
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct evidence of polyamorphism in paracetamol
Yen
Nguyen Thi
ab,
Klaus
Rademann
b and
Franziska
Emmerling
*a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstätter-Str. 11, 12489 Berlin, Germany. E-mail: franziska.emmerling@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, 12489 Berlin, Germany
First published on 9th October 2015
Abstract
While polymorphism is a common phenomenon in the crystallization processes of organic compounds, polyamorphism has gained importance only recently. Using sophisticated sample environments and applying in situ scattering methods and vibrational spectroscopy, the complete crystallization process of organic compounds from solution can be traced and characterized. Diffuse scattering from amorphous intermediates can be investigated by analyzing the atomic pair distribution function (PDF) to gain further insights into molecular pre-orientation. The crystallization behavior of paracetamol was studied exemplarily under defined, surface-free conditions. Based on the choice of the solvent, the formation of different polymorphs is promoted. The thermodynamically stable form I and the metastable orthorhombic form II could be isolated in pure form directly from solution. For both polymorphs, the crystallization from solution proceeds via a distinct amorphous precursor phase. PDF analyses of these different amorphous states indicate a specific pre-orientation of the analyte molecules introduced by the solvent. The resulting crystalline polymorph is already imprinted in these proto-crystalline precursors. Direct experimental evidence for the polyamorphism of paracetamol is provided.
Introduction
The crystallization of solids is of great importance in many geological, biological and industrial processes and has been investigated for centuries.1 Crystallization from solution is the most widely applied method for the isolation and purification of compounds in fine chemistry and pharmacy. Despite considerable efforts, the understanding of nucleation and crystal growth,2 and external influences on the crystallization progress is still limited. For organic compounds the propensity to form polymorphs, e.g. different crystalline structures of the same compound, is facilitated by weak directional and non-directional intermolecular interactions like hydrogen bonds, van der Waals interaction, or electrostatic dipole interactions.3–5 For amorphous states, polyamorphism,6–8 the existence of more than one amorphous phase, receives increasing attention. Polyamorphism was reported for ice,9 inorganic compounds,10,11 and alloys.12,13 A correlation between polymorphism and polyamorphism is postulated for organic compounds, assuming that each amorphous phase yields a specific crystalline form.14 A definition of polyamorphism given by Hancock et al. states that the presence of true polyamorphism prerequisites the existence, or usually coexistence, of two or more condensed amorphous phases separated by a clear phase transition.15 Since the different crystal structures of a given organic substances differ only slightly in their energies, minor changes of the experimental conditions like surfaces, temperature, or humidity can influence the crystallization pathway and the resulting product.An understanding of the early stages of the crystallization is essential to control the resulting crystalline phase, size, and morphology. The early stages of the phase transformation process is difficult to observe on the atomic and meso scale.16
Typically, the classical nucleation theory is used to describe the crystallization from solution. The classical nucleation process was found for numerous systems both in experiments and simulations.17–20 Theoretical prediction by simulation has been increasingly applied as a valuable possibility to characterize organic crystal structures and their packing motifs.21 New crystal forms of 5-fluorocytosine were found using crystal energy landscape and taking into account the structurally characteristic hydrogen-bonding ribbon of 5-fluorocytosine.22 Further studies of hydrochlorothiazide with the same method showed different polymorphs with a variety of structural motifs.23 Theoretical predictions bear limitations regarding the intricacy of the molecular system and the variety of influencing factors on the crystallization process. Therefore, polymorph screening and experiments with sophisticated analytical techniques are still equally important as complementary methods.
Recent experimental observations indicate that both the aggregation of primary units such as pre-nucleation clusters or nanometric building blocks as well as amorphous precursors24–26 can trigger nucleation and growth.2,27–29 Further detailed insight into this topic and the crystallization pathways can be gained using in situ analytics allowing experiments with high time resolution. Time-resolved Raman microspectroscopy studies supported the finding of an unusually fast crystallization phenomenon of nifedipine suggesting a pre-ordered physical arrangement of amorphous nifedipine as the reason for the deviation from classical diffusion theory.30 Seefeldt and co-workers studied the crystallization process and kinetics of carbamazepin–nicotinamide cocrystals from the amorphous phase under isothermal conditions in separated in situ experiments. In situ hot-stage polarized microscopy, hot stage Raman microscopy, thermomicroscopy, calorimetry, and X-ray powder diffraction were applied to gain further insights into the crystallization process.31 These tools facilitated the identification of two different crystallization pathways proceeding via different intermediate states depending on the heating rate. The authors could show that the thermal history has a significant influence on the crystallization.
Studies of the complete crystallization process require analytical methods which are able to detect and characterize non-crystalline and crystalline states. The atomic pair distribution (PDF) analysis is capable of determining the local structure of non-crystalline materials. This method is typically applied for the structural characterization of glasses, liquids, nanocrystalline, and disordered compounds,32–34 whereas only few studies focus on crystalline and amorphous organic compound materials.35,36 From an experimental perspective, rapid acquisition PDF (RAPDF) measurements proved to be ideal for time-resolved and parametric measurements of local structure through phase transitions.37 Repeated investigations of the crystallization process under the same controlled experimental conditions and the suitable analytics, in the best case, in situ simultaneously applied methods, offer an excellent opportunity to gain a better understanding of this process.
In this study, we examine the crystallization process of paracetamol (acetaminophen) from solution by combined in situ Raman spectroscopy and in situ synchrotron X-ray diffraction. The crystallization conditions were defined using an acoustic levitator and an enveloping gas stream to control environment, temperature, and humidity. The crystallization progress triggered by the evaporation of the solvent was followed completely. The formation of amorphous intermediates (proto-crystalline precursors) which finally crystallized to either form I or form II was elucidated. Investigations of the PDF pattern allowed a detailed analysis of the polyamorphism. Based on these data, it can be concluded, that the solvent triggers the pre-ordering of the amorphous precursor and determines the fate of the resulting crystal structure.
Results and discussion
Paracetamol was chosen as a model system for our studies. While three crystalline modifications of paracetamol are known to exist,38,39 only the structure of the thermodynamically stable monoclinic polymorph I and the metastable orthorhombic polymorph II are well examined.40 In a typical crystallization experiment 5 μL of a paracetamol- solution was injected in an acoustic levitator. This device offers a surface-free sample handling, controlled temperature, and humidity (see Fig. 1). As a result of the solvent evaporation, the concentration of the analyte increased leading to a supersaturated solution. At this point, the crystallization of the analyte can be observed. The crystallization process was studied simultaneously using synchrotron XRD and Raman spectroscopy at a high time resolution (approx. 15 s per XRD pattern/Raman spectrum). These measurement conditions enable the detection and unambiguous assignment of phases (solution, amorphous, crystalline) and transformations arising during the crystallization from solution to the solid final form. In a second step, total scattering experiments were performed under the same experimental conditions to gain more information about the amorphous intermediates. | ||
| Fig. 1 Experimental setup with the acoustic levitator and climatic chamber for the control of crystallization conditions. Setup for the crystallization investigation with simultaneously recorded synchrotron X-ray pattern (λX = 1 Å = 0.1 nm) and Raman spectra (λL = 785 nm). Similar setup for the pair distribution function analysis with synchrotron X-ray radiation (λX = 0.158 Å = 0.0158 nm). Raman spectra were not measured. | ||
Repeated experiments using different solvents led to the crystallization of either the thermodynamically stable monoclinic form I or the metastable orthorhombic form II at room temperature. The purity of the crystalline products is documented by the X-ray patterns and the Raman spectra showing exclusively characteristic signals of one of the two polymorphs (see Fig. 2, bottom left and middle).41
 | ||
| Fig. 2 The crystallization process of form I (top) and form II (middle) detected by combined in situ synchrotron XRD (top and middle left) and Raman spectroscopy (top and middle right). The X-ray patterns of the crystallized polymorphs are compared with the data entries HXACAN27 (form I) and HXACAN31 (form II) of the CSD (bottom left). The Raman spectra of the amorphous phases (bottom right) and final products (bottom middle) during the crystallization to form I and form II are presented. The enlargement of the vibration signal of the methyl group in methanol solution is shown in the range of 2850–3060 cm−1 (bottom right). | ||
Mixtures of both forms, I and II, were never obtained. In Table 1 the used solvents and obtained polymorphs are summarized. The formation of pure form I from solution and melt has been described in literature.42 The isolation of the metastable modification II from solution is difficult due to the instability and the fast transformation mechanism to the stable polymorph I. The growth of pure form II crystals was only possible from saturated solution and using vaccination crystals of form II.42 These experiments were conducted in glass containers. As a consequence low nucleus contamination of form I could still be traced, which accelerates the conversion to form I.43Fig. 2 shows two typical examples for the crystallization progress of the modification I from 1-propanol (see Fig. 2, top) and modification II from methanol (see Fig. 2, middle). Initially, the broad amorphous X-ray scattering of the solution with the solvents is detected (0.30 min and 0.15 min). The corresponding Raman spectra show the typical signals for 1-propanol and methanol. 1-propanol can be identified by the methyl rocking and C–C stretching vibration at 859 cm−1 (see Fig. 2, top, marked yellow). The methyl stretching bands for 1-propanol are at 2878 cm−1 (symmetric) and 2964 cm−1 (asymmetric); the corresponding vibrations for methanol can be found at 2835 cm−1 and 2941 cm−1 (see Fig. 2, middle, marked yellow).44 In comparing the positions of these Raman signals with the Raman spectra of pure 1-propanol and methanol only the asymmetric methyl stretch band of methanol is shifted to larger wave number 2945 cm−1 (see Fig. 2, bottom right, enlargement). This fact indicates that the C–H bond strength of the methanol methyl group is weaker in solution with paracetamol molecules. With increased reaction time, the intensity of the signals attributed to the solvents decrease and the characteristic vibration signals of amorphous paracetamol at 1236 cm−1, 1280 cm−1, 1326 cm−1, 1376 cm−1 and 1618 cm−1 increase (10.00 min and 18.30 min).45 At this time, the X-ray patterns exhibit no reflexes. After the start of the crystallization, first reflexes of the polymorphs appear (10.20 min and 18.40 min) increasing in intensity. The polymorphs were identified based on their diffractograms and Raman spectra (see Table 2).
| Solvents | Observed polymorphs |
|---|---|
| Acetic acid, acetone, acetonitrile, chloroform, ethanol, 1,4-dioxane, 1-propanol, 2-propanol, tetrahydrofuran | Form I |
| 2-Butanone, ethyl acetate, methanol, water | Form I, form II |
| Assignment of the vibrations | Raman bands of form I [cm−1] | Raman bands of form II [cm−1] |
|---|---|---|
| C–H bending (phenyl ring) | 652 | 651 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bending O bending |
798 | 799 |
| Phenyl ring | 858, 1169, 1278, 1324, 1372 | 861, 1170, 1280, 1327, 1375 |
| O–H bending | 1611, 1618 | 1609, 1624 |
| CO stretching |
1649 | 1642, 1651 |
| N–H bending | 1649 | 1649 |
| Symmetrical methyl stretching | 2932 | 2935 |
The comparison of the two progresses shows distinct similarities: after the solvent evaporates, an amorphous phase is formed from which the crystallization of the corresponding polymorph proceeds. Thorough analysis of the amorphous X-ray patterns arising after solvent evaporation displays a shift of 1° in 2θ of the diffuse scattering backgrounds (10.05 min and 18.25 min). The simultaneously recorded Raman spectra of the amorphous states (see Fig. 2, bottom right) show characteristic vibration signals with almost identical relative intensities and broad bands, based on the distribution range of bond lengths and bond angles within these disordered phases. Differences in band positions and relative intensities can be indicated from 2800 to 3100 cm−1. This range can be assigned to the symmetric and asymmetric vibrations of the methyl group and the C–H stretching vibrations of the phenyl ring. The characteristic symmetric methyl stretch vibration of the corresponding solvent with low intensity can be found in both Raman spectra. Small traces of solvent are detectable next to the amorphous phases. These residues vanish completely at the onset of the crystallization. Obviously, the two amorphous phases differ which is expressed in the minor differences in diffractograms and Raman spectra. Previous studies showed that the polymorphic structures of paracetamol are determined by the intermolecular hydrogen bonds of the hydroxyl and acetamido group.46 An influence of the hydrogen bond strength on the torsion between acetamido group and the phenyl plane is described. The authors also confirmed a strong correlation between the length of the hydrogen bonds and the dynamic and rotation behavior of the methyl unit. Taking into account those results and our findings, hydrogen bonds also define the amorphous phases with the effect on the paracetamol molecule characteristics including methyl group interaction and angle between phenyl plane and acetamido group.47,48 We assume that the amorphous states already contain polymorph-specific (proto-crystalline) information controlled by the nature of the solvent. To obtain structural information on these amorphous precursors, PDF analyses based on high resolution synchrotron data were used. The same experimental conditions were chosen for the total scattering experiments. The time-resolved diffraction patterns and the corresponding converted PDFs are shown in Fig. 3. The atomic pair distribution function represents the probability to find two atoms separated by a distance r.49 Based on the diffraction patterns, solution and amorphous phase can be distinguished from the crystalline state. The start of the crystallization can be determined from the appearance of Bragg reflexes. At the beginning, the solution exhibits the scattering maximum at q = 14.46 nm−1 (20.42° 2θ, Cu) in 1-propanol and q = 16.88 nm−1 (23.89° 2θ, Cu) in methanol. With progressive evaporation the maximum shifts by 0.16 nm−1 (0.23° 2θ, Cu) to higher scattering vectors prior to the crystallization of form I. During the formation of polymorph II, a shift of 1.62 nm−1 (2.32° 2θ, Cu) to smaller q is detected during the formation of the amorphous phase. For the PDF analysis, scattering data up to qmax = 170 nm−1 were used to generate the G(r) functions. The evaluation of the converted PDFs requires the differentiation between intra- and intermolecular atom–atom distances. We define the distances within a paracetamol molecule as i ≤ 8 Å, whereas j ≥ 2.5 Å describes the atom–atom distance between paracetamol molecules. The overlapping interval represents intra- and intermolecular distances (see Fig. 3, shaded in grey). The PDFs of the solution and the amorphous states lack significant intermediate range order. Therefore, the PDFs can be considered as PDFs of single molecules. During the evaporation of the solvent, the intramolecular distances of a paracetamol molecule remain unaltered, the analyses focus on changes in the range in which intra- and intermolecular distances co-occur. The polymorphic structures of paracetamol are characterized by hydrogen bonds between the hydroxyl group and oxygen atom of the carbonyl group forming chains. These chains are bound by further hydrogen bonds between the N–H unit and the hydroxyl group resulting in folded layers (form I) and flat layers (form II).
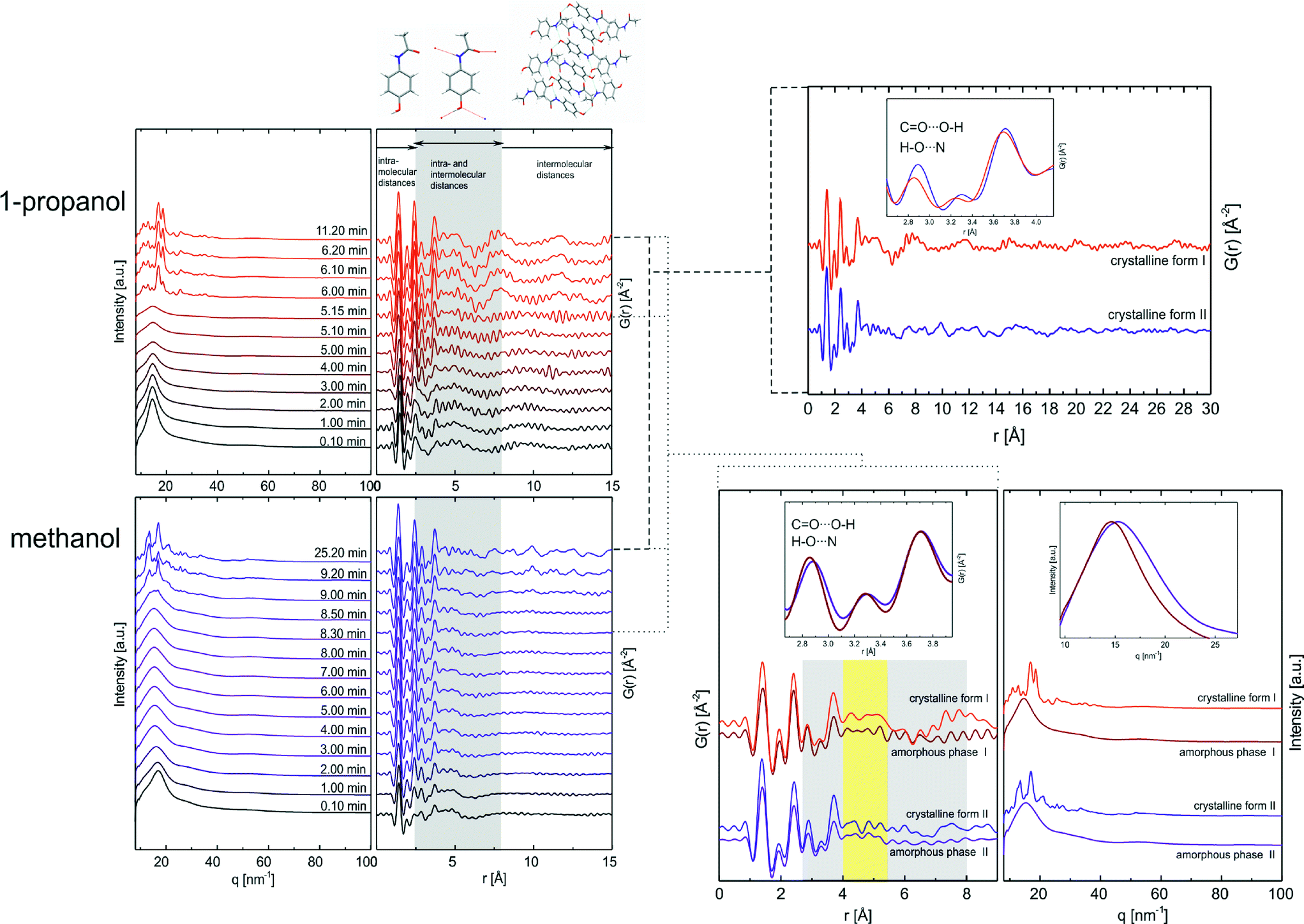 | ||
| Fig. 3 Time-resolved crystallization process of monoclinic form I (top left) from 1-propanol and orthorhombic form II (bottom left) from methanol are analyzed by high energy synchrotron X-ray to gain high q-range data. The PDFs of crystalline products are compared and structurally characteristics are highlighted in the enlargement (top right). The graphic below left displays the PDFs of amorphous phases shortly before crystallization starts compared with the PDFs of the corresponding crystalline forms. The inset shows a characteristic range from 2.65 to 4.0 Å. To the right, there are the corresponding X-ray patterns of the amorphous and crystalline phases. The inset highlights the scattering shift of the two different amorphous phases. | ||
The CO⋯O–H and H–O⋯N hydrogen bonds in the range of 2.65–2.93 Å lead to a pronounced peak in the PDF pattern (see Table 3). At higher distances the existence of these hydrogen bonds is additionally reflected in a distribution generated by the adjacent atoms to those involved in the hydrogen bonds (3.46–4.0 Å). When looking at the PDFs of the crystallization products of both processes (see Fig. 3, top right), two characteristic atom–atom distance peaks can be found in these areas. The enlargement reveals a slight peak shift of these peaks to smaller distances in the PDF of monoclinic form I (red) when comparing with the PDF of polymorph II (blue). This displacement reflects differences in the hydrogen bond strength and length between the polymorphic crystalline structures (see Table 3). Several diffraction and spectroscopic studies of crystalline paracetamol at low and high temperature confirm the described phenomenon by demonstrating larger strength of individual hydrogen bonds between hydroxyl and acetamido group in form I resulting in higher stability.41,48 The presence of long-range order in both crystalline forms is evident in the PDFs. The intensity decrease and the peak broadening of the patterns in both PDFs can be explained by the limited q-range used for the Fourier transformation and the low motion correlation with increasing distance. The PDFs in the early stages of crystallization process show atom distance distributions up to about 8 Å. In solution and in the amorphous stages distances within molecules and no long-range order can be detected. At the beginning of crystallization two significant peaks representing hydrogen bonds in the range of 2.65 to 2.93 Å and 3.46 to 4.0 Å have low intensities, which can be explained by the lack of hydrogen bonding network between analyte molecules. During the evaporation of the solvent, the intensity of these peaks increases indicating the formation of first structurally defining hydrogen bonds (after 5 min in 1-propanol or 1 min in methanol). An analysis of the PDFs shortly before the crystallization (5.15 min and 8.30 min) (see Fig. 3, left) suggests a similar distance distribution compared to the corresponding crystalline forms (see Fig. 3, insets top and bottom left). At this point, the characteristic hydrogen bonds between hydroxyl and acetamido group determining the paracetamol structure are formed. This is an indication that the nature of the solvent can influence the structure of the formed amorphous phase. The distribution shift of characteristic peaks to a lower range in the amorphous state of form I highlight the development of stronger and shorter hydrogen bonds between functional groups in contrast to form II. Closer examination of the PDFs in the overlapping intra- and intermolecular indicates major differences of peak positions and relative intensities in the region of 4–5.4 Å (see Fig. 3, marked yellow), reflecting differences in the arrangement of the amorphous phases (amorphous phase I and amorphous phase II). These differences occur prior to the crystallization of form I and form II.
| Intermolecular distances (D⋯A) | Form I [Å] | Form II [Å] |
|---|---|---|
| CO⋯O–H |
2.65 | 2.71 |
| H–O⋯N | 2.90 | 2.93 |
| CO⋯C–OH |
3.46 | 3.48 |
| OC⋯O–H |
3.69 | 3.76 |
| HO–C⋯N | 3.73 | 4.00 |
Comparing the PDFs of the amorphous phases with the PDFs of the resulting crystalline products in the range of 4.0–5.4 Å following observations are important: i) the PDF of the amorphous form I directly before the crystallization and the PDF of the crystalline form I show a pronounced resemblance. A similar, but stronger resemblance can be observed for the PDFs of the amorphous form II and crystalline form II. ii) For both polymorphs, the key features of the final crystalline phase is already pre-ordered and imprinted in the amorphous (proto-crystalline) precursor. The crystallization of the metastable form II proceeds from an amorphous precursor in which the key structure motifs of the crystalline form II are already formed to a great extent facilitating the fast crystallization of form II. Summarizing, the crystallization studies of paracetamol from different solvents and under controlled temperature and humidity result in two different pathways which involve two different amorphous states (see Fig. 4).
 | ||
| Fig. 4 Top: Overview of the crystallization of paracetamol from solution in different solvents and amorphous precursors during the crystallization of form I and form II. Bottom: Corresponding X-ray patterns of the different stages. | ||
Summary and conclusions
The influence of the solvent on the crystallization of different polymorphs of paracetamol was investigated in situ under controlled conditions minimizing further influencing factors. Based on the choice of the solvent, it was possible to crystallize selectively the monoclinic form I and the metastable form II.Two different amorphous states were identified as structure defining precursors during the crystallization. The two amorphous states are discussed in terms of an intrinsic structure directly related to the crystalline polymorphs I and II. These proto-crystalline structures were investigated by atomic pair distribution function analysis. Obviously, different solvents govern the local order of the amorphous intermediates selectively. For the first time, direct evidence for the occurrence of polyamorphism during the crystallization of an organic compound from solution could be derived.
We conclude that the crystallization process of different polymorphs I and II of paracetamol from solutions proceeds via amorphous states. It is central that two different amorphous precursor states are formed.
Our findings indicate the possibility to control the crystallization pathways for the selective isolation of a distinct crystalline structure.
Materials and methods
Materials
Paracetamol (≥99% purity, Sigma-Aldrich) was used without purification. Based on powder X-ray diffraction (Bruker AXS, D8 Discover, Cu Kα1 radiation) and Raman spectroscopy the compound was identified as the thermodynamically stable monoclinic form I. Paracetamol was dissolved in different organic solvents. Acetone (≥99.5%), ethanol (≥99.8%) and 2-propanol (≥99.7%) were purchased from Th. Geyer GmbH & Co. KG (Renningen, Germany), tetrahydrofuran (≥99.8%), 1,4-dioxane (≥99.5%), 1-propanol (≥99.5%), and acetic acid (100%) from Merck KGaA (Darmstadt, Germany), ethyl acetate (≥99.5%), and 2-butanone (≥99.5%) from Fluka Chemie GmbH (Buchs, Switzerland), acetonitrile (≥99.8%) from J. T. Baker (Deventer, Netherlands), methanol (≥99.9%) from Carl Roth GmbH + Co. KG (Karlsruhe, Germany), and chloroform (≥99.0%) from AppliChem GmbH (Darmstadt, Germany).Crystallization experiments
Solutions with half the saturation concentration of paracetamol in the corresponding solvent were prepared.50 In a typical experiment, a droplet (5–7 μL) was positioned with an Eppendorf pipette in a custom-made acoustic levitator equipped with a climate control device.51 This setup enables the investigation of crystallization processes without any influence of surfaces under controlled temperature and humidity.25,52,53 The temperature was kept at 22.5 ± 0.5 °C and the humidity was reduced to 17.5 ± 2.5% RH. The crystallization was analyzed by synchrotron X-ray diffraction combined with Raman spectroscopy. Total scattering experiments for pair distribution analysis were collected for the crystal formation from methanol and 1-propanol solution.Raman spectroscopy
Raman spectra were detected using a Raman RXN1™ Analyzer (Kaiser Optical Systems, Inc., Ecully, France) with NIR excitation radiation at 785 nm, equipped with a CCD camera (1024 × 256 pixels). A non-contact probe head (working distance 1.5 cm, spot size 1 mm ∅) was used for the experiments. Spectra were recorded with an irradiance of 6.6 W cm−2 on the sample and an acquisition time of 1 × 8 s, resulting in a time resolution of 15 s.Scattering experiments
XRD experiments were performed at the μSpot beamline (BESSY II, Helmholtz Centre Berlin for Materials and Energy). A beam diameter of 100 μm at a photon flux of 1 × 109 s−1 and a ring current of 300 mA was used.54 The beam wavelength was 1.000 Å. Diffraction patterns were collected with a MarMosaic CCD detector (3072 × 3072 pixels) and a time resolution of 15 s. The entries HXACAN27 and HXACAN31 of the Cambridge Structural Database were used for the assignment of the monoclinic modification I and orthorhombic II.Total scattering data were performed at the beamline ID11 at the ESRF (European Synchrotron Radiation Facility) with a time resolution of 1 s and 10 s using PDF method for the acquisition. A Frelon CCD detector (47.2 × 47.2 pixels) located 85.8 mm behind the sample was used. A wavelength of 0.158154 Å and a beam size of 300 × 300 μm were used. The measurements were conducted with a data acquisition time of 10 s until the crystallization process was completed. All synchrotron 2D pattern are converted into the equivalent 1D profiles (intensity versus scattering vector q).55
Pair distribution function (PDF) analyses based on experimental total scattering data were generated using PDFgetX3.56 The Fourier transformation calculates G(r),
| G(r) = 4πr[ρ(r) − ρ0] |
where ρ(r) is the microscopic pair density, ρ0 is the average number density and Q the momentum transfer. S(Q) is the normalized structure function determined from the experimental diffraction intensity. Further information about the PDF method can be found in the literature.49 For qualitative analyses the PDFs were compared.

Acknowledgements
We gratefully acknowledge financial support by the Deutsche Forschungsgemeinschaft through SPP 1415 (crystalline non-equilibrium phases and polymorphs): Ra494/15-1, and Em198/4-1.References
- I. Sunagawa, J. Cryst. Growth, 1977, 42, 214–223 CrossRef CAS.
- D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergstrom and H. Colfen, Chem. Soc. Rev., 2014, 43, 2348–2371 RSC.
- J. P. Brog, C. L. Chanez, A. Crochet and K. M. Fromm, RSC Adv., 2013, 3, 16905–16931 RSC.
- A. Gavezzotti and G. Filippini, J. Am. Chem. Soc., 1995, 117, 12299–12305 CrossRef CAS.
- T. L. Threlfall, Analyst, 1996, 121, 269–269 RSC.
- P. H. Poole, T. Grande, F. Sciortino, H. E. Stanley and C. A. Angell, Comput. Mater. Sci., 1995, 4, 373–382 CrossRef CAS.
- T. Loerting, V. V. Brazhkin and T. Morishita, Adv. Chem. Phys., 2009, 143, 29–82 CrossRef CAS.
- J. Wiedersich, A. Kudlik, J. Gottwald, G. Benini, I. Roggatz and E. Rossler, J. Phys. Chem. B, 1997, 101, 5800–5803 CrossRef CAS.
- M. M. Koza, B. Geil, K. Winkel, C. Kohler, F. Czeschka, M. Scheuermann, H. Schober and T. Hansen, Phys. Rev. Lett., 2005, 94 Search PubMed.
- R. J. Hemley, J. Badro and D. M. Teter, Phys. Meets Mineral., 2000, 173–204 CAS.
- J. H. E. Cartwright, A. G. Checa, J. D. Gale, D. Gebauer and C. I. Sainz-Diaz, Angew. Chem., Int. Ed., 2012, 51, 11960–11970 CrossRef CAS PubMed.
- X. Gang and C. L. Chien, J. Appl. Phys., 1987, 61, 3246–3248 CrossRef.
- C. A. Hoel, S. Xie, C. Benmore, C. Malliakas, J.-F. Gaillard and K. R. Poeppelmeier, Z. Anorg. Allg. Chem., 2011, 637, 885–894 CrossRef CAS.
- K. Winkel, W. Hage, T. Loerting, S. L. Price and E. Mayer, J. Am. Chem. Soc., 2007, 129, 13863–13871 CrossRef CAS PubMed.
- B. C. Hancock, E. Y. Shalaev and S. L. Shamblin, J. Pharm. Pharmacol., 2002, 54, 1151–1152 CrossRef CAS PubMed.
- C. Sun, D. Xu and D. Xue, CrystEngComm, 2013, 15, 7783–7791 RSC.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- H. Y. Yoshikawa, Y. Hosokawa and H. Masuhara, Cryst. Growth Des., 2006, 6, 302–305 CAS.
- B. C. Knott, V. Molinero, M. F. Doherty and B. Peters, J. Am. Chem. Soc., 2012, 134, 19544–19547 CrossRef CAS PubMed.
- E. Sanz, C. Vega, J. R. Espinosa, R. Caballero-Bernal, J. L. F. Abascal and C. Valeriani, J. Am. Chem. Soc., 2013, 135, 15008–15017 CrossRef CAS PubMed.
- S. L. Price, Acc. Chem. Res., 2009, 42, 117–126 CrossRef CAS PubMed.
- A. T. Hulme and D. A. Tocher, Cryst. Growth Des., 2006, 6, 481–487 CAS.
- A. Johnston, A. J. Florence, N. Shankland, A. R. Kennedy, K. Shankland and S. L. Price, Cryst. Growth Des., 2007, 7, 705–712 CAS.
- L. C. Jacobson, W. Hujo and V. Molinero, J. Am. Chem. Soc., 2010, 132, 11806–11811 CrossRef CAS PubMed.
- S. E. Wolf, J. Leiterer, M. Kappl, F. Emmerling and W. Tremel, J. Am. Chem. Soc., 2008, 130, 12342–12347 CrossRef CAS PubMed.
- B. Xie, T. J. Halter, B. M. Borah and G. H. Nancollas, Cryst. Growth Des., 2014, 14, 1659–1665 CAS.
- P. Gruene, D. M. Rayner, B. Redlich, A. F. G. van der Meer, J. T. Lyon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS PubMed.
- A. Dey, P. H. H. Bomans, F. A. Mueller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 CrossRef CAS PubMed.
- D. Gebauer, A. Voelkel and H. Coelfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- T. Gnutzmann, K. Rademann and F. Emmerling, Chem. Commun., 2012, 48, 1638–1640 RSC.
- K. Seefeldt, J. Miller, F. Alvarez-Nunez and N. Rodriguez-Hornedo, J. Pharm. Sci., 2007, 96, 1147–1158 CrossRef CAS PubMed.
- H. W. Sheng, H. Z. Liu, Y. Q. Cheng, J. Wen, P. L. Lee, W. K. Luo, S. D. Shastri and E. Ma, Nat. Mater., 2007, 6, 192–197 CrossRef CAS PubMed.
- R. B. Neder and V. I. Korsunskiy, J. Phys.: Condens. Matter, 2005, 17, S125–S134 CrossRef CAS.
- S. J. L. Billinge and M. G. Kanatzidis, Chem. Commun., 2004, 749–760 RSC.
- K. Nollenberger, A. Gryczke, C. Meier, J. Dressman, M. U. Schmidt and S. Bruehne, J. Pharm. Sci., 2009, 98, 1476–1486 CrossRef CAS PubMed.
- D. Prill, P. Juhas, M. U. Schmidt and S. J. L. Billinge, J. Appl. Crystallogr., 2015, 48, 171–178 CAS.
- P. J. Chupas, X. Y. Qiu, J. C. Hanson, P. L. Lee, C. P. Grey and S. J. L. Billinge, J. Appl. Crystallogr., 2003, 36, 1342–1347 CrossRef CAS.
- M. Haisa, S. Kashino and H. Maeda, Acta Crystallogr., Sect. B: Struct. Sci., 1974, 30, 2510–2512 CrossRef.
- M. A. Perrin, M. A. Neumann, H. Elmaleh and L. Zaske, Chem. Commun., 2009, 3181–3183 RSC.
- P. DiMartino, A. M. GuyotHermann, P. Conflant, M. Drache and J. C. Guyot, Int. J. Pharm., 1996, 128, 1–8 CrossRef.
- B. A. Kolesov, M. A. Mikhailenko and E. V. Boldyreva, Phys. Chem. Chem. Phys., 2011, 13, 14243–14253 RSC.
- G. Nichols and C. S. Frampton, J. Pharm. Sci., 1998, 87, 684–693 CrossRef CAS PubMed.
- K. Kachrimanis, D. E. Braun and U. J. Griesser, J. Pharm. Biomed. Anal., 2007, 43, 407–412 CrossRef CAS PubMed.
- R. L. Sobocinski and J. E. Pemberton, Langmuir, 1992, 8, 2049–2063 CrossRef CAS.
- A. Luczak, L. J. Jallo, R. N. Dave and Z. Iqbal, Powder Technol., 2013, 236, 52–62 CrossRef CAS.
- I. D. H. Oswald, D. R. Allan, P. A. McGregor, W. D. S. Motherwell, S. Parsons and C. R. Pulham, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 1057–1066 Search PubMed.
- I. G. Binev, P. Vassileva-Boyadjieva and Y. I. Binev, J. Mol. Struct., 1998, 447, 235–246 CrossRef CAS.
- E. B. Burgina, V. P. Baltakhinov, E. V. Boldyreva and T. P. Shakhtschneider, J. Struct. Chem., 2004, 45, 64–73 CrossRef CAS.
- T. B. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Pergamon Materials Series, 2003 Search PubMed.
- R. A. Granberg and A. C. Rasmuson, J. Chem. Eng. Data, 1999, 44, 1391–1395 CrossRef CAS.
- M. C. Schlegel, K. J. Wenzel, A. Sarfraz, U. Panne and F. Emmerling, Rev. Sci. Instrum., 2012, 83 Search PubMed.
- J. Leiterer, F. Emmerling, U. Panne, W. Christen and K. Rademann, Langmuir, 2008, 24, 7970–7978 CrossRef CAS PubMed.
- T. Gnutzmann, Y. N. Thi, K. Rademann and F. Emmerling, Cryst. Growth Des., 2014, 14, 6445–6450 CAS.
- O. Paris, C. H. Li, S. Siegel, G. Weseloh, F. Emmerling, H. Riesemeier, A. Erko and P. Fratzl, J. Appl. Crystallogr., 2007, 40, S466–S470 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Hausermann, High Pressure Res., 1996, 14, 235–248 CrossRef.
- P. Juhas, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |