
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A new small molecule gelator and 3D framework ligator of lead(II)†
Jane V.
Knichal
,
William J.
Gee
,
Andrew D.
Burrows
*,
Paul R.
Raithby
* and
Chick C.
Wilson
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: C.C.Wilson@bath.ac.uk
First published on 24th August 2015
Abstract
Reacting equimolar quantities of 5-allenyl-1,3-benzenedicarboxylic acid (H2abd) with lead(II) acetate trihydrate in N,N-dimethylformamide (DMF) under solvothermal conditions results in formation of a metallogel with a critical gelation percentage of 1% w/v. Elemental analysis performed on the gel provided a molecular composition ratio of [Pb(abd)(H2O)]n (1). Viewing the gel by scanning electron microscopy (SEM) identified an entangled network of cross-linked nano-fibres. 1H-NMR aliquots of hydrated lead(II) acetate added to a solution of H2abd in deuterated DMF allows inferences to be made about solution-state behaviour that occurs during the initial gel aggregation stage. Under non-solvothermal conditions, combining H2abd and hydrated lead(II) acetate resulted in formation of single crystals suitable for X-ray diffraction, which were identified as a 3D coordination polymer with composition [Pb(abd)(DMF)] (2). Structural features observed within this 3D coordination polymer provide the basis for assigning the molecular structure to the fibrils present within gel 1. This assertion is supported by comparable vibrational profiles taken from a sample of dried gel 1 to that of crystalline 2, and the matching of early solution-state 1H-NMR spectroscopic trends to later solid-state observations.
Introduction
Supramolecular gels formed by low molecular weight gelators are a class of material in which interest is rapidly expanding to provide a diverse range of applications to commerce, industry and medicine.1 This interest has grown from the founding work developing organic gelators2 to include metallogelators, inorganic species capable of sustaining hydro- and organo-gels.1a,3 This development takes advantage of the diverse coordination modes provided by metal nodes, which allow new options for influencing the gelation process. Typically two strategies are employed to generate metallogels; these target either coordination polymeric gelators, or discrete metal complexes able to adopt fibrillar constructs with the aid of metallophilic, van der Waals, and other non-covalent interactions.1a,3 Examples of the former include copper and silver pyridyl gelators,4 coordination-driven metallocyclic platinum species,5 lanthanide coordination polymers,6 and discrete zinc clusters.7 Examples utilising non-covalent interactions encompass Au⋯Au or Pt⋯Pt interactions,8 and typically such complexes tether several long alkyl chains to aid facilitation of the gelation process.Recently, metallogels have been applied as luminescent media8 or hosts for luminescent guests,4a as templates for nanoparticle formation,9 as means of controlling crystalline polymorph growth,10 for redox and chiral molecule responsiveness,11 and in catalysis,12 among a range of innovative applications. Of particular relevance to this work is the recent identification of a lead(II) metallogel ligated by a conjugated pyridyl species that is capable of purifying wastewater by selectively removing toxic metal ions and dyes.13
Formation of gels as opposed to solid or crystalline materials is governed by a largely unpredictable balance of non-covalent interactions that include pH, pKa and solubility,14 as well as choice of metal, anion and solvent.15 Perturbation of this balance may lead to collapse of gel states to precipitate or crystalline species,16 in some cases with unique outcomes such as chiral resolution.17 Studies of the conversion of coordination polymers to polymeric gels have recently highlighted structural links between the former and the latter.18 Similarly, changes to the pore shape of covalent polymers have been shown to be templated by the presence of different gel morphologies.19 Interest in metallogels has also been shown to link strongly to the area of MOFs, with amorphous MOF aerogels and xerogels reported, and shown to exhibit high degrees of micro- and macroporosity.20 Short-range MOF structures of tripodal pyridyl ligands have also recently been identified as gelators for rigid fibrous metallogel networks, analogous to the present work.6,21
Our interest in lead(II) coordination polymers as potential responsive functional materials has stemmed from a study of facile single crystal to single crystal (SCSC) transitions driven by interconversion of secondary interactions and primary bonds about the flexible coordination sphere of lead(II).22 This, coupled with the propensity for weak hydrogen bonding within lead(II) coordination polymers,23 suggests that judicious functionalised ligand choice may yield materials beyond the scope of typical lead-1,3-benzenedicarboxylate (mbdc) coordination polymers.24 This work forms part of an ongoing programme to develop switchable functional metal-ligand systems of MOFs25 and coordination polymers.22
Herein, we report the synthesis of a new example of a lead(II) metallogel derived from solvothermal reaction of lead(II) acetate trihydrate with a previously unreported 5-allenyl-1,3-benzenedicarboxylic acid (H2abd) in N,N-dimethylformamide (DMF). Analysis of the air-dried gel yielded an elemental composition consistent with an empirical formula [Pb(abd)(H2O)]n (1), supported by further analyses including IR and 1H-NMR spectroscopy, and scanning electron microscopy (SEM). Mixing H2abd and lead(II) acetate trihydrate in DMF under ambient conditions yielded a crystalline 3D coordination polymer product after two days, [Pb(abd)(DMF)] (2), that was analysed by single-crystal X-ray diffraction methods. This solid-state analysis provides a postulated model for the fibrillar structure of the gel.
Results and discussion
Synthesis
Commercially available dimethyl 5-ethynylisophthalate was converted to the allene species by a one-step addition of p-formaldehyde mediated by copper(I) iodide,26 followed by ester deprotection to the diacid form (H2abd) with sodium hydroxide and an acidic workup (Scheme 1). | ||
| Scheme 1 Synthesis of H2abdvia Me2abd. a) CuI (0.5 equiv.), (CH2O)n (2.5 equiv.), iPr2NH (1.8 equiv.), dioxane, reflux; b) NaOH (10 equiv.), THF, 4 h, RT, acidic workup. | ||
The overall yield for this two-step process was 31%. Both new organic compounds (Me2abd, H2abd) were characterised by IR, 1H- and 13C-NMR spectroscopy, and high resolution ESI mass spectrometry.
The 1H-NMR spectrum (CDCl3) of Me2abd contains a prominent singlet at δ 3.96 ppm that corresponds to the methyl ester groups, and a characteristic doublet and triplet at δ 5.27 ppm and 6.24 ppm that are assigned to the allene group. The remaining aryl resonances were observed at δ 8.14 and 8.51 ppm. The diacid ligand H2abd showed a similar pattern of resonances albeit with shifts mediated by solvent effects and the absence of the methyl group signal (vide infra). In both the protected and deprotected forms, the allene functionality was observed using IR spectroscopy at 1935 cm−1. Deprotection effected a shift of the carbonyl band from 1722 cm−1 to 1685 cm−1.
Isolation of either the metallogel 1, or crystalline form 2, from equimolar amounts of H2abd and lead(II) acetate trihydrate was dependent upon the temperature of the reaction mixture. In the absence of heating, gelation does not occur and, upon standing, crystalline material was isolated after two weeks. Heating the reaction mixture at 100 °C for 72 h favoured formation of a thermodynamic metallogel product. Optimisation of the process identified the critical gelation concentration as 1% w/v of H2abd and hydrated lead(II) acetate as gauged by the tube-inversion test. This gel forming concentration compares favourably to low-molecular-weight organogelators (LMOGs) which are known to form gels a very low concentrations (<2% w/v).27 Upon shaking, the formed gel became a slow flowing liquid in which the gel structure was partly fragmented. This material failed all future inversion tests. Such behaviour is indicative of the initial gel possessing a rigid internal structure.21
Solid-state analysis of the dried gel by powder X-ray diffraction (PXRD) methods found the material possessed amorphous character, precluding this known means of matching crystalline and gel structural characteristics.28
Scanning electron microscopy of 1
The nanoscale morphology of a freeze-dried sample of metallogel 1 was visualised using SEM, which revealed an extended network of fibrous structures that aggregate into large entangled worm-like morphologies (Fig. 1). Well-defined fibres were observed at the periphery of the image, and are analogous to fibrils present in similar gel materials.29 The average diameter of these fibres was found to be 40 nm, while the densely entangled worm-like structures were found to be larger in size, varying in diameter from 40–160 nm. A higher magnification view of the worm-like structure may be found in the ESI† (Fig. S5). | ||
| Fig. 1 SEM image of a 1% w/v metallogel of 1 showing the two most prominent morphological features: rod-like fibres are present at the periphery around a large agglomeration of worm-like morphologies in the centre. | ||
1H-NMR spectroscopic titrations of Pb(OAc)2·3H2O to H2abd
To gain insights into the initial solution-state behaviour of H2abd and hydrated lead(II) acetate during gel formation, the aggregation process was monitored using 1H-NMR spectroscopy in DMF-d7. A neat spectrum of H2abd [29.2 mM] was first collected, which showed the expected allene doublet and triplet at δ 5.43 and 6.63 ppm respectively, as well as aromatic signals at δ 8.21 and 8.49 ppm. Seven aliquots of 0.125 molar equivalents of Pb(OAc)2·3H2O were successively added and the spectra compared (Fig. 2). The results show a net upfield shift for all non-acidic hydrogen atoms of H2abd, with the most pronounced effect seen for the ArH2 aromatic hydrogens and the CH allene hydrogen (Fig. 2, top). These shifts can be rationalised in terms of increased nucleophilicity in the local ligand environment once carboxylate formation mediated by chelation to lead(II) occurs, as well as from structural insights obtained from the crystalline form 2 (vide infra). The ligands chelating the lead(II) aggregates are labile based on the observed gradual migration of peaks, as opposed to the appearance of new peaks upon lead(II) acetate addition. The lack of new peaks consistent with coordinated DMF also suggests that any solvent interactions within the gel are labile in nature. The incremental increase in lead(II) acetate effected a broadening and loss of splitting patterns associated with abd. This can be attributed to self-assembly of fibrillar nanoscale structures that tumble slower in solution than the NMR timescale upon reaching a critical size. This leads to enhanced spin–spin interactions and faster relaxation of transverse magnetisation, giving a short T2.30 The final titre yielded an equimolar mixture in which the signal broadening was too great for meaningful 1H-NMR spectroscopic interpretation. While these spectroscopic observations are consistent with the anticipated early events of self-assembly during the gelation process, only by heating at 100 °C could the mixture be ‘set’ into the gel form, in which reliable spectroscopic data could not be obtained. | ||
| Fig. 2 Titration isotherm and 1H-NMR spectroscopic titration stack plot for the interaction between H2abd and Pb(OAc)2·3H2O in DMF-d7. | ||
Crystallographic analyses
To gain insights into the coordinative behaviour of abd following reaction with hydrated lead(II) acetate, Pb(OAc)2·3H2O and H2abd were combined under ambient conditions. Slow diffusion of dilute anhydrous DMF solutions of H2abd and lead(II) acetate trihydrate yielded colourless needle-like crystals suitable for X-ray structure determination after two days (see Experimental section). The crystals were found to have a molecular composition of [Pb(abd)(DMF)] (2).The asymmetric unit of 2 contains a single Pb(II) atom coordinated by a carboxylate group of a bridging abd ligand and a single molecule of DMF. The metal centre is eight-coordinate based on a limiting value for a Pb–O bond of 3.30 Å.31 The coordination sphere is comprised of four carboxylates, each of which bridges between two metal centres in a μ-(κ2O:κ1O') fashion, and two DMF molecules that bridge between two metal centres (Fig. 3). This culminates in a 1D chain of Pb(II) atoms propagating along the crystallographic c axis (Fig. 4, top). The abd ligands link these chain motifs together to provide a (4,4) network comprised of squares and diamonds when viewed down the c axis (Fig. 4, bottom). The square channels are occupied by the allene groups of the adb ligands while the diamond channels are filled by coordinated DMF molecules that propagate in a contiguous fashion.
| Structure | 2 |
| Formula | C14H13NO5Pb |
| FW | 482.44 |
| Space group |
P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c 21c |
| a [Å] | 19.0391(2) |
| b [Å] | 19.0391(2) |
| c [Å] | 8.0073(2) |
| α [°] | 90 |
| β [°] | 90 |
| γ [°] | 90 |
| Vol [Å3] | 2902.55(9) |
| Z | 8 |
| D calc [g cm−3] | 2.208 |
| μ [mm−1] | 11.645 |
| F(000) | 1808 |
| No. of indep. Reflns (Rint) | 3182 (0.0283) |
| R 1, wR2 (I > 2σ) | 0.0283, 0.0409 |
 | ||
| Fig. 3 Coordination environment about the Pb(II) atom in 2. Thermal ellipsoids are shown with 50% probability. Symmetry operators: (') = 1/2 + y, x − 1/2, z − 1/2, ('') = 1 − y, x, −z, (''') = 1/2 + x, 1/2 − y, 1/2 − z. | ||
 | ||
| Fig. 4 (Top) Bridging carboxylates link Pb(II) atoms into a chain motif. (Bottom) Bridging abd ligands link the cluster chains into a complete 3D network. | ||
A crystallographic comparison of this work to other known lead(II) mbdc coordination polymers serves to display the diversity of secondary building units (SBUs) and structural motifs that are known. The complex with the parent ligand, H2bdc, forms a 3D coordination polymer comprising linked tetranuclear [Pb4(μ4-O)] SBUs under mildly basic solvothermal conditions.24a As observed in the present work, π–π stacking interactions are widespread, causing ligands to bridge between SBUs in pairs. Changing to hydrothermal conditions in synthesis of this parent complex yielded a zeolite-type topology that exhibits uni-dimensional pores running along the a, b and c axes.24b This framework possesses an interesting purely inorganic skeleton, which plausibly may result from the scarcity of coordinating solvent molecules other than water (i.e. DMF), which promotes the formation of the observed Pb–O–Pb linkage. The inclusion of additional ligand–ligand attractive interactions, such as I⋯I interactions, appears to influence the motif,24c with formation of a tetranuclear I4 unit within a 5-iodo-1,3-benzenedicarboxylato coordination polymer imparting an ordered rod-shaped SBU, superseding any π–π interactions.
Molecular structure of fibrillar gel 1
Elemental analysis on an air dried sample of 1 yielded a carbon and hydrogen content of 30.91% and 1.89% respectively, which is consistent with an empirical formula of [Pb(abd)(H2O)]n and provides the starting point for a proposed structure.As with a number of previous reports, the crystalline characteristics determined from a polymorphic form provide the basis for our molecular assignment.4,6,21 As no nitrogen content was found to be present, displacement of the coordinated DMF molecules in 2 for water molecules would yield a framework with a ratio of [Pb(abd)(H2O)], which matches (C: 30.71, H: 1.93) the experimentally determined elemental percentages. Solid-state evidence for the location of coordinated DMF within linear channels of 2 suggests a facile means whereby water may easily penetrate and displace the DMF molecules from within such a structure. A comparison between the infrared spectra for 1 and 2 identifies a prevalence of matching vibrational modes (Table 2). While weak, the asymmetric stretching vibration for the allene group in the dried gel was observed at 1941 cm−1. One possible explanation for a decrease in intensity of this vibration is an electron withdrawing effect influencing the allene group upon gel formation. Similar observations of very weak or absent vibrations have been noted for the nitrile functionality.32 The terminal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 allene wagging mode was also inferred in gel 1 as a shoulder present at 799 cm−1. No band was observed at 1635 cm−1, the expected location for the CO vibration for coordinated DMF,33 which is consistent with the observed lack of nitrogen by elemental analysis.
CH2 allene wagging mode was also inferred in gel 1 as a shoulder present at 799 cm−1. No band was observed at 1635 cm−1, the expected location for the CO vibration for coordinated DMF,33 which is consistent with the observed lack of nitrogen by elemental analysis.
Dissolving a portion of gel 1 in DMSO-d6 spiked with DCl revealed resonances attributable to the allene group by 1H-NMR spectroscopy (Fig. S6, ESI†), suggesting that the gelation process does not require degradation of the allene group at elevated temperatures.
The upfield shifting of all the non-acidic ligand hydrogen atoms in the 1H-NMR titration experiment, in particular for the allene CH and aromatic ArH2 resonances (Fig. 2), likely results from nucleophilic interactions from localised carboxylate groups that surround abd during self-assembly. Such interactions can be inferred in the solid-state structure of 2. In addition to the expected upfield shift in the aromatic hydrogens of abd upon carboxylate formation, each of the ArH2 hydrogens is well-aligned to the carboxylate group of an adjacent abd ligand (Fig. 5), including interactions within the range of van der Waals interaction (<2.72 Å). Similarly, in the solid-state the allene CH hydrogen is aligned to bifurcate two adjacent carboxylate groups. In both instances a self-assembly process that resembles the solid-state structure of 2 would be expected to enhance shielding of these hydrogens. By contrast, the terminal allene CH2 groups protrude into a hydrophobic channel (Fig. 4) that limits nucleophilic interactions to weaker inter-allene C–H⋯π interactions, and the aromatic ArH hydrogen is poorly aligned to be influenced by any inter-ligand nucleophilic carboxylate groups, implying that additional nucleophilic interactions upon self-assembly are unlikely. The nucleophilic interactions determined for each protic region in solid-state 2 are remarkably consistent with the magnitude of the observed Δδ changes that occur during the solution-state self-assembly visualised by the 1H-NMR spectroscopic study.
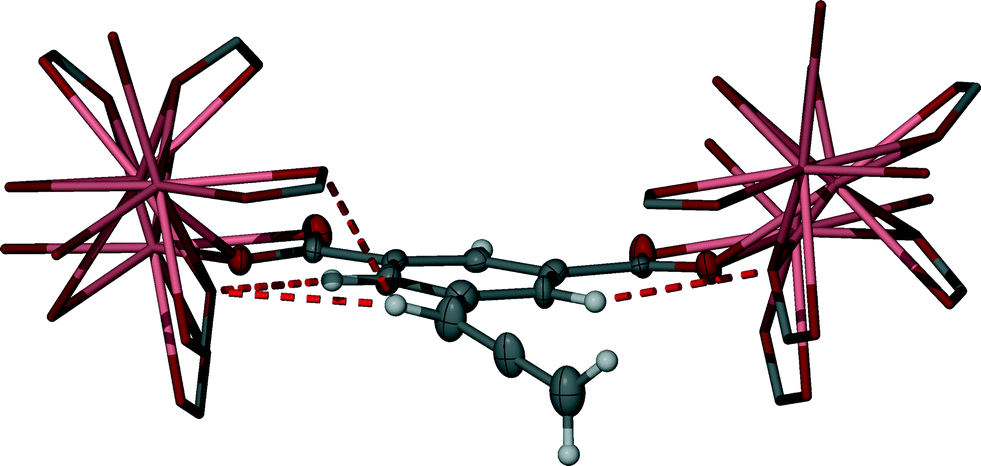 | ||
| Fig. 5 Example of the local environment of abd in 2 showing the primary additional nucleophilic interactions, represented here as red dashed lines. Thermal ellipsoids are shown with 50% probability. | ||
While care must be taken when comparing solution- and solid-state observations, as well as gel and crystalline materials, these results indicate that the framework characteristics observed in 2 persist within gel 1, albeit in an amorphous form. There are known examples in which MOF-type structures are incorporated into gel materials,6,21 and spectroscopic analyses similarly support similarities in the local environments on the molecular level between 1 and 2, allowing a similar approach to be used to deduce the molecular structure for 1 proposed here. These results provide the first solid-state evidence relating to the structure of a lead(II) metallogel, and a new example of a 3D coordination polymeric gel species for which only a few examples are known.
Experimental
General remarks
Starting materials and solvents were purchased from commercial sources and used as received. NMR spectra were recorded on a Bruker Advance 300 MHz Ultrashield NMR spectrometer. Infrared spectra were recorded on a PerkinElmer Spectrum 100 spectrometer equipped with an ATR sampling accessory. Abbreviations for IR bands are s = strong, m = medium, w = weak, br = broad. Elemental analyses (CHN) were performed on a CE-440-Elemental Analyzer (Exeter Analytical).Synthesis
CCH2, J = 6 Hz), 6.25 (t, 1H, HCCCH2, J = 6 Hz), 8.14 (s, 2H, ArH), 8.51 (s, 1H, ArH). 13C-NMR (CDCl3, 298 K, 75 MHz): 52.4 (CH3), 80.0 (HCCCH2), 92.9 (HCCCH2), 128.9 (ArC), 131.0 (ArC), 131.7 (ArC), 135.3 (ArC), 166.2 (CO), 210.1 (HCCCH2). HRMS (ESI): calcd. for C13H13O4 [M + H]+: 233.0814, found: 233.0832. IR (ATR): 3009 (w), 2954 (w), 1935 (m), 1722 (s), 1599 (w), 1432 (s), 1347 (w), 1322 (m), 1301 (m), 1252 (m), 1237 (s), 1203 (s), 1126 (m), 1082 (m), 996 (s), 853 (m), 863 (m), 751 (s), 724 (m), 689 (m), 664 (m) cm−1.
CCH2, J = 6 Hz), 6.59 (t, 1H, HCCCH2, J = 6 Hz), 8.07 (s, 2H, ArH), 8.29 (s, 1H, ArH). 13C-NMR (CD3OD, 298 K, 75 MHz): 79.8 (HCCCH2), 93.6 (HCCCH2), 130.3 (ArC), 132.7 (ArC), 133.1 (ArC), 136.9 (ArC), 168.8 (CO), 211.4 (HCCCH2). HRMS (ESI): calcd. for C10H9NaO4 [M + Na]+: 227.0320, found: 227.0426. IR (ATR): 2829 (br m), 2553 (br m), 1936 (m), 1684 (s), 1598 (s), 1420 (s), 1272 (s), 1250 (s), 1210 (m), 1113 (m), 912 (s), 856 (s), 757 (s), 685 (m), 661 (m) cm−1.
X-ray crystallography
X-ray diffraction data for framework 2 was collected on an Agilent Gemini A-Ultra diffractometer37 at the University of Bath using Mo-Kα radiation, the crystal being cooled to 150 K by an Agilent Cryojet.38 Empirical absorption correction was applied using the multiscan program in SCALE3 ABSPACK. Crystal data and details of the structure refinement and data collection are shown in Table 1. Structural solution and refinement was carried out using SHELXL-9739 utilising the graphical interface X-Seed.40Conclusions
The solvothermal reaction of a new diacid allene, H2abd, with hydrated lead(II) acetate has yielded a metallogel with a critical gelation point of 1% w/v in DMF, with a worm-like morphology composed of nanoscale fibres. Combining H2abd and hydrated lead(II) acetate in DMF in the absence of solvothermal conditions yielded crystalline needles after two days that were determined crystallographically to be composed of [Pb(abd)(DMF)] (2). This 3D coordination polymer forms the basis for structural assignment of gel 1. The initial aggregation behaviour of H2abd with hydrated lead acetate was evaluated in the solution-state by a 1H-NMR titration experiment, which identified the growth of nanoscale structures and local trends in hydrogen shielding that can be related to solid-state 2. A molecular structure for the lead(II) gel has been developed consistent with these observations, which matches the elemental composition of [Pb(abd)(H2O)]n determined for 1 and reflects the local architecture in crystalline structure 2, in which displacement of coordinated DMF for water molecules occurs. Infrared spectroscopic analysis comparing a dried sample of gel 1 with crystalline form 2 identified broad similarities between the vibration band profiles.Acknowledgements
We are grateful to the EPSRC for financial support of the project (EP/K004956/1) and the University of Bath for a studentship to JVK.Notes and references
- (a) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC; (b) M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed; (c) A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- J. Zhang and C.-Y. Su, Coord. Chem. Rev., 2013, 257, 1373 CrossRef CAS PubMed.
- (a) W. J. Gee and S. R. Batten, Chem. Commun., 2012, 48, 4830 RSC; (b) B. Li, L. Tang, L. Qiang and K. Chen, Soft Matter, 2011, 7, 963 RSC; (c) X. de Hatten, N. Bell, N. Yufa, G. Christmann and J. R. Nitschke, J. Am. Chem. Soc., 2011, 133, 3158 CrossRef CAS.
- X. Yan, T. R. Cook, J. B. Pollock, P. Wei, Y. Zhang, Y. Yu, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 4460 CrossRef CAS PubMed.
- G. Nandi, H. M. Titi, R. Thakuria and I. Goldberg, Cryst. Growth Des., 2014, 14, 2714 CAS.
- J. G. Hardy, X. Cao, J. Harrowfield and J.-M. Lehn, New J. Chem., 2012, 36, 668 RSC.
- (a) A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179 CrossRef CAS; (b) I. Odriozola, I. Loinaz, J. A. Pomposo and H. J. Grande, J. Mater. Chem., 2007, 17, 4843 RSC; (c) A. Y. Y. Tam, K. M. C. Wong, G. X. Wang and V. M. M. Yam, Chem. Commun., 2007, 2028 RSC; (d) F. Camerel, R. Ziessel, B. Donnio, C. Bourgogne, D. Guillon, M. Schmutz, C. Iacovita and J. P. Bucher, Angew. Chem., Int. Ed., 2007, 46, 2659 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Soft Matter, 2011, 7, 2412 RSC.
- D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154 RSC.
- W. G. Miao, L. Zhang, X. F. Wang, H. Cao, Q. X. Jin and M. H. Liu, Chem. – Eur. J., 2013, 19, 3029 CrossRef CAS PubMed.
- B. Xing, M.-F. Choi and B. Xu, Chem. – Eur. J., 2002, 8, 5028 CrossRef CAS.
- S. Sengupta and R. Mondal, J. Mater. Chem. A, 2014, 2, 16373 CAS.
- (a) A. Mallick, E.-M. Schön, T. Panda, K. Sreenivas, D. Díaz Díaz and R. Banerjee, J. Mater. Chem., 2012, 22, 14951 RSC; (b) I. Kapoor, E.-M. Schön, J. Bachl, D. Kühbeck, C. Cativiela, S. Saha, R. Banerjee, S. Roelens, J. J. Marrero-Tellado and D. Díaz Díaz, Soft Matter, 2012, 8, 3446 RSC; (c) K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797 CrossRef CAS PubMed.
- (a) G. O. Lloyd and J. W. Steed, Soft Matter, 2011, 7, 75 RSC; (b) D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154 RSC; (c) J. Gao, S. Wu, T. J. Emge and M. A. Rogers, CrystEngComm, 2013, 15, 4507 RSC; (d) C. D. Jones, J. C. Tan and G. O. Lloyd, Chem. Commun., 2012, 48, 2110 RSC.
- (a) S. Saha, E.-M. Schön, C. Cativiela, D. Díaz Díaz and R. Banerjee, Chem. – Eur. J., 2013, 19, 9562 CrossRef CAS; (b) M. Pyzalska, M. Smal and R. Luboradzki, J. Non-Cryst. Solids, 2011, 357, 3184 CrossRef CAS PubMed; (c) Y. Xu, C. Kang, Y. Chen, Z. Bian, X. Qiu, L. Gao and Q. Meng, Chem. – Eur. J., 2012, 18, 16955 CrossRef CAS PubMed; (d) S. Samai, P. Ghosh and K. Biradha, Chem. Commun., 2013, 49, 4181 RSC.
- S. P. Anthony, L. Wang, S. Varughese and S. M. Draper, CrystEngComm, 2013, 15, 6602 RSC.
- T. Ishiwata, Y. Furukawa, K. Sugikawa, K. Kokado and K. Sada, J. Am. Chem. Soc., 2013, 135, 5427 CrossRef CAS PubMed.
- J. A. Forster, D. W. Johnson, M.-O. M. Pipenbrock and J. W. Steed, New J. Chem., 2014, 38, 927 RSC.
- M. R. Lohe, M. Rose and S. Kaskel, Chem. Commun., 2009, 6056 RSC.
- (a) Y.-R. Liu, L. He, J. Zhang, X. Wang and C.-Y. Su, Chem. Mater., 2009, 21, 557 CrossRef CAS; (b) H. H. Lee, S. H. Jung, S. Park, K.-M. Park and J. H. Jung, New J. Chem., 2013, 37, 2330 RSC.
- J. V. Knichal, W. J. Gee, A. D. Burrows, P. R. Raithby, S. J. Teat and C. C. Wilson, Chem. Commun., 2014, 50, 14436 RSC.
- J. V. Knichal, W. J. Gee, A. D. Burrows, P. R. Raithby and C. C. Wilson, Cryst. Growth Des., 2015, 15, 465–474 CAS.
- (a) E.-C. Yang, J. Li, B. Ding, Q.-Q. Liang, X.-G. Wang and X.-J. Zhao, CrystEngComm, 2008, 10, 158 RSC; (b) L. Zhang, Y.-Y. Qin, Z.-J. Li, Q.-P. Lin, J.-K. Cheng, J. Zhang and Y.-G. Yao, Inorg. Chem., 2008, 47, 8286 CrossRef CAS PubMed; (c) S.-H. Luo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 494 CAS.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482 CrossRef CAS PubMed.
- J. Kuang and S. Ma, J. Org. Chem., 2009, 74, 1763 CrossRef CAS PubMed.
- M. Llusar and C. Sanchez, Chem. Mater., 2008, 20, 782 CrossRef CAS.
- (a) P. Byrne, G. O. Lloyd, L. Applegarth, K. M. Anderson, N. Clarke and J. W. Steed, New J. Chem., 2010, 34, 2261 RSC; (b) J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem. – Eur. J., 2014, 20, 279 CrossRef CAS.
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Springer, 2006 Search PubMed.
- M. P. Foster, C. A. McElroy and C. D. Amero, Biochemistry, 2007, 46, 331 CrossRef CAS PubMed.
- R. L. Davidovich, V. Stavila, D. V. Marinin, E. I. Voit and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef CAS PubMed.
- (a) R. E. Kitson and N. E. Griffith, Anal. Chem., 1952, 24, 334 CrossRef CAS; (b) A. Urbatsch, W. J. Gee, G. B. Deacon and S. R. Batten, Tetrahedron Lett., 2013, 54, 2661 CrossRef CAS PubMed.
- J. R. Shapley, Inorganic Syntheses, Wiley-Interscience, 2004, vol. 34, p. 120 Search PubMed.
- (a) L. V. Daimay, B. C. William and G. G. Feanete, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, Boston, 1991 Search PubMed; (b) J. H. Wotiz and D. E. Mancuso, J. Org. Chem., 1957, 22, 207 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
- B. M. Trost and A. McClory, Org. Lett., 2006, 8, 3627 CrossRef CAS.
- Agilent Gemini A-Ultra Diffractometer, http://www.chem.agilent.com/en-US/Products/Instruments/x-raycrystallography/smallmoleculesystem/geminiultra/pages/default.aspx.
- Agilent CryojetXL, http://www.chem.agilent.com/en-US/Products/Instruments/x-raycrystallography/accessories/cryojetxl/pages/default.aspx.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Barbour, XSEED: A graphical interface for use with the SHELX97 program suite, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
Footnote |
† Electronic supplementary information (ESI) available: 1H and 13C-NMR spectra for Me2abd and H2abd (S1–S4), 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnification of dried gel (S5), 1H-NMR characterisation of the allene group in a digested gel sample (S6) and an IR spectra for both gel 1, crystalline 2 (S7–S8). CCDC 1416616 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01689d 000× magnification of dried gel (S5), 1H-NMR characterisation of the allene group in a digested gel sample (S6) and an IR spectra for both gel 1, crystalline 2 (S7–S8). CCDC 1416616 for 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01689d |
| This journal is © The Royal Society of Chemistry 2015 |