
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved in situ studies on the formation mechanism of iron oxide nanoparticles using combined fast-XANES and SAXS†
Anke
Kabelitz
a,
Ana
Guilherme
a,
Maike
Joester
ab,
Uwe
Reinholz
a,
Martin
Radtke
a,
Ralf
Bienert
a,
Katrin
Schulz
c,
Roman
Schmack
c,
Ralph
Kraehnert
c and
Franziska
Emmerling
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstätter Strasse 11, D-12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
bHumboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Strasse 2, D-12489 Berlin, Germany
cTechnische Universität Berlin, Department of Chemistry, Strasse des 17. Juni 124, D-10623 Berlin, Germany
First published on 30th September 2015
Abstract
The reaction of iron chlorides with an alkaline reagent is one of the most prominent methods for the synthesis of iron oxide nanoparticles. We studied the particle formation mechanism using triethanolamine as reactant and stabilizing agent. In situ fast-X-ray absorption near edge spectroscopy and small-angle X-ray scattering provide information on the oxidation state and the structural information at the same time. In situ data were complemented by ex situ transmission electron microscopy, wide-angle X-ray scattering and Raman analysis of the formed nanoparticles. The formation of maghemite nanoparticles (γ-Fe2O3) from ferric and ferrous chloride was investigated. Prior to the formation of these nanoparticles, the formation and conversion of intermediate phases (akaganeite, iron(II, III) hydroxides) was observed which undergoes a morphological and structural collapse. The thus formed small magnetite nanoparticles (Fe3O4) grow further and convert to maghemite with increasing reaction time.
Introduction
Iron oxide nanoparticles (FeOx NPs) are intensively studied because of their broad range of applications in different areas like sensing,1 catalysis,2 magnetic storage media,3 and biomedicine.4–6 The most prominent iron oxides are maghemite (γ-Fe2O3) and magnetite (Fe3O4) with a spinel structure.7 To synthesize these FeOx NPs, the most common synthesis involves precipitating an iron precursor in an alkaline, aqueous solution.8–10 Changing parameters like pH, iron precursor ratio (Fe2+/Fe3+), iron concentration, ionic strength, temperature, alkaline agent, and stabilization agent, the composition and size of the nanoparticles can be varied.11–13 Stabilization agents like phosphates,14 carboxy-dextran,15 and triethanolamine (TREA)16,17 are used to alter the magnetic properties and water solubility of FeOx NPs and to achieve a narrow size distribution.To gain deeper insight into the mechanism of a given NP synthesis procedure, many studies rely on ex situ methods. Baumgartner et al. elucidated the formation mechanism of magnetite nanoparticles based on ex situ cryo-transmission electron microscopy (TEM).18 Their study reveals that preliminary formed nanometric ferrihydrite particles agglomerate and transform into magnetite nanoparticles. Ahn et al. observed the formation of different iron oxide hydroxide intermediates during the formation of magnetite nanoparticles using ex situ X-ray diffraction (XRD) and TEM.10Ex situ analysis of FeOx NPs typically requires sample preparation. Steps like centrifugation and drying could lead to artifacts, e.g. a change in the particle size.19,20
In situ studies can provide information on the particle formation, the formation of intermediates, and phase changes directly in the reaction media without the need for such a sample preparation.21–24 To investigate the formation mechanism in real time, combinations of different analytical tools like scattering methods (small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS)) are available. In situ studies on metal oxide systems were reported, coupling scattering and spectroscopic techniques.25 The formation mechanism of SnO2 NPs is illustrated by Caetano et al. coupling Raman spectroscopy and extended X-ray absorption fine structure (EXAFS), which were supported by SAXS data.26 Bremholm et al. investigated the formation of magnetite nanoparticles using in situ SAXS and WAXS in supercritical water (Pcritical = 221 bar, Tcritical = 374 °C).27 In their study, an ammonium iron(III) citrate precursor was injected into a preheated reactor. The authors observed the formation of amorphous iron(III) hydroxide nanoclusters, which dissolve or decompose completely before magnetite nanoparticles crystallize. Jensen et al. described the formation of maghemite NPs from the ammonium iron(III) citrate precursor at high temperatures and under high pressure by using total scattering data and atomic pair distribution function (PDF) analysis.28 In both mentioned FeOx NPs syntheses only Fe3+ precursors are used in the reaction. For detailed knowledge about the formation mechanisms, methods for determining the changes in the oxidation state during the reaction are required.
Here, we present the first in situ study of the formation process of maghemite NPs in aqueous solution coupling X-ray absorption near edge spectroscopy (XANES) and SAXS investigation. We followed the occurrence of intermediates during the co-precipitation reaction by combining these methods. XANES analysis provides information on the oxidation state and the local structure of the iron atoms. SAXS data provide information about the size of the particles. We study the formation of maghemite nanoparticles from ferric and ferrous chloride using TREA in situ supplemented by complementary XRD, TEM, Raman and selected area electron diffraction (SAED). The advantage of the chosen synthesis is the small number of reaction partners and the favorable properties of TREA, which react as stabilizing and alkaline agent. In the following, the experimental part, the characterization of the final products, and intermediates is shown, followed by the results of the in situ characterization. In addition, we studied the influence of HCl solution of the reaction mechanism of maghemite NPs to propose a formation mechanism.
Experimental
Synthesis of iron oxide nanoparticles
The synthesis used for the in situ reactions was modified based on the synthesis procedure described by Peng et al.16 Peng synthesized γ-Fe2O3 NPs with a diameter of 8 nm starting from a solution of ferrous and ferric chloride and adding TREA. The solution was heated and refluxed for 3 h. In our synthesis, we fixed the temperature to 115 °C to slow down the reaction.A typical synthesis is depicted in Fig. 1a. The iron precursor solution was prepared by dissolving ferric chloride hexahydrate (FeCl3·6H2O, Aldrich, >97% purity) and ferrous chloride tetrahydrate (FeCl2·4H2O, Aldrich, ≥99% purity) in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in water (Millipore, 18.2 MΩ cm, TOC ≤ 5 ppb, flushed with nitrogen) to reach a total iron concentration of 0.31 M. All reagents were used without further purification. TREA (Acros Oganics, 99+% purity) was mixed with water in a ratio of 3:1 (v/v) (TREA solution). Both solutions were filled into separate vials, sealed, and preheated to 115 °C (oil bath) for 10 min. 16 mL of TREA solution was added to 4 mL of iron precursor solution. The synthesis was carried out under continuous magnetic stirring after addition of the TREA solution to the iron precursor solution. The solution was treated at 115 °C (oil bath) for 90 min. In a second type of experiment, HCl was added to the iron precursor solution (final concentration 0.125 M) (see Table 1).
:1 in water (Millipore, 18.2 MΩ cm, TOC ≤ 5 ppb, flushed with nitrogen) to reach a total iron concentration of 0.31 M. All reagents were used without further purification. TREA (Acros Oganics, 99+% purity) was mixed with water in a ratio of 3:1 (v/v) (TREA solution). Both solutions were filled into separate vials, sealed, and preheated to 115 °C (oil bath) for 10 min. 16 mL of TREA solution was added to 4 mL of iron precursor solution. The synthesis was carried out under continuous magnetic stirring after addition of the TREA solution to the iron precursor solution. The solution was treated at 115 °C (oil bath) for 90 min. In a second type of experiment, HCl was added to the iron precursor solution (final concentration 0.125 M) (see Table 1).
 | ||
| Fig. 1 a) Procedure for the synthesis of γ-Fe2O3 NPs. The two solutions were preheated to 115 °C and mixed afterwards. The heating of the iron chloride solutions (iron precursor solution) leads to an increased turbidity, which is marked in dark yellow. The iron oxide nanoparticles were obtained after heating the mixed solution for 90 min. b) Experimental setup for coupled XANES and SAXS investigations using an acoustic levitator as sample holder. | ||
| Experiment type | Iron precursor solution | TREA solution | |
|---|---|---|---|
| Iron precursor | FFe3+ | ||
| 1 | FeCl2, FeCl3 | 0.5 | TREA |
| 2 | FeCl2, FeCl3, HCl | 0.5 | TREA |
Sample environment for synchrotron experiments
An acoustic levitator was used as a sample holder for the SAXS and XANES experiments (see Fig. 1b).29 In this device a stationary ultrasonic field allows to levitate liquid samples and solids in a contact-free environment.30 The acoustic levitator was integrated in the μSpot beamline BESSY II synchrotron (Helmholtz Centre Berlin for Materials and Energy, Germany) as described elsewhere.31 Time-resolved SAXS and XANES data were obtained simultaneously.32,33Time-resolved in situ XANES and SAXS experiments
The experimental setup is depicted in Fig. 1b. During the synthesis, samples (4 μL) were extracted from the reaction solution at different reaction times, placed immediately in the acoustic levitator, and investigated by time-resolved XANES and SAXS experiments. The X-ray beam was monochromatized using a Double Crystal Monochromator (DCM) Si (111). A beam size of 100 μm was used for the XANES and SAXS experiments.For XANES analysis, the monochromatized beam has an energy resolution of E/ΔE = 5000, which corresponds to an energy resolution of about 1.4 eV for Fe-K edge (7112 eV). The fluorescence of the Fe-K line was detected with a silicon drift detector (AXA, KETEK, Munich, Germany) at a working distance of 10 mm. A Fe foil (12.5 μm thick) was used to provide an accurate internal calibration of the monochromator for all spectra. The associated uncertainty was experimentally determined by measuring the foil 10 times. A value of ±0.3 eV was obtained.
The XANES analysis was adjusted to meet the requirements of the time-resolved experiments. The excitation energy was tuned between 7107 eV and 7134 eV. An energy step size of 0.5 eV was applied between 7017 eV and 7125 eV and 1.5 eV between 7125 eV and 7134 eV. The time per step was four seconds, resulting in acquisition time of 270 s for a XANES spectrum. XANES data were processed by ATHENA.34 This GUI program belongs to the main package IFEFFIT (v. 1.2.11). The AutoBK background subtraction procedure was used with the Rbkg parameter set to 1.0 Å. All spectra were normalized to 1. The position of the absorption edge can be derived from the maximum of the first derivative of the XANES data. Simulations of the XANES spectra for the molecule FeCl3·6H2O were performed using FEFF9 software.35 Further information is found in the ESI† (Fig. S1).
For SAXS analysis, a two-dimensional MarMosaic CCD X-ray detector with 3072 × 3072 pixels was used to record the scattering intensity at a sample-detector distance of 819.8 mm. In a typical experiment, the corresponding SAXS data were collected for 300 s every seven minutes. The obtained scattering images were processed and converted into diagrams of scattered intensities versus the scattering vector q employing an algorithm from the FIT2D software.36q is defined by q = 4π/λ·sinθ with θ being half of the scattering and λ being the wavelength. (The small shift of the wavelength required for the XANES measurement can be neglected in the evaluation of the SAXS data.) All SAXS data were evaluated using a global scattering function described by Beaucage et al.,37 to achieve the particle size in terms of the radius of gyration and the power law contribution at higher q values.
The curve fitting was done using IRENA38 implemented in the software package IGOR PRO. Further information of the background correction and fitting procedure is given in the ESI† (S2). A separate SAXS experiment with a higher time resolution was performed with a wavelength of 1.0000 Å (12398 eV). The SAXS data were collected for 20 s every three minutes.
Samples for the Raman measurements were taken from the reaction solution. Raman spectra were measured on a LabRam HR800 (Horiba Jobin Yvon, Bensheim, Germany) equipped with a 633 nm HeNe laser (Horiba Jobin Yvon, Bensheim, Germany). The instrument was coupled to a BX41 microscope (Olympus, Hamburg, Germany) with a 60× immersion objective. The backscattered Raman light was detected by a liquid nitrogen-cooled CCD detector (1024 × 256 pixels, Horiba). The laser intensity was 5.09·105 W cm−2 at the liquid sample.
Further characterization
TEM samples were prepared by adding acetone to the reaction solution. The resulting precipitate was dispersed in ethanol. 15 μL of the solution were placed onto carbon-coated 200 mesh copper grids (EMS) and dried at room temperature. TEM and high resolution TEM (HR-TEM) images of the nanoparticles were acquired using a FEI Tecnai G2 20S-TWIN (FEI, USA) with an acceleration voltage of 200 keV, equipped with a LaB6 electron source. The particle diameter was evaluated by averaging over 100 measured nanoparticles. Environmental scanning electron microscopy (ESEM) analysis of the dried samples was performed on an XL30 ESEM-FG instrument (FEI Company, Hillsboro, USA) operating at 15 kV.For XRD analysis, the colloidal solutions were cooled to room temperature. After the addition of acetone, the precipitate was centrifuged (2500 rpm, 4 min), washed three times with acetone, and dried for 10 h at room temperature. XRD patterns were collected using a wavelength of 10000 Å (12398 eV). The dried final FeOx NPs were analyzed using a standard powder sample holder. In a typical experiment, XRD data were collected for 120–180 s. The crystallite size D is estimated based on the Scherrer equation D = K·λ/β·cosθ, with λ being the wavelength of the X-ray radiation, β being the full width at half maximum, K being the Scherrer shape factor, and θ half of the scattering angle.39
Results and discussion
In the following sections, we present the characterization of the final products and first intermediate followed by the results of the in situ characterization. Further experiments with HCl solution were performed and a formation mechanism is proposed based on the resulting data.Characterization of the final particles without adding HCl
Fig. 2 shows the result of the characterization of the final particles for the synthesis with ferric(II) and ferrous(III) chloride in the presence of TREA. The final nanoparticles were characterized using XRD, TEM, and SAXS. Fig. 2a displays the XRD data of the dried final nanoparticles. All detected reflections are consistent with data base entry for maghemite (JCPDS, PDF: 39-1346, red). The diffraction pattern of the product shows broad reflections, which reflect the small crystallite size of the nanoparticles around 7.7 nm.39 | ||
| Fig. 2 a) XRD analysis of the final FeOx NPs in comparison to the JCPDS database entry (PDF: 39-1346) for maghemite (red) and a simulated pattern for maghemite with a crystallite size of 7 nm (grey). The XRD pattern are calculated for Cu Kα radiation. b) TEM image of the maghemite NPs (scale bar 20 nm). c) HR-TEM image of maghemite NPs with parallel lines marking the observed lattice fringes as well as FFT images corresponding to the two marked regions of the image (scale bar 5 nm). The nanoparticles have a spherical shape and appear to be dominantly monocrystalline, since lattice fringes (marked with red lines) extend through the full particle diameter. The Fourier transformations show individual spots corresponding to the periodical distances of the lattice planes of 0.21 nm and 0.25 nm. d) SAXS data of the final particles in comparison to a theoretical fit of spherical particles with a gyration radius of 4.2 nm. | ||
The TEM image of the maghemite nanoparticles (see Fig. 2b and c) shows spherical particles with an average particle diameter of 6.8 ± 1.3 nm. The HR-TEM and the corresponding Fourier transformations of the maghemite NPs document that the whole particles are crystalline. The lattice fringes correspond well to the crystal structure obtained from XRD.
The small-angle X-ray scattering curve of the maghemite NPs shows a Gaussian decay in the high q-region and a plateau in the low q-region (see Fig. 2d). The power-law decay of the scattering curve is equal to q−4, indicating a smooth surface of the nanoparticles. The scattering curve was fitted by a unified fit model (Fig. 2d, red line). The evaluation of the SAXS data results in a radius of gyration of 4.2 nm, corresponding to a particle diameter DP of 10.8 nm. The particle diameter DP is related to the radius of gyration RG by  . The differences in the determined diameter can be attributed to a small amount of agglomerates.
. The differences in the determined diameter can be attributed to a small amount of agglomerates.
Characterization of the first intermediate without adding HCl
Simultaneous in situ XANES and SAXS analyses were performed on the iron chloride solution and the first intermediate. In Fig. 3a, the XANES spectra (Fe K-edge) are presented as a function of time. Fig. 3b shows the evaluation of the integrated pre-edge peak area (IPA) and the position of the absorption edge during the reaction. The XANES spectrum of the yellow iron precursor solution, containing ferrous and ferric chloride with a fraction of Fe3+ ions (FFe3+) = 0.5, is shown as grey curve (Fig. 3a). The spectrum exhibits an absorption edge at 7124.1 eV and a low IPA (see Fig. 3b). During the reaction at 115 °C, the iron chloride solution turns into a dark yellow, turbid solution (experiment type 1). The position of the absorption edge and the IPA show no significant change during heating of the iron chloride solution, which indicates that both Fe2+ and Fe3+ ions are still present.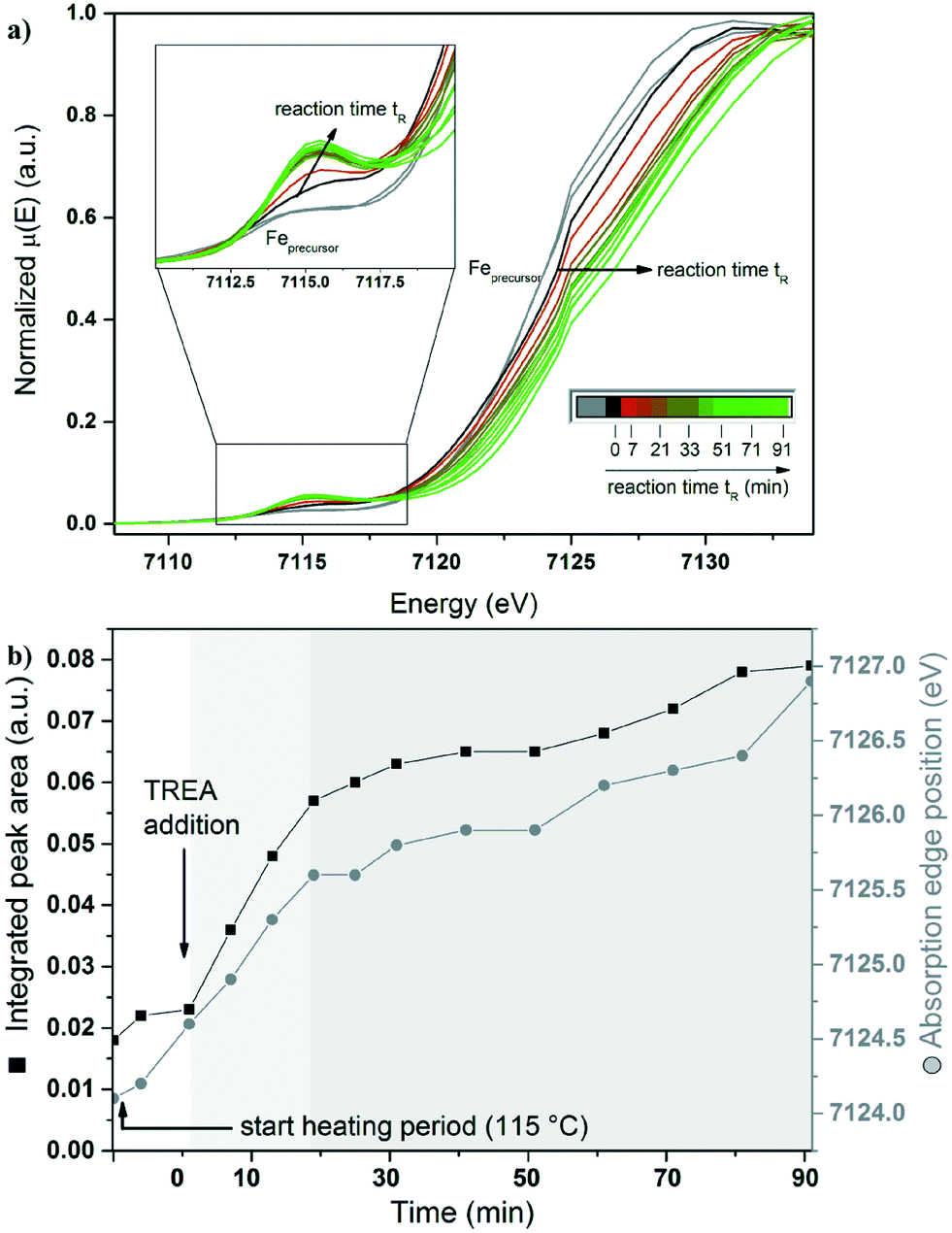 | ||
| Fig. 3 a) In situ XANES data (Fe K-edge) as a function of time. Spectra of the iron chloride solution and those obtained during heating are marked in grey. The spectrum taken one minute after the addition of TREA is depicted in black. Spectra taken shortly after the addition of TREA are indicated by a colour change from dark red (7 min) to light green (91 min). b) Evaluation of the normalized integrated pre-edge peak area (black squares) and absorption edge position (grey circles) of the XANES data as a function of time. The arrow at 0 min visualizes the addition of TREA to the ferric and ferrous solution. The light and dark grey shaded backgrounds illustrate different stages during the reaction. | ||
Fig. 4a displays the corresponding series of SAXS data obtained during the same synthesis. By heating the iron chloride solution, the scattering intensity increases in the low q-range (grey line). This indicates the formation of large particles or aggregates above the size limit of the SAXS experiment (see ESI† Fig. S3).
 | ||
| Fig. 4 a) SAXS scattering curves as a function of time. Scattering curves of the iron chlorides precursor solutions obtained during heating at 115 °C are marked in grey. The scattering pattern taken one minute after the addition of TREA is marked in black. Scattering patterns collected shortly after the addition of TREA are marked by a colour change from dark red (7 min) to light green (91 min). b) Evaluation of the radius of gyration as a function of time. No evaluation of a radius of gyration was possible until a reaction time of 7 min. The dashed line at 0 min visualizes the addition of TREA to the heated iron chlorides. The light and dark grey shaded backgrounds illustrate different stages during the reaction. | ||
The first intermediate formed during heating the precursor solution was identified by XRD as the iron oxide hydroxide akaganeite (β-Fe(III)O(OH, Cl) with a crystallite size of 20 nm (Fig. S4a†).39 The obtained results are backed by Raman (Fig. S5†) and ESEM (Fig. S4b†) data.
Time-resolved in situ investigation of the reaction of γ-Fe2O3 NPs without adding HCl
To elucidate the formation process of the maghemite nanoparticles, time-resolved investigations combining XANES and SAXS were performed. This investigation starts with the addition of TREA to the akaganeite solution and corresponds to the reaction time “0”. After adding TREA, the dark yellow akaganeite solution turns dark green immediately and the absorption edge position shifts slightly to higher energies (Fig. 3a, black line). The integrated pre-edge peak area does not change significantly. During the next 21 min the absorption edge position shifts to higher energies and the IPA increases by a factor of three (light grey area in Fig. 3b).After heating the reaction mixture for 90 min, the colloidal solution turns dark brown. During that period the absorption edge shifts to higher energies until reaching a value of 7126.9 eV for the final particles (see Fig. 3a, light green line). Furthermore, the IPA changes as the reaction time increases. Between 21 and 91 minutes after the addition of TREA (dark grey area), the change in both features is smaller in contrast to the first 21 min. The IPA increases by a factor of four compared to the broad pre-edge peak of the iron chloride solution.
All XANES spectra reveal a relatively low pre-edge peak intensity. The change of the pre-edge peak indicates a distortion of a small amount of the centrosymmetric octahedral coordination geometry of the iron to a tetrahedral coordination geometry. A pure tetrahedral coordination of the iron atoms would be characterized by an intense pre-edge peak.40 Most iron oxides are coordinated octahedrally, resulting in a small integrated pre-edge peak area. Only magnetite and maghemite exhibit tetrahedral coordinated Fe in addition to octahedral coordinated Fe. Fig. 3b shows the shift of the position of the absorption edge with increasing reaction time. This change in the pre-edge peak indicates a structural transition of the geometry of the iron during the reaction and the oxidation of Fe2+ to Fe3+. During the reaction, the increase in the integrated pre-edge peak area is accompanied by a shift of the absorption edge position. These changes indicate that the variation in the oxidation state and the structural changes in the iron species are correlated.
Pre-edge peak XANES spectra of iron chloride mixtures for different fractions of ferric chloride (FFe3+ = 0 to 1) in solution are shown in Fig. S6a and b.† The position of the absorption edge and the pre-edge peak shift to higher energies with increasing Fe3+ content in the solution.
The difference between the centroid of the pre-edge peak for pure Fe2+ and Fe3+ is 1.5 eV, in accordance with the literature.40–42 The centroid position of the iron chloride solution pre-edge peak is shifted to higher energies compared to the iron oxides pre-edge peak (see Fig. S6a†). This can be explained by a stronger binding of the Cl− anion to the iron cation compared to oxygen.40,43 Simulations of the XANES spectra of FeCl3·6H2O support this result (see Fig. S1†).
The corresponding series of SAXS data shows that a high intensity in the low q-range of the SAXS curve is still detectable, directly after adding TREA (Fig. 4a, black line). Within the first 7 min of the reaction, the scattering intensity increases in the high q-range. Small particles with a radius of gyration of 3.0 nm are formed (Fig. 4b). SAXS experiments performed with a higher time resolution (without simultaneous acquisition of XANES data) proof that particles with a gyration radius of 1.6 nm are already formed 4 min after the addition of TREA (Fig. S3†). Small and large particles are present at the same time. Raman spectroscopic investigations hint at the presence of magnetite as dominant phase, besides the signals for TREA (Fig. S5†). SAED data in Fig. S7† of the small nanoparticles supports the formation of iron oxides with a spinel structure (maghemite or magnetite). The large particles could be described by the already formed akaganeite or by the formation of iron(II, III) hydroxides.44 The corresponding XANES spectrum is characterized by an increased pre-edge peak. Within the first 7 min the position of the adsorption edge does not change significantly, indicating that Fe2+ cations are still present in the solution.
After 14 min the Gaussian decay shifts to a lower q-range in the SAXS curve, indicating a growth of the nanoparticles (light grey area, Fig. 4a). At the same time, the strong intensity in the very low q-range vanishes, indicating the complete conversion of the first formed large particles to nanoparticles with a gyration radius of 4.1 nm. The corresponding XANES data suggest that iron oxide nanoparticles are already present in this period (14–21 min). Raman spectroscopic investigations indicate magnetite as dominant phase.
With increasing reaction time, the shape of the scattering curves shows no significant changes (dark grey area, see Fig. 4b). The final radius of gyration amounts to 4.2 nm. The Raman spectra indicate the absence of magnetite Fe3O4 as dominant phase (669 cm−1, A1g mode) after a reaction time of 80 min.
Formation studies of γ-Fe2O3 NPs with HCl
To probe whether the formation of large akaganeite particles is crucial for the nanoparticle particle formation, an experiment in 0.125 M HCl solution was carried out (experiment type 2). The SAXS data are shown in Fig. 5a. In contrast to the experiment without HCl solution, no scattering contribution of the heated ferric and ferrous chloride solution can be detected. This indicates the absence of akaganeite as particles. | ||
| Fig. 5 a) SAXS data as a function of time in 0.125 M HCl solution. The scattering curve 1 min after the addition of TREA is marked in black. The scattering curves obtained shortly after the addition of TREA are indicated by a colour change from dark red (7 min) to light green (91 min). b, c) Evaluation of the normalized integrated pre-edge peak areas and absorption edge positions of the XANES data as a function of time in 0.125 M HCl solution. Data for the experiment, without adding HCl (black) and with adding HCl to the iron chloride solution (blue) are depicted. After the collection of the data for iron chlorides at room temperature, the solution was treated at 115 °C (black arrow). The arrow at 0 min visualizes the addition of TREA to the ferric and ferrous solution. The light and dark grey shaded backgrounds illustrate different stages during the reaction. | ||
After adding TREA, the intensity of the SAXS curve increases in the low q-range (Fig. 5a, black line). This scattering contribution can be related to the formation of larger particles or aggregates above the size limit of the SAXS experiment. With increasing reaction time, final nanoparticles with a radius of gyration of 3.3 nm can be obtained, corresponding to a diameter of 8.5 nm.
The use of a 0.125 M HCl solution for the iron precursor solution shows a moderate effect on the XANES data (see Fig. 5b, c and S8†). The position of the absorption edge and the IPA for the maghemite NPs obtained with and without adding HCl solution are comparable. Thus in comparison with experiment type 1, the detection of large particles after the addition of TREA can be explained by the formation of iron(II, III) hydroxides.44 The formation of akaganeite can be excluded due to the knowledge that akaganeite only form at a pH lower than 6.45 This indicates that the first intermediate phase akaganeite is not required for the formation of the γ-Fe2O3 NPs. The difference in particle size can be explained by the higher ionic strength after adding HCl to the iron chloride precursor.13
Proposed formation mechanism for the γ-Fe2O3 NPs from all experiments
Considering the obtained results, a formation mechanism can be proposed (Fig. 6). The iron chlorides dissolve in aqueous solution to ferric and ferrous chloride. After heating the iron chloride salts at 115 °C, akaganeite is formed. The formation of akaganeite results from the hydrolysis of FeCl3 in aqueous solution under thermal treatment.46 The incorporation of the Cl− ions stabilizes the tunnel structure of akaganeite. At the same time, Fe2+ ions are still present in the solution. The dissolution of TREA in water release OH− ions, which can replace the Cl− ions in the akaganeite structure at higher pH.10 The high temperature and the basic media can lead to a collapse of the akaganeite.45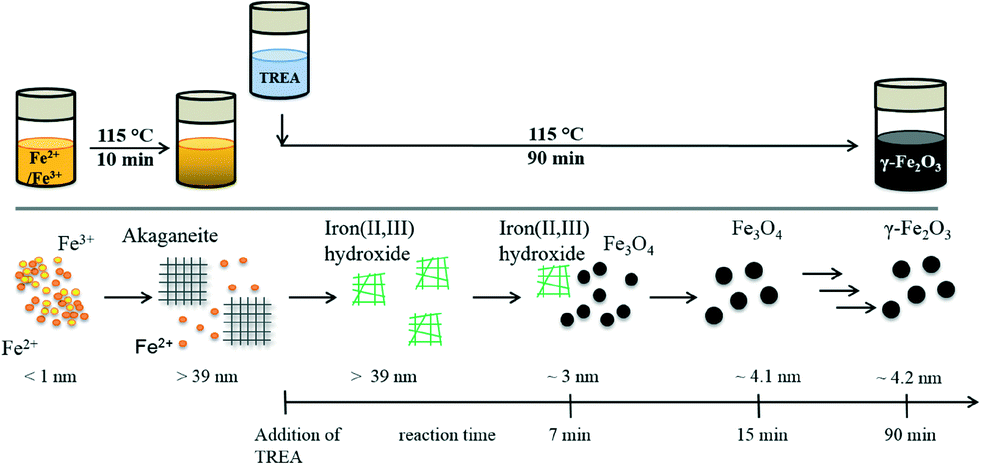 | ||
| Fig. 6 Synthesis procedure and the proposed mechanism for maghemite nanoparticle synthesis. The dissolved iron chlorides are depicted as small orange (Fe2+) and yellow (Fe3+) spheres. Akaganeite is presented as grey grids. The formation of the iron(II, III) hydroxides is depicted as green disordered shapes. The nanoparticles with a spinel structure are presented as black spheres. The radii of the particles are given. | ||
The formation of akaganeite is not required for the formation of maghemite nanoparticles as seen in experiment type 2. Dissolving the iron precursor in a 0.125 M HCl solution suppresses the formation of akaganeite and simplifies the reaction mechanism.47 The green solution, seen after the addition of TREA, is usually explained by the formation of iron(II, III) hydroxides.44
The formed phases were studied using Raman investigations. Raman analysis indicates the absence of iron oxides like maghemite and magnetite as dominant phase during the first minute. The formation of small nanoparticles with a radius of gyration of 3 nm can be detected after a reaction time of 7 min. Raman analysis shows a strong indication for magnetite (signal at 669 cm−1) (see Fig. S5†). In literature the transformation of the iron(II, III) hydroxides to magnetite is described by a dissolution/recrystallization process.48,49 Particle growth is detected up to a radius of gyration of 4.2 nm during the reaction. The absence of the magnetite signal in the Raman spectrum for the final particles can be explained by the oxidation of the magnetite nanoparticles to maghemite during the last reaction period (20 minutes). These results show that the formation process of maghemite nanoparticles can be studied in situ using TREA as stabilization and alkaline agent.
Conclusions
The combination of time-resolved in situ SAXS and XANES experiments allows investigating the formation of γ-Fe2O3 nanoparticles in aqueous solution on a structural and chemical level. Based on the data, a formation mechanism consisting of four phases is proposed. In the first stage akaganeite is formed as an intermediate after heating the iron chloride solutions. Fe2+ ions are still present during this process. After addition of TREA, iron(II, III) hydroxides are formed. During the next 7 min the formation of magnetite particles in the low nm range could be detected (RG = 3 nm). Afterwards, the growth of magnetite NPs to the final particle size was observed. Finally, maghemite NPs with a radius of gyration of 4.2 nm were formed. The addition of HCl solution leads to γ-Fe2O3 NPs, although akaganeite as large particles are not formed. This indicates that the intermediate formation of akaganeite is not mandatory for the formation mechanism of the γ-Fe2O3 NPs. The presence of both, ferric and ferrous ions is important for the formation mechanism of FeOx NPs. The benefit of TREA is its property to react as alkaline and stabilization agent. Therefore, the critical point in the FeOx NP synthesis, the addition of the base, can be investigated by the addition of a stabilization agent and intermediates can be detected. Thus the addition of a stabilization and alkaline agent results in an enhanced control of the particle size. The obtained knowledge will allow controlling the formation of the NPs in solution and further to tune the properties of the final product.Acknowledgements
The authors acknowledge ZELMI at Technical University Berlin for support in TEM analysis. The authors acknowledge Ines Feldmann for support in ESEM analysis. RK thanks in particular Einstein Foundation Berlin for generous support provided by an Einstein-Junior-Fellowship (EJF-2011-95). FE und RK thank for the generous support provided by the CRC 1109.Notes and references
- V. Urbanova, M. Magro, A. Gedanken, D. Baratella, F. Vianello and R. Zboril, Chem. Mater., 2014, 26, 6653 CrossRef CAS.
- P. Li, D. E. Miser, S. Rabiei, R. T. Yadav and M. R. Hajaligol, Appl. Catal., B, 2003, 43, 151 CrossRef CAS.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS.
- E. Amstad, M. Textor and E. Reimhult, Nanoscale, 2011, 3, 2819 RSC.
- A. Meddahi-Pellé, A. Legrand, A. Marcellan, L. Louedec, D. Letourneur and L. Leibler, Angew. Chem., Int. Ed., 2014, 53, 6369 CrossRef PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, 2003 Search PubMed.
- I. Martínez-Mera, M. E. Espinosa-Pesqueira, R. Pérez-Hernández and J. Arenas-Alatorre, Mater. Lett., 2007, 61, 4447 CrossRef PubMed.
- M. Fang, V. Strom, R. T. Olsson, L. Belova and K. V. Rao, Nanotechnology, 2012, 23 Search PubMed.
- T. Ahn, J. H. Kim, H.-M. Yang, J. W. Lee and J.-D. Kim, J. Phys. Chem. C, 2012, 116, 6069 CAS.
- H.-C. Roth, S. P. Schwaminger, M. Schindler, F. E. Wagner and S. Berensmeier, J. Magn. Magn. Mater., 2015, 377, 81 CrossRef CAS PubMed.
- L. Babes, B. Denizot, G. Tanguy, J. J. Le Jeune and P. Jallet, J. Colloid Interface Sci., 1999, 212, 474 CrossRef CAS PubMed.
- X.-P. Qiu, Chin. J. Chem., 2000, 18, 834 CrossRef CAS PubMed.
- Y. Sahoo, H. Pizem, T. Fried, D. Golodnitsky, L. Burstein, C. N. Sukenik and G. Markovich, Langmuir, 2001, 17, 7907 CrossRef CAS.
- K. Tao, S. Song, J. Ding, H. J. Dou and K. Sun, Colloid Polym. Sci., 2011, 289, 361 CAS.
- Q. Peng, J. Gan, S. Wang, L. Kong, G. Chen, Y. Yang and G. Huang, Ind. Eng. Chem. Res., 2013, 52, 7713 CrossRef CAS.
- T. Xia, J. P. Wang, C. L. Wu, F. C. Meng, Z. Shi, J. Lian, J. Feng and J. Meng, CrystEngComm, 2012, 14, 5741 RSC.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310 CrossRef CAS PubMed.
- J. Tuoriniemi, A. Johnsson, J. P. Holmberg, S. Gustafsson, J. A. Gallego-Urrea, E. Olsson, J. B. C. Pettersson and M. Hassellov, Sci. Technol. Adv. Mater., 2014, 15 Search PubMed.
- M. Baalousha, Y. Ju-Nam, P. A. Cole, B. Gaiser, T. F. Fernandes, J. A. Hriljac, M. A. Jepson, V. Stone, C. R. Tyler and J. R. Lead, Environ. Toxicol. Chem., 2012, 31, 983 CrossRef CAS PubMed.
- J. Polte, M. Herder, R. Erler, S. Rolf, A. Fischer, C. Wurth, A. F. Thunemann, R. Kraehnert and F. Emmerling, Nanoscale, 2010, 2, 2463 RSC.
- J. Polte, X. Tuaev, M. Wuithschick, A. Fischer, A. F. Thuenemann, K. Rademann, R. Kraehnert and F. Emmerling, ACS Nano, 2012, 6, 5791 CrossRef CAS PubMed.
- M. Wuithschick, B. Paul, R. Bienert, A. Sarfraz, U. Vainio, M. Sztucki, R. Kraehnert, P. Strasser, K. Rademann, F. Emmerling and J. Polte, Chem. Mater., 2013, 25, 4679 CrossRef CAS.
- J. F. Kettemann, M. Wuithschick, G. Caputo, R. Krahnert, N. Pinna, K. Rademann and J. Polte, CrystEngComm, 2015, 17, 1865 RSC.
- M. Staniuk, O. Hirsch, N. Kränzlin, R. Böhlen, W. van Beek, P. M. Abdala and D. Koziej, Chem. Mater., 2014, 26, 2086 CrossRef CAS.
- B. L. Caetano, F. Meneau, C. V. Santilli, S. H. Pulcinelli, M. Magnani and V. Briois, Chem. Mater., 2014, 26, 6777 CrossRef CAS.
- M. Bremholm, M. Felicissimo and B. B. Iversen, Angew. Chem., Int. Ed., 2009, 48, 4788 CrossRef CAS PubMed.
- K. M. O. Jensen, H. L. Andersen, C. Tyrsted, E. D. Bojesen, A. C. Dippel, N. Lock, S. J. L. Billinge, B. B. Iversen and M. Christensen, ACS Nano, 2014, 8, 10704 CrossRef CAS PubMed.
- M. C. Schlegel, K.-J. Wenzel, A. Sarfraz, U. Panne and F. Emmerling, Rev. Sci. Instrum., 2012, 83, 055101 CrossRef CAS PubMed.
- J. Leiterer, W. Leitenberger, F. Emmerling, A. F. Thunemann and U. Panne, J. Appl. Crystallogr., 2006, 39, 771 CrossRef CAS.
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342 CrossRef CAS PubMed.
- O. Paris, C. Li, S. Siegel, G. Weseloh, F. Emmerling, H. Riesemeier, A. Erko and P. Fratzl, J. Appl. Crystallogr., 2007, 40, s466 CrossRef CAS.
- J. Polte, T. T. Ahner, F. Delissen, S. Sokolov, F. Emmerling, A. F. Thunemann and R. Kraehnert, J. Am. Chem. Soc., 2010, 132, 1296 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503 RSC.
- A. P. Hammersley, K. Brown, W. Burmeister, L. Claustre, A. Gonzalez, S. McSweeney, E. Mitchell, J.-P. Moy, S. O. Svensson and A. W. Thompson, J. Synchrotron Radiat., 1997, 4, 67 CrossRef CAS PubMed.
- G. Beaucage, H. K. Kammler and S. E. Pratsinis, J. Appl. Crystallogr., 2004, 37, 523 CrossRef CAS.
- J. Ilavsky and P. R. Jemian, J. Appl. Crystallogr., 2009, 42, 347 CrossRef CAS.
- P. Scherrer, Nachr. Ges. Wiss. G6ttingen, 1918, 98 Search PubMed.
- M. Wilke, F. Farges, P. E. Petit, G. E. Brown and F. Martin, Am. Mineral., 2001, 86, 714 CrossRef CAS.
- S. Fujieda, A. Yoshino, K. Shinoda, Y. Takahashi and S. Suzuki, ISIJ Int., 2014, 54, 125 CrossRef CAS.
- D. Testemale, J. Brugger, W. Liu, B. Etschmann and J.-L. Hazemann, Chem. Geol., 2009, 264, 295 CrossRef CAS PubMed.
- M. J. Apted, G. A. Waychunas and G. E. Brown, Geochim. Cosmochim. Acta, 1985, 49, 2081 CrossRef CAS.
- P. Refait and J. M. R. Génin, Corros. Sci., 1993, 34, 797 CrossRef CAS.
- J. Cai, J. Liu, Z. Gao, A. Navrotsky and S. L. Suib, Chem. Mater., 2001, 13, 4595 CrossRef CAS.
- J. D. Bernal, Clay Miner., 1959, 4, 15 CAS.
- R. M. Cornell and R. Giovanoli, Clays Clay Miner., 1988, 36, 385 CAS.
- A. Sumoondur, S. Shaw, I. Ahmed and L. G. Benning, Mineral. Mag., 2008, 72, 201 CrossRef CAS PubMed.
- P. H. Refait, M. Abdelmoula and J. M. R. GÉnin, Corros. Sci., 1998, 40, 1547 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ce01585e |
| This journal is © The Royal Society of Chemistry 2015 |