
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stress-mediated formation of nanocrystalline calcitic microlens arrays†
Ingo
Schmidt
a,
Emil
Zolotoyabko
b,
Peter
Werner
c,
Kyubock
Lee
d,
Manfred
Burghammer
e,
Peter
Fratzl
a and
Wolfgang
Wagermaier
*a
aDepartment of Biomaterials, Max Planck Institute of Colloids and Interfaces, Potsdam 14424, Germany. E-mail: Wolfgang.Wagermaier@mpikg.mpg.de
bDepartment of Materials Science and Engineering, Technion - Israel Institute of Technology, Haifa 32000, Israel
cExperimental Department II, Max Planck Institute of Microstructure Physics, 06120 Halle, Germany
dBiomass and Waste Energy Laboratory, Korea Institute of Energy Research, 152 Gajeong-ro, Yuseong-gu, Daejeon 305-343, Korea
eEuropean Synchrotron Radiation Facility, BP220, 38043 Grenoble Cedex 9, France
First published on 20th October 2015
Abstract
Organic additives are known to help minerals grow into complex shapes, which are beneficial for achieving various functional (e.g. optical) properties. Calcite-based microlens arrays (MLAs) are good examples of functional materials produced via the self-assembly of amorphous calcium carbonate (ACC) nanoparticles followed by a heat-mediated phase transformation into calcite. The optical transparency of the MLAs is preserved due to the nanocrystalline nature of the calcite formed. In this paper, we investigate the corresponding structural changes by mapping the local lattice parameters and size of calcite crystallites within individual microlenses. We find that the driving force for producing calcite with a crystallite size of 10 nm is the minimization of residual strains and related elastic energy by plastic deformation, which includes grain boundary formation and twinning. Local strains/stresses originate from transformation-associated macroscopic volume changes, which arise because of the differences in specific volume (per CaCO3 molecule) of ACC and calcite, firstly due to water loss and then to short-order atomic rearrangements. MLAs fabricated in this way represent a striking example for a stress-engineered nanocrystalline material produced with almost no energy cost through phase transformation, as compared to grain refinement by mechanical processing.
Introduction
Calcium carbonate-based minerals are among the most abundant minerals on earth and include materials of both geological and biogenic (i.e. produced by organisms) origin. The most thermodynamically stable polymorph of calcium carbonate under normal conditions is calcite, which has a rhombohedral symmetry (point group![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and space group Rc (No. 167)). Often, this symmetry is expressed in a hexagonal setting with lattice parameters a = 4.98964 Å and c = 17.0673 Å.1 Optically, calcite is birefringent, with the main optical axis coinciding with the [001] crystallographic direction, along which the birefringence effect disappears. This property is used by some organisms, e.g. in the design of calcitic MLA which is a part of the photoreceptor system of the brittle star Ophiocoma wendtii. The alignment of the c-axis of each calcite crystal with the propagation direction of light allows the elimination of the birefringence effect.2
m and space group Rc (No. 167)). Often, this symmetry is expressed in a hexagonal setting with lattice parameters a = 4.98964 Å and c = 17.0673 Å.1 Optically, calcite is birefringent, with the main optical axis coinciding with the [001] crystallographic direction, along which the birefringence effect disappears. This property is used by some organisms, e.g. in the design of calcitic MLA which is a part of the photoreceptor system of the brittle star Ophiocoma wendtii. The alignment of the c-axis of each calcite crystal with the propagation direction of light allows the elimination of the birefringence effect.2
Bio-inspired fabrication of complicated calcitic structures and, especially, MLAs attracts more and more attention from research groups worldwide3–11 since MLAs may find potential applications in directional displays, artificial compound eyes, and signal-routing connectors.12
This approach makes use of the fact that in the presence of certain organic additives (or even without them13) minerals can spontaneously grow into complex shapes, which are beneficial for achieving various functional (e.g. optical) properties.14–19 Recently, it was reported that micron-sized hemispherical lenses could be produced via the self-assembly of ACC particles, which are stabilized by an organic surfactant.6 Heat-induced phase transformation converts hydrated ACC into anhydrous nanocrystalline calcite while keeping the hemispherical morphology.20 The calcitic MLA shows polarization-dependent optical properties.20 Moreover, in spite of the strong birefringence of calcite, the optical transparency of the MLA is preserved due to the resultant 10–20 nm crystal size. The latter assists in diminishing parasitic light scattering at grain boundaries and suppressing the undesirable birefringence effect.20
In this regard, controlling the nanostructure of polycrystalline MLAs (which is directly related to their optical performance) via amorphous/crystalline transformation is of great importance. For example, single-crystalline calcitic MLAs with concave lenses and tunable crystal orientations have been fabricated by template-assisted epitaxial growth.11 The orientation-dependent imaging performance and polarization-dependent optical properties of MLAs have been demonstrated.11 At the same time, the optical performance is preserved for nanocrystalline calcitic MLAs, transformed from ACC lenses by a temperature-induced phase transformation.6,20 In order to elucidate the physical mechanisms responsible for this nanostructuring, we carried out detailed synchrotron X-ray diffraction studies utilizing a focused X-ray beam, polarization optical microscopy (POM), and transmission electron microscopy (TEM).
Results and discussion
As grown, individual ACC lenses have hemispherical shapes with a diameter of about 5 microns and show a hexagon-type two-dimensional long-range order within the MLA (see Fig. 1a). However, when water is lost, ACC is crystallized into calcite even at room temperature. Therefore, the optical and mechanical characteristics of the MLA can potentially be affected by this transformation and, namely, such considerations stand behind our study. Note that the crystallization of ACC is accompanied by significant volume shrinkage and related strain/stress evolution. Nevertheless, despite this fact, the spherical shape of individual lenses is kept after crystallization (see the optical image in Fig. 1b). Note that the bright/dark contrast in Fig. 1b depends on the orientation of the calcite c-axis with respect to the optical axes of the polarizer and analyzer in POM. It is clearly seen that the crystallization of ACC starts in the center of the red square region in Fig. 1b and propagates radially. The POM images of the crystallized samples look like those produced by spherulites in crystallized polymers, revealing characteristic Maltese crosses20 (see also Fig. S1a in the ESI† taken from another investigated sample and showing several Maltese crosses).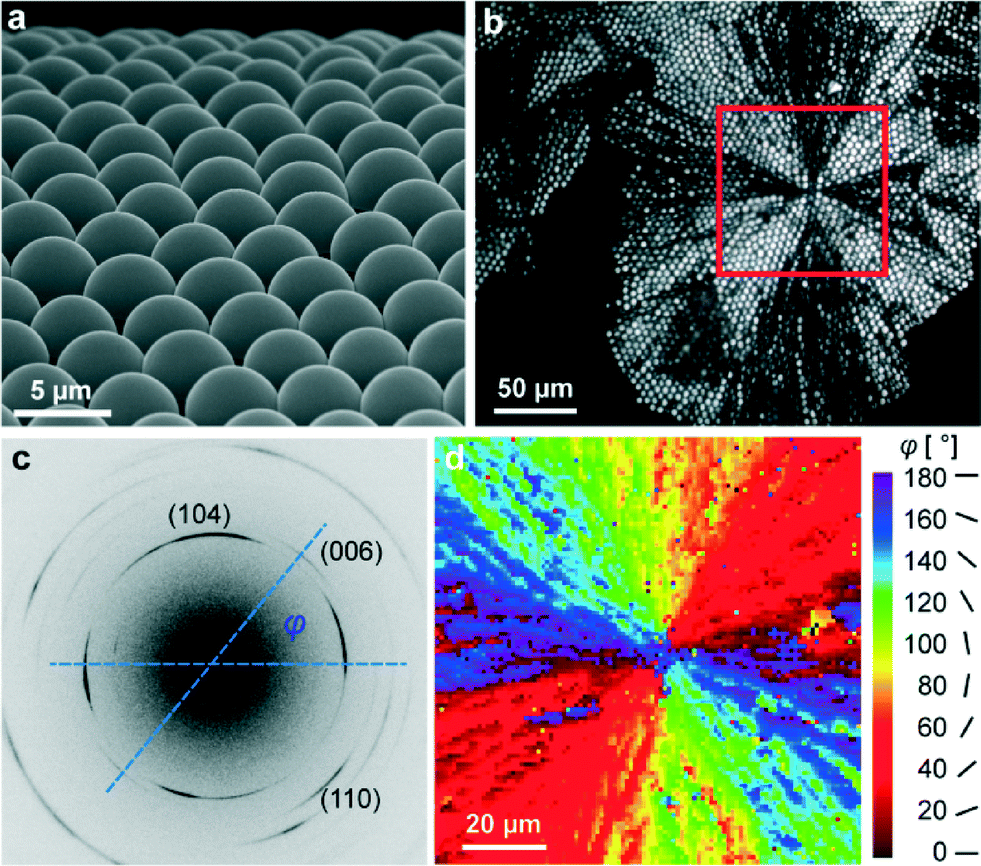 | ||
| Fig. 1 (a) SEM image of the ACC MLA; (b) POM image of the crystallized spherical lens array. Crystallization started in the center of the red square region. (c) Typical diffraction pattern taken from calcitic lenses with a 1 μm focused X-ray beam. The image has been optimized to maximize the contrast to illustrate all relevant peaks. The orientation of the c-axis within the MLA plane with respect to the horizontal axis of the detector plane is indicated by angle φ. (d) Mapping of the orientation angle φ in the red square region in (b). | ||
More detailed information on the orientation of the optically unique c-axis is extracted from the X-ray diffraction patterns produced by the X-ray beam with a diameter of 1 μm, which is much smaller than individual lenses composing the MLA. In these measurements, the incident X-ray beam was perpendicular to the plane of the MLA. Typical diffraction patterns (see Fig. 1c) show diffraction arcs (well-restricted parts of the Debye rings) originating from different atomic planes. Taking the line which connects the intensity maxima of two oppositely located (006)-arcs as the local orientation of the c-axis, we find the angle φ between the c-axis and the horizontal axis in the detector plane (see Fig. 1c). We can plot a two-dimensional distribution of angles φ (see the colored map in Fig. 1d), which complements the optical image in Fig. 1b. Similar information regarding the second investigated sample is graphically depicted in Fig. S1b.† These data show that many calcite crystallites, comprising our lenses, have an optical axis situated close to the MLA plane (i.e. not parallel to the normal to the array plane, as in brittle stars).
Another important piece of information comes from diffraction measurements of calcite lattice parameters. Crystallization processes from amorphous phases are intensively studied in biogenic minerals grown by organisms (see e.g.ref. 17, 18, 21–23). In our case, individual lenses within the MLA are in contact with each other (see below), so the development of inhomogeneous strain/stresses acting against volume shrinkage is expected, as a result of the interaction between different parts of the system.24,25 Note that the relative volume change (of about 0.35%), which takes place when cooling calcite from 300 °C down to room temperature, is much lower than the volume change (few percent) during amorphous/crystalline transformation. Correspondingly, only the latter process is considered in our paper.
Bearing these considerations in mind, we analyzed the diffraction patterns, taken from thousands of individual lenses, and extracted the local a- and c- lattice parameters by using the (110) and (006) reflections. Regions with highly significant differences (P < 0.001) for a and c values could be observed (see Fig. S2 and respective statistical analysis in the ESI†). After that, we compared the extracted lattice parameters with those of pure geological calcite1 and plotted the relative distortions, e3 = Δc/c and e1 = Δa/a, as two-dimensional maps (see Fig. 2 and Fig. S1c and d†). Fig. 2a and b represent maps containing regions with tensile-type distortions (of the positive sign, red-colored) reaching a maximum at emt ≈ 0.4%, and compression-type distortions (of the negative sign, blue-colored) reaching up to emc ≈ −0.4%. In these distorted regions, the calcite optical axis is oriented nearly horizontally (φ = 0 or 180°) or vertically (φ = 90°) in the plane of the map (compare Fig. 1 and 2). Distorted regions are separated by domains, in which both e1 and e3 are close to zero (green-yellow color). Note that the e1 and e3 maps complement each other: regions with compression-like e1 distortions exhibit tensile-like e3 distortions and vice versa. The maps are almost symmetric; the averaged (over 9227 sampling points) distortions are: <e3 = Δc/c> = −0.04% and <e1 = Δa/a> = −0.13%. Note that these relative distortions have a negative sign, i.e. are compressive in nature. Under compression, calcite is much stronger than under tension, so the above-mentioned values are really low as compared to the onset of plastic deformation in calcite (about 1%26). All these general conclusions remain valid regarding several other investigated samples (see e.g. Fig. S1c and d†).
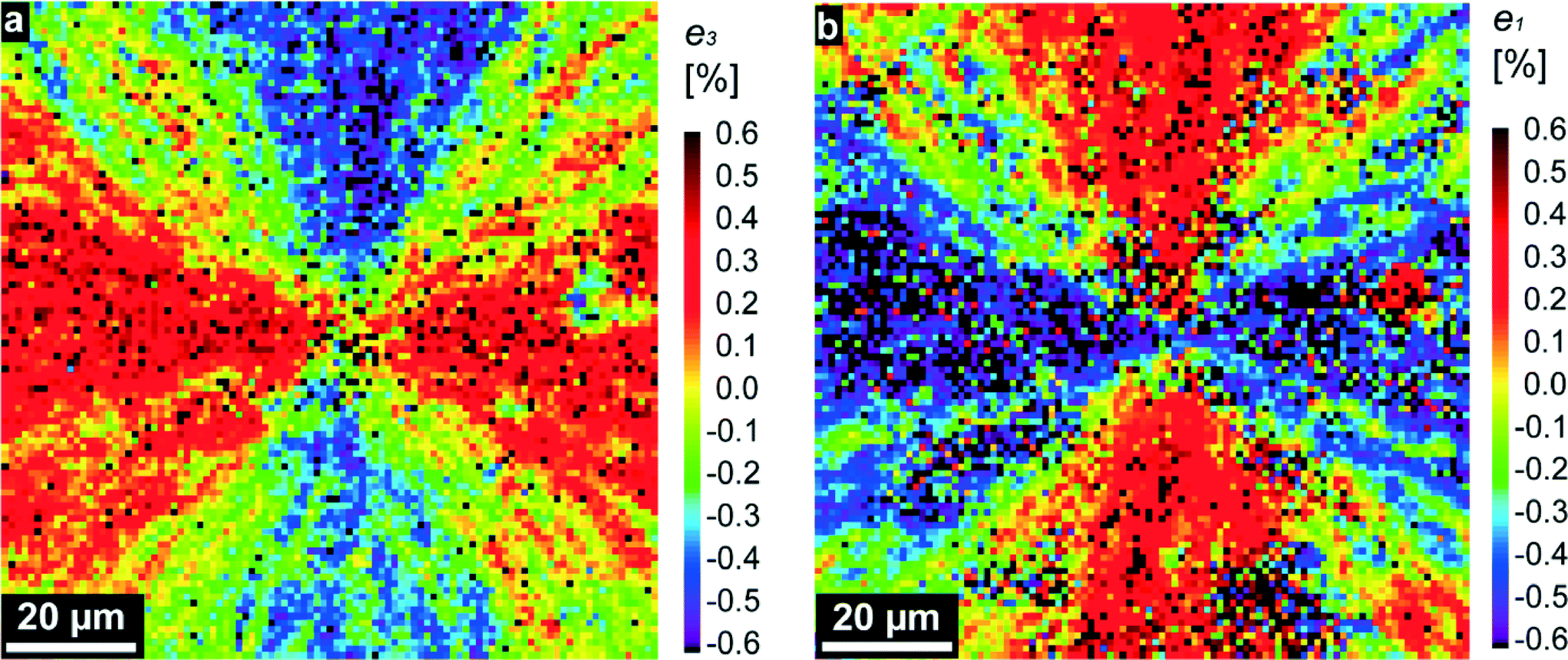 | ||
| Fig. 2 Maps of the relative lattice distortions (strains, in percent), e3 = Δc/c (a) and e1 = Δa/a (b), showing the relative differences in the corresponding lattice parameters, c and a, measured locally in the red squared region in Fig. 1b, and those tabulated for pure geological calcite in ref. 1. | ||
For further analysis, we consider the measured lattice distortions as primarily elastic deformations (residual strains), which remain after crystallization is ended. As we see in Fig. 1b and S1a,† crystallization starts from some nucleation event and proceeds in radial directions, producing in the beginning a circular crystallization front.19 Amorphous/crystalline transformation reduces the bulk free energy, which is in competition with the growing interface energy between the amorphous and the crystalline phase. A circular shape of the crystallization front is thermodynamically favorable since a circle has a minimal border length for a given area surrounded by it. However, radial front propagation from a point source means that the contacting individual crystals, located along slightly misoriented propagation beams, will experience mismatched strains, contributing to the undesirable elastic energy of the sample.
Another source of elastic strains is contact forces between adjacent lenses, which oppose the above-mentioned volume shrinkage during calcite crystallization. TEM imaging techniques were applied to analyze the crystalline structure especially in the transition region between two adjacent lenses. A representative example is shown in Fig. 3a, in which the cross-section of two adjacent hemispheres is seen. The cross-sectional sample was prepared by the FIB technique (see the “Experimental” section). For such a kind of TEM micrograph, we used the “bright-field” TEM mode, where an image is generated by the non-diffracted beam (“0” beam). The image in Fig. 3a indicates that the hemispheres consist of small crystallites (<50 nm), which can be regarded as building blocks of the spherical lenses. Due to slight local variations in the orientation of the crystallites (<5°), they appear with different intensities. Under the chosen imaging (Bragg-excited) conditions, regions ‘1’ and ‘2’ in Fig. 3a are darker than the surrounding matrix. However, the corresponding Fast Fourier transform (FFT) diffractograms (Fig. 3b and c) demonstrate that the adjacent regions in both hemispheres have nearly the same orientation.
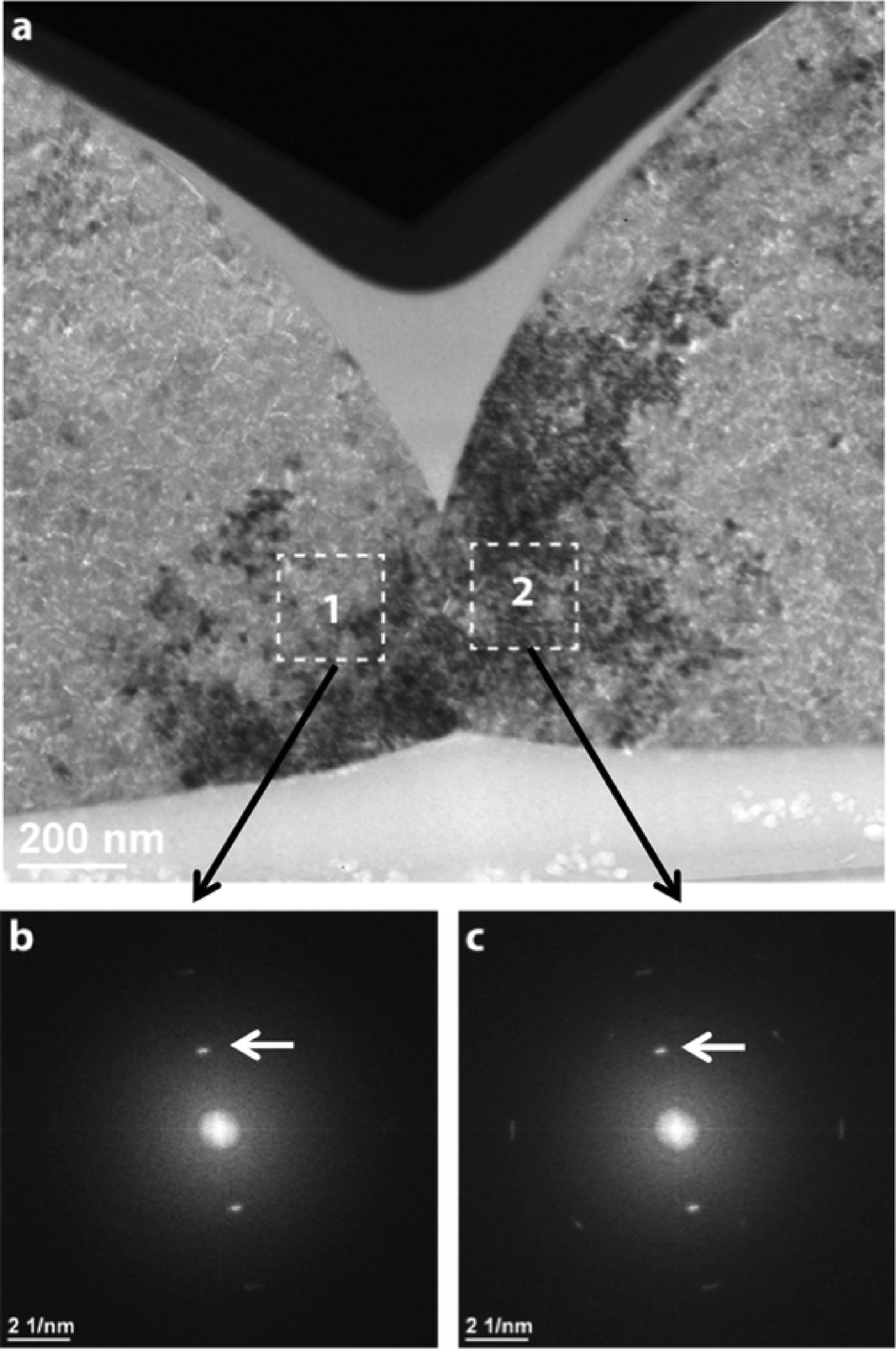 | ||
| Fig. 3 (a) Bright-field TEM micrograph showing two contacting spherical lenses. Since for imaging some kind of the “diffraction contrast technique” was applied, the dark crystal regions (marked as 1 and 2) indicate closely related crystallographic orientations of the crystallites. (b) and (c) FFT images generated from the lattice plane micrographs, the latter being taken, respectively, from regions 1 and 2 in (a). The FFT images reveal a similar disposition of the diffraction spots (indicated by arrows) which provides clear-cut evidence of nearly the same crystallographic orientations in the contacting regions. | ||
This finding indicates that the lenses strongly interact with each other, trying to transfer information on crystal orientation. On the other hand, due to the above discussed reasons, this interaction is the source of elastic strains mapped in Fig. 2a and b.
High enough strains are removed in the course of plastic deformation, which proceeds by grain boundary formation and twinning.20,26 Simulations have shown27 that among the four well-established twin laws in calcite,28 the (001)-twin has the lowest energy formation and is most favorable, being generated by a two-dimensional heterogeneous nucleation mechanism. In fact, twinning of this type was found before in our MLA, based on the analysis of space distribution of different crystallographic orientations extracted from X-ray diffraction patterns.20 In regions where twinning proceeded, elastic strains are greatly reduced and we find close to zero values of e1 and e3 in our distortion maps (yellow-colored regions in Fig. 2a and b, Fig. S1c and S1d†). Moreover, in these regions due to the arising twin boundaries, we expect a smaller size of crystallites which coherently scatter X-rays.29
To verify this point, we measured the width of the strongest (104) diffraction line across the MLA and extracted the crystallite size using the Scherrer equation (see the “Experimental” section). The obtained results for two investigated samples are presented as maps in Fig. 4a and S1e.† We see that the crystallite size is globally in the range of 10–20 nm which fits well the TEM results.20 However, its distribution across the MLA is strongly non-homogeneous, revealing areas with larger grains (closer to 20 nm, yellow-colored) and smaller grains (closer to 10 nm, blue-colored). The grain size of about 20 nm is comparable to that of the initial amorphous particles (visualized by TEM) before crystallization.20 Grain boundaries are formed during crystallization to reduce the elastic energy of the system. Seemingly, some grains are also rotated, which is evidenced by observing the diffraction arcs in X-ray diffraction patterns (Fig. 1c).
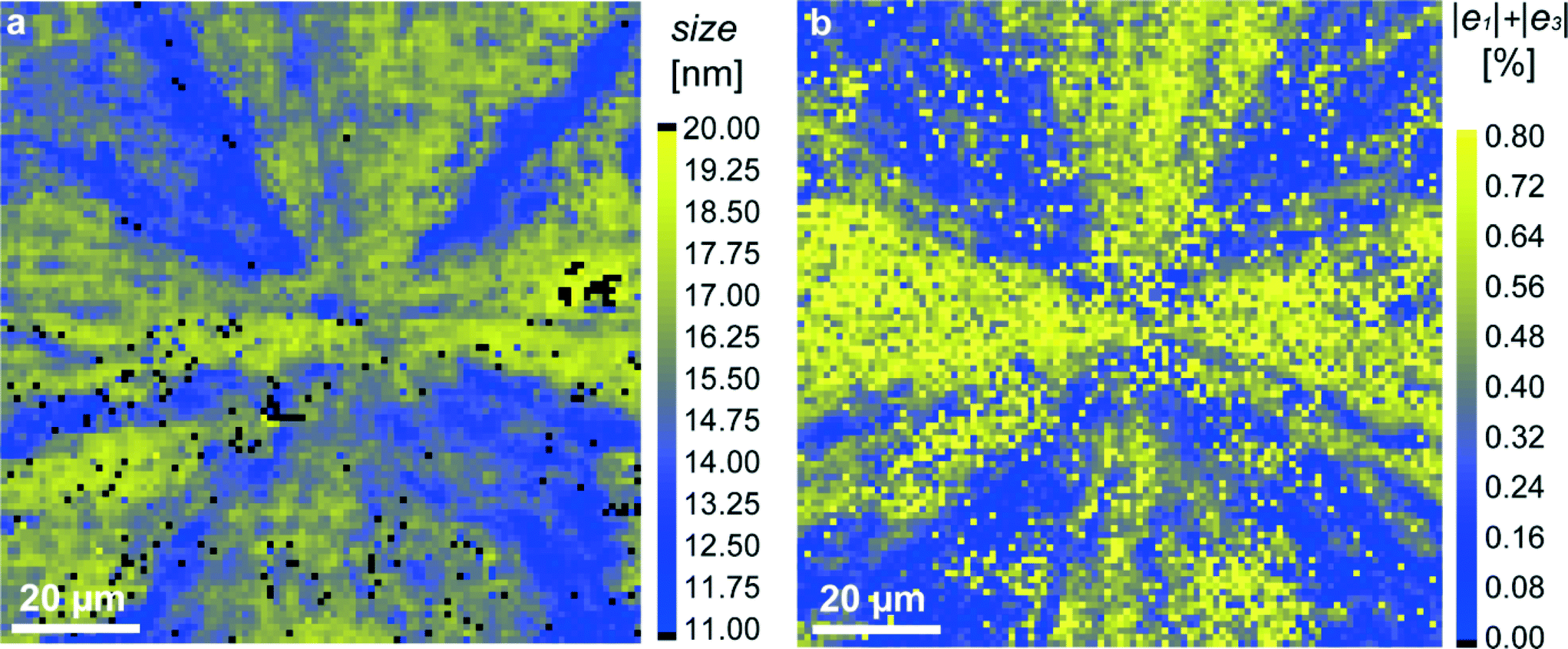 | ||
| Fig. 4 Mapping of structural parameters in the red-squared region in Fig. 1b: (a) crystallite size, extracted from the width of the (104) diffraction profiles; (b) magnitude of the sum |e1| + |e3| (relative lattice distortions). | ||
Additional reduction of elastic energy takes place by twinning.20 As expected, smaller grains, separated by twin boundaries, are located in regions in which twinning proceeded and hence, elastic strains are close to zero. In order to emphasize this important correlation, we build supplementary maps for the sum |e1| + |e3|, in which the regions with the simultaneous disappearance of strains e1 and e3i.e. with |e1| + |e3| ≈ 0, are clearly seen (blue color in Fig. 4b and S1f†). When comparing the grain-size maps and (|e1| + |e3|) maps, we see that the spatial distribution of the blue-colored features in both maps looks rather similar.
Finally, we make some estimates for residual stresses remaining after crystallization of the MLA. Considering relative lattice distortions, e1 and e3, as elastic residual strains, we find that maximal residual stress components, σ1 and σ3, in our system do not exceed |σ1m| = E1|e1m| and |σ3m| = E3|e3m|, where E1 and E3 are the Young moduli along the a- and c-axis, respectively, σ1m and σ3m are the corresponding maximal residual stress components, and e1m and e3m are the maximal residual strain values. Taking the elastic constants from ref. 30, we calculate E1 = 91.6 GPa and E3 = 57.6 GPa. Using the maximum strain magnitude, |e1m| ≈ |e3m| ≈ 0.4%, from the measured maps (Fig. 2), we find |σ1m| ≈ 370 MPa and |σ3m| = E3|e3| ≈ 230 MPa. Nanoindentation measurements show hardness values of about 2.3–2.5 GPa for geological calcite and 3.5–4.6 GPa for biogenic calcite.26,31,32 The onset of plastic deformation under compression is assumed to be of about 0.5–1 GPa.31,33 Therefore, calcite under compression will endure even upon the above-mentioned stresses (overestimated) between 230 and 370 MPa. However, practically the stresses will be much smaller since, as can be deduced from the strain maps, the σ1 and σ3 components locally have different signs (like strain components, e1 and e3), i.e. act in opposite ways. Taking this fact into account, we estimate the maximal tensile stress, σ, acting normally to the calcite cleavage plane (104). The latter is inclined by angle, α ≈ 45°, with respect to the (001) and (100) crystallographic directions. Therefore, in the case of biaxial stresses:
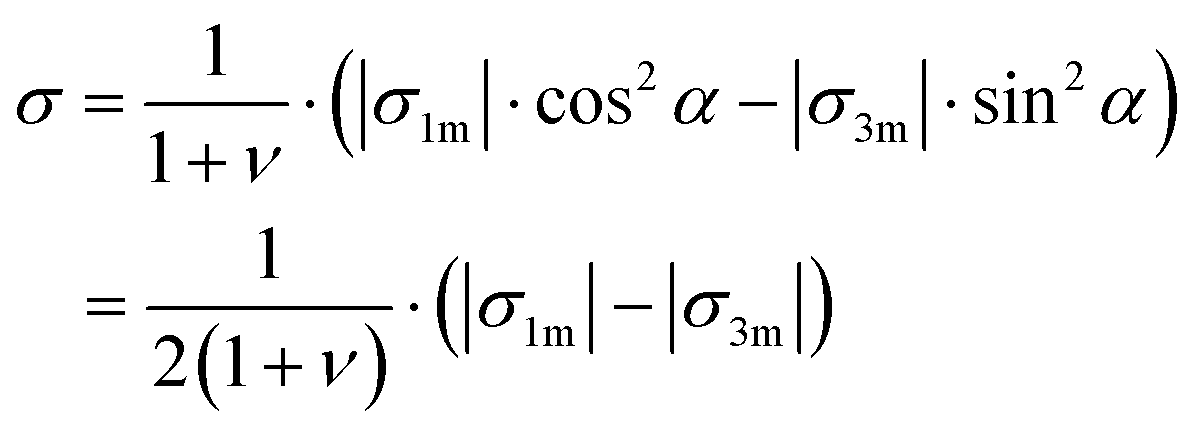 | (1) |
Using a Poisson ratio of ν = 0.32,34 we find a rather small value of σ ≈ 50 MPa. It seems that the calcite samples can accommodate such stresses even under tension.33,35 Resistance to fracture is greatly enhanced by reducing the grain size. The stress, σf, needed to cause intra-crystalline brittle fracture can be estimated as 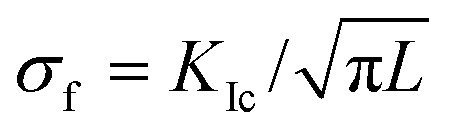 , where KIc is the fracture toughness and L is the initial crack length equal in our case to the average grain size. In calcite, fracture toughness is strongly orientation-dependent. Taking L ≈ 20 nm (as grain size estimate) and the lowest value of KIc = 0.2 MPa m½ for the (104) cleavage plane36 yields σf ≈ 800 MPa, which is much higher than the above stress estimations based on the lattice distortion data.
, where KIc is the fracture toughness and L is the initial crack length equal in our case to the average grain size. In calcite, fracture toughness is strongly orientation-dependent. Taking L ≈ 20 nm (as grain size estimate) and the lowest value of KIc = 0.2 MPa m½ for the (104) cleavage plane36 yields σf ≈ 800 MPa, which is much higher than the above stress estimations based on the lattice distortion data.
Conclusions
In summary, we carried out systematic structural studies of calcitic MLAs by using synchrotron X-ray diffraction in order to elucidate their nano-crystalline design. Analyzing the diffraction patterns taken from individual lenses, we built detailed two-dimensional maps of the crystal orientation, crystallite size, and relative lattice distortions, e1 = Δa/a and e3 = Δc/c along the a- and c-axis, respectively (as compared to pure geological calcite). The spatial distribution of the relative lattice distortions (strains) is strongly non-homogeneous: there are regions with positive and negative strains, which are separated by regions in which the magnitudes of e1 and e3 are close to zero. In areas with non-zero strains, the e1 and e3 components have opposite signs, so the averaged values (over the entire map) are close to zero. The observed lattice strains arise during amorphous/crystalline phase transformation mainly because of the difference in the specific volume per molecule in the amorphous and crystalline phases. A comparison of the maps revealed a clear correlation between the magnitude of lattice strains and crystallite size; the smallest crystalline size (about 10 nm) is observed in regions with elastic strains close to zero. Diminishing of elastic strains is a result of plastic deformation, which proceeds in these regions by twinning of the (001)-type. Correspondingly, the final reduction of crystallite size is related to the arising twin boundaries.29 The nanocrystallinity of the calcite microlenses, which is caused by inhomogeneous strains/stresses arising during amorphous/crystalline phase transformation, is the basis of the good optical performance20 of thus produced MLAs.Severe plastic deformation has already been proved to be an effective processing strategy for fabricating nanocrystalline metals and ceramics by using e.g. equal channel angular pressing (ECAP)37 or high-energy ball milling.38 The crystallized MLA is another remarkable example of utilizing mechanical stresses to produce a nanocrystalline material, but in this case with virtually no energy cost. Stress-mediated nanostructuring of calcite presents an alternative strategy for fabricating MLAs (compared with single-crystal calcitic microlens systems) while still enabling their optical functionality.
Experimental
Sample preparation
The ACC MLAs were synthesized by self-assembly under ambient conditions at the air–water interface but without a template by using a Ca(OH)2 solution and the organic surfactant polysorbate 20.6 The ACC MLAs were then dried on a glass slide with no glue used for attachment. Crystallization was accomplished for 1 h at 300 °C with another glass slide on top of the MLA. Potential cracks across the MLA due to water loss are eliminated by crystallizing the ACC MLA without rigid attachment to the substrate. After crystallization, confocal Raman imaging characterization was performed (see Fig. S3†) and no signal from organic molecules (polysorbate 20) was detected. The crystallized MLAs were transferred to a pre-wetted X-ray sample holder (silicon nitride membrane) and dried again or attached to a TEM sample holder (silicon) with polydimethylsiloxane (PDMS).Synchrotron X-ray diffraction
X-ray diffraction experiments with high spatial resolution were carried out at the ID13 beamline of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) using a focused X-ray beam (nearly 1 μm in diameter) with a wavelength of λ = 0.88571 Å. Two-dimensional wide-angle X-ray diffraction patterns were collected by using a FReLoN4M detector (2048 × 2048 pixels, ~50 μm pixel size) installed at a distance of 104.91 mm behind the sample. For these measurements, the crystallized MLAs were transferred onto a 1 μm silicon nitride membrane (Norcada NX5200F, 2 × 2 mm2 window) with the curved lens surface on the top. The scanning of the MLA in the plane perpendicular to the incident X-ray beam was realized by using the precise xyz-scanning stage of the beamline.X-ray data analysis was performed by using the DPDAK software package.39 In order to determine the calcite lattice parameters, the two-dimensional intensity distributions were first azimuthally integrated (i.e. along the Debye rings) and then thus obtained one-dimensional intensity profiles (as a function of the scattering vector, q) were analyzed to determine the peak positions of the different calcite reflections (hkl). The peak positions for the (110) and (006) diffraction profiles were determined by applying Voigt profile fitting. The obtained peak positions in q-space are converted to d-spacings and then, lattice parameters a and c are determined using the relationship:
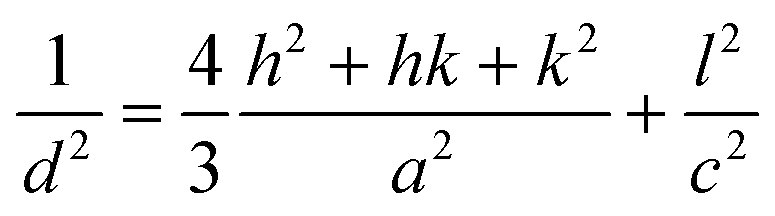 | (2) |
The average size, L, of the crystallites, which coherently scatter X-rays, was derived by using the Scherrer equation
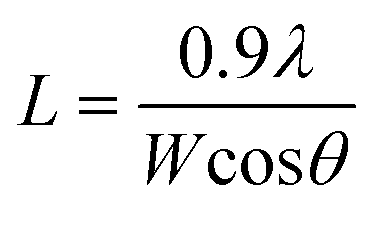 | (3) |
Transmission electron microscopy (TEM)
Samples for TEM were prepared from amorphous MLAs fixed on a silicon sample holder by a 2 μm thick coating of PDMS and crystallized by the same heat-treatment procedure as the samples for the X-ray experiments. Then, the samples were carbon-coated and cut by a focused ion beam (FIB, FEI “Nova NanoLab 600”) along the diameter of the circular lens shape in the direction of shortest distance to the neighboring lenses. For TEM/STEM, a FEI TITAN 80-300 operating at 300 keV was used.Polarization optical microscopy (POM)
The POM images of the MLA before and after heat treatment were obtained by using a Leica DM RXA2 and treated with a digital color camera, Leica DFC 480, to define the regions of interest for subsequent synchrotron experiments, these regions typically being between 100 × 100 μm2 and 150 × 150 μm2.Acknowledgements
We would like to thank H. Blumtritt for sample preparation by the focused ion-beam technique (FIB) and E. Di Cola for support at the ESRF. E. Z. thanks the Shore Fund for Advanced Composites (Technion) and K. L. thanks the International Cooperation Program managed by the National Research Foundation of Korea (NRF-2014K2A7A1044321) for partial financial support.References
- E. Zolotoyabko, E. N. Caspi, J. S. Fieramosca, R. B. Von Dreele, F. Marin, G. Mor, L. Addadi, S. Weiner and Y. Politi, Cryst. Growth Des., 2010, 10, 1207–1214 CAS.
- J. Aizenberg, A. Tkachenko, S. Weiner, L. Addadi and G. Hendler, Nature, 2001, 412, 819–822 CrossRef CAS PubMed.
- J. Aizenberg, D. A. Muller, J. L. Grazul and D. R. Hamann, Science, 2003, 299, 1205–1208 CrossRef CAS PubMed.
- P. K. Ajikumar, L. G. Wong, G. Subramanyam, R. Lakshminarayanan and S. Valiyaveettil, Cryst. Growth Des., 2005, 5, 1129–1134 CAS.
- H. Colfen, Angew. Chem., Int. Ed., 2008, 47, 2351–2353 CrossRef PubMed.
- K. Lee, W. Wagermaier, A. Masic, K. P. Kommareddy, M. Bennet, I. Manjubala, S. W. Lee, S. B. Park, H. Colfen and P. Fratzl, Nat. Commun., 2012, 3, 725–727 CrossRef PubMed.
- C. Li and L. M. Qi, Angew. Chem., Int. Ed., 2008, 47, 2388–2393 CrossRef CAS PubMed.
- R. J. Park and F. C. Meldrum, Adv. Mater., 2002, 14, 1167–1169 CrossRef CAS.
- M. H. Wu, C. Park and G. M. Whitesides, Langmuir, 2002, 18, 9312–9318 CrossRef CAS.
- S. Yang, C. K. Ullal, E. L. Thomas, G. Chen and J. Aizenberg, Appl. Phys. Lett., 2005, 86 CAS.
- X. Ye, F. Zhang, Y. Ma and L. Qi, Small, 2015, 14, 1677–1682 CrossRef PubMed.
- R. Sambles, Nature, 2001, 412, 783–783 CrossRef CAS PubMed.
- W. L. Noorduin, A. Grinthal, L. Mahadevan and J. Aizenberg, Science, 2013, 340, 832–837 CrossRef CAS PubMed.
- H. Colfen, Curr. Opin. Colloid Interface Sci., 2003, 8, 23–31 CrossRef CAS.
- P. Fratzl, J. R. Soc., Interface, 2007, 4, 637–642 CrossRef CAS PubMed.
- F. C. Meldrum and H. Colfen, Chem. Rev., 2008, 108, 4332–4432 CrossRef CAS PubMed.
- S. Weiner and L. Addadi, J. Mater. Chem., 1997, 7, 689–702 RSC.
- S. Weiner and L. Addadi, Annu. Rev. Mater. Res., 2011, 41, 21–40 CrossRef CAS.
- K. L. Yu, T. X. Fan, S. Lou and D. Zhang, Prog. Mater. Sci., 2013, 58, 825–873 CrossRef.
- I. Schmidt, K. Lee, E. Zolotoyabko, P. Werner, T. S. Shim, Y.-K. Oh, P. Fratzl and W. Wagermaier, ACS Nano, 2014, 8, 9233–9238 CrossRef CAS PubMed.
- L. Addadi, S. Raz and S. Weiner, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- B. Hasse, H. Ehrenberg, J. C. Marxen, W. Becker and M. Epple, Chem. – Eur. J., 2000, 6, 3679–3685 CrossRef CAS.
- S. Weiner, I. Sagi and L. Addadi, Science, 2005, 309, 1027–1028 CrossRef CAS PubMed.
- P. Fratzl, F. D. Fischer, J. Svoboda and J. Aizenberg, Acta Biomater., 2010, 6, 1001–1005 CrossRef PubMed.
- E. Zolotoyabko and B. Pokroy, CrystEngComm, 2007, 9, 1156–1161 RSC.
- L. Li and C. Ortiz, Nat. Mater., 2014, 13, 501–507 CrossRef CAS PubMed.
- M. Bruno, F. R. Massaro, M. Rubbo, M. Prencipe and D. Aquilano, Cryst. Growth Des., 2010, 10, 3102–3109 CAS.
- B. Pokroy, M. Kapon, F. Marin, N. Adir and E. Zolotoyabko, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7337–7341 CrossRef CAS PubMed.
- E. Zolotoyabko, Basic Concepts of X-Ray Diffraction, Wiley-VCH, Weinheim, 2014 Search PubMed.
- C. C. Chen, C. C. Lin, L. G. Liu, S. V. Sinogeikin and J. D. Bass, Am. Mineral., 2001, 86, 1525–1529 CrossRef CAS.
- M. E. Kunitake, L. M. Mangano, J. M. Peloquin, S. P. Baker and L. A. Estroff, Acta Biomater., 2013, 9, 5353–5359 CrossRef CAS PubMed.
- T. H. Metzger, Y. Politi, G. Carbone, B. Bayerlein, I. Zlotnikov, E. Zolotoyabko and P. Fratzl, Cryst. Growth Des., 2014, 14, 5275–5282 CAS.
- F. J. Turner, D. T. Griggs and H. Heard, Geol. Soc. Am. Bull., 1954, 65, 883–934 CrossRef CAS.
- C. C. Lin, Phys. Chem. Miner., 2013, 40, 157–166 CrossRef CAS.
- E. Rybacki, B. Evans, C. Janssen, R. Wirth and G. Dresen, Tectonophysics, 2013, 601, 20–36 CrossRef.
- B. K. Atkinson, J. Geophys. Res., 1984, 89, 4077–4114 CrossRef CAS.
- R. Z. Valiev and T. G. Langdon, Prog. Mater. Sci., 2006, 51, 881–981 CrossRef CAS.
- C. C. Koch and Y. S. Cho, Nanostruct. Mater., 1992, 1, 207–212 CrossRef CAS.
- G. Benecke, W. Wagermaier, C. H. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Muller-Buschbaum, M. Trebbin, S. Forster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ce01757b |
| This journal is © The Royal Society of Chemistry 2015 |