers by 4 kJ mol−1 in opposite directions. The maximum change in overall rate coefficients was 5%, 1% and no change for MA, DMA and TMA respectively. The room temperature branching ratio for the MA reaction was found to be more sensitive to the barrier heights as lowering the barrier for C-abstraction and raising the barrier for N-abstraction gave 33C:67N while shifting the barriers in opposite directions gave 2C:98N. For the DMA no change was exposed.
Eckart tunnelling was included in the master equation model. The imaginary frequencies for all H-shift reactions are below 450 cm−1 (Table S9, ESI†), and, consequently, tunnelling was found to have negligible influence on the calculated rate coefficients and branching ratios.
Discussion
Literature comparisons
Rudić et al.12 carried out a theoretical study of the MA + Cl reaction employing a variant of G2-model chemistry,37 and found essentially the same PES as derived in the present study. In particular, they identified the strongly bound pre-reactive complex as a 2-center-3-electron bond involving the nitrogen lone pair and the unpaired electron on Cl. The magnitude of the Cl–N bond strengths calculated in this study are larger than the one measured for Cl–pyridine,38 where a 2-center-3-electron bond is also formed. The trend in Cl–N bond strengths (Cl–TMA > Cl–DMA > Cl–MA > Cl–pyridine) makes good physical chemical sense since it can be attributed to the methyl groups donating electron density to the N lone pair.The MA + Cl reaction dynamics study shows a roughly 50:50 branching in the initial abstraction.12 It should be noted that the reactants are far from being thermalized in the study: the translational collision energy is about 2000 cm−1, and there is very little rotational or vibrational energy in the methylamine reactant (2000 cm−1 corresponds to a translational temperature of ∼2900 K). The present calculations show an increase in the C–H abstraction yield from 0.09 at 300 K to 0.22 at 600 K, so the 48% yield reported in the reaction dynamics study12 appears to be in reasonable agreement with the theoretical findings of this study.
Implications for atmospheric chemistry of amines
Consideration of the rate coefficients reported in this study in conjunction with rate coefficients for OH + amine reactions that were reported recently by Onel et al.,39 suggests that the Cl rate coefficients are faster at 298 K by factors of 16, 6, and 6 for MA, DMA, and TMA, respectively, and that these rate coefficient ratios change very little as a function of temperature. In the marine boundary layer, Cl concentrations are typically 1–10 percent of OH concentrations.9 Hence, it appears that reaction with Cl is a minor but significant sink for amines in marine environments.The calculated branching ratios in the MA and DMA reactions with Cl suggest that N–H abstraction dominates in the chlorine reactions in contrast to the corresponding OH reactions, where C–H abstraction dominates.40–42 In areas with elevated chlorine atom concentrations the Cl reactions may therefore contribute significantly to nitramine and nitrosamine formation (i.e. RR′N + NO2 → RR′NNO2). An experimental determination of the branching ratios for the MA and DMA + Cl reactions is clearly needed.
Conclusions
The rate coefficients for the chlorine atom reactions with methylamine, dimethylamine and trimethylamine have been determined using the laser flash photolysis – resonance fluorescence technique. The reactions are extremely fast with nearly temperature independent rate coefficients close to the gas kinetic collision limit. Quantum chemical calculations show that the reactions are dominated by strongly bound pre-reaction complexes and submerged barriers, and statistical rate theory confirms that the reactions are collision controlled. Reaction with Cl appears to make a small but non-negligible contribution to destruction of amines in marine atmospheric environments. Unlike OH reactions with mono- and di-methyl amine, the Cl reactions are predicted theoretically to proceed predominantly by abstraction of hydrogen from the N atom, thus making Cl + amine reactions a potentially important source of atmospheric nitramines and nitrosamines.Acknowledgements
This work is part of the technology qualification of amines for the CO2 Capture Mongstad Project (CCM) funded by the Norwegian state through Gassnova SF. Additional support to the University of Oslo from the Research Council of Norway through a Centre of Excellence Grant (Grant No. 179568/V30) is acknowledged. Additional support to Georgia Tech from NASA (Grant No. NNX12AE02G) is also acknowledged.Notes and references
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 524–546 CrossRef CAS.
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 561–577 CrossRef CAS.
- C. J. Nielsen, H. Herrmann and C. Weller, Chem. Soc. Rev., 2012, 41, 6684–6704 RSC.
- D. Lee and A. S. Wexler, Atmos. Environ., 2013, 71, 95–103 CrossRef CAS.
- R. Atkinson, Chem. Rev., 1986, 86, 69–201 CrossRef CAS.
- P. Paasonen, T. Olenius, O. Kupiainen, T. Kurten, T. Petaja, W. Birmili, A. Hamed, M. Hu, L. G. Huey, C. Plass-Duelmer, J. N. Smith, A. Wiedensohler, V. Loukonen, M. J. McGrath, I. K. Ortega, A. Laaksonen, H. Vehkamaki, V. M. Kerminen and M. Kulmala, Atmos. Chem. Phys., 2012, 12, 9113–9133 CAS.
- J. Almeida, S. Schobesberger, A. Kurten, I. K. Ortega, O. Kupiainen-Maatta, A. P. Praplan, A. Adamov, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, J. Dommen, N. M. Donahue, A. Downard, E. Dunne, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, R. Guida, J. Hakala, A. Hansel, M. Heinritzi, H. Henschel, T. Jokinen, H. Junninen, M. Kajos, J. Kangasluoma, H. Keskinen, A. Kupc, T. Kurten, A. N. Kvashin, A. Laaksonen, K. Lehtipalo, M. Leiminger, J. Leppa, V. Loukonen, V. Makhmutov, S. Mathot, M. J. McGrath, T. Nieminen, T. Olenius, A. Onnela, T. Petaja, F. Riccobono, I. Riipinen, M. Rissanen, L. Rondo, T. Ruuskanen, F. D. Santos, N. Sarnela, S. Schallhart, R. Schnitzhofer, J. H. Seinfeld, M. Simon, M. Sipila, Y. Stozhkov, F. Stratmann, A. Tome, J. Trostl, G. Tsagkogeorgas, P. Vaattovaara, Y. Viisanen, A. Virtanen, A. Vrtala, P. E. Wagner, E. Weingartner, H. Wex, C. Williamson, D. Wimmer, P. Ye, T. Yli-Juuti, K. S. Carslaw, M. Kulmala, J. Curtius, U. Baltensperger, D. R. Worsnop, H. Vehkamaki and J. Kirkby, Nature, 2013, 502, 359–363 CrossRef CAS PubMed.
- M. Rozenberg, A. Loewenschuss and C. J. Nielsen, J. Phys. Chem. A, 2014, 118, 1004–1011 CrossRef CAS PubMed.
- O. W. Wingenter, B. C. Sive, N. J. Blake, D. R. Blake and F. S. Rowland, J. Geophys. Res.: Atmos., 2005, 110, D20308 CrossRef.
- J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dube, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander and S. S. Brown, Nature, 2010, 464, 271–274 CrossRef CAS PubMed.
- J. D. Raff, B. Njegic, W. L. Chang, M. S. Gordon, D. Dabdub, R. B. Gerber and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13647–13654 CrossRef PubMed.
- S. Rudić, C. Murray, J. N. Harvey and A. J. Orr-Ewing, Phys. Chem. Chem. Phys., 2003, 5, 1205–1212 RSC.
- B. J. Finlayson-Pitts and J. N. Pitts, Jr., Chemistry of the upper and lower atmosphere, Academic Press, San Diego, 2000, p. 146 Search PubMed.
- R. E. Stickel, J. M. Nicovich, S. Wang, Z. Zhao and P. H. Wine, J. Phys. Chem., 1992, 96, 9875–9883 CrossRef CAS.
- J. M. Nicovich, S. Wang and P. H. Wine, Int. J. Chem. Kinet., 1995, 27, 359–368 CrossRef CAS.
- J. J. Orlando, C. A. Piety, J. M. Nicovich, M. L. McKee and P. H. Wine, J. Phys. Chem. A, 2005, 109, 6659–6675 CrossRef CAS PubMed.
- J. M. Nicovich, S. Parthasarathy, F. D. Pope, A. T. Pegus, M. L. McKee and P. H. Wine, J. Phys. Chem. A, 2006, 110, 6874–6885 CrossRef CAS PubMed.
- Z. Zhao, D. T. Huskey, J. M. Nicovich and P. H. Wine, Int. J. Chem. Kinet., 2008, 40, 259–267 CrossRef CAS.
- M. Marinkovic, M. Gruber-Stadler, J. M. Nicovich, R. Soller, M. Mülhäuser, P. H. Wine, L. Bache-Andreassen and C. J. Nielsen, J. Phys. Chem. A, 2008, 112, 12416–12429 CrossRef CAS PubMed.
- A. I. Chichinin, Chem. Phys. Lett., 1993, 209, 459–463 CrossRef CAS.
- S. A. Sotnichenko, V. C. Bokun and A. I. Nadkhin, Chem. Phys. Lett., 1988, 153, 560–568 CrossRef CAS.
- A. I. Chichinin, Khim. Fiz., 1996, 15, 49–63 CAS.
- Y. Matsumi, K. Izumi, V. Skorokhodov, M. Kawasaki and N. Tanaka, J. Phys. Chem. A, 1997, 101, 1216–1221 CrossRef CAS.
- E. Tannenbaum, E. M. Coffin and A. J. Harrison, J. Chem. Phys., 1953, 21, 311–318 CrossRef CAS.
- M. J. Hubin-Franskin, J. Delwiche, A. Giuliani, M. P. Ska, F. Motte-Tollet, I. C. Walker, N. J. Mason, J. M. Gingell and N. C. Jones, J. Chem. Phys., 2002, 116, 9261–9268 CrossRef CAS.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schuetz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2012.1, a package of ab initio programs, 2012 Search PubMed.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- NIST Computational Chemistry Comparison and Benchmark Database. NIST Standard Reference Database Number 101, R. D. Johnson III, Release 16a, August 2013. http://cccbdb.nist.gov/.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221–7230 CrossRef CAS.
- Z. Zhao, D. T. Huskey, K. J. Olsen, J. M. Nicovich, M. L. McKee and P. H. Wine, Phys. Chem. Chem. Phys., 2007, 9, 4383–4394 RSC.
- L. Onel, L. Thonger, M. A. Blitz, P. W. Seakins, A. J. C. Bunkan, M. Solimannejad and C. J. Nielsen, J. Phys. Chem. A, 2013, 117, 10736–10745 CrossRef CAS PubMed.
- C. R. C. Lindley, J. G. Calvert and J. H. Shaw, Chem. Phys. Lett., 1979, 67, 57–62 CrossRef CAS.
- C. J. Nielsen, B. D'Anna, M. Karl, M. Aursnes, A. Boreave, R. Bossi, A. J. C. Bunkan, M. Glasius, A.-M. K. Hansen, M. Hallquist, K. Kristensen, T. Mikoviny, M. M. Maguta, M. Müller, Q. Nguyen, J. Westerlund, K. Salo, H. Skov, Y. Stenstrøm and A. Wisthaler, Summary Report: Photo-oxidation of Methylamine, Dimethylamine and Trimetahylamine. Climit project no. 201604 NILU OR 2/2011, ISBN 978-82-425-2357-0, NILU, 2011.
- L. Onel, M. Blitz, M. Dryden, L. Thonger and P. Seakins, Environ. Sci. Technol., 2014, 48, 9935–9942 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Figures of reactants, products and stationary points on the reaction surfaces. Tables with rate coefficients, energies of reactants, products and stationary points on the reaction surfaces. See DOI: 10.1039/c4cp03801k |
| ‡ Present address: Mercury Experts LLC, 11710 Sterling Brook St., Pearland, TX 77584, USA. |
| This journal is © the Owner Societies 2015 |