
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intermolecular interaction energies in transition metal coordination compounds†
Andrew G. P.
Maloney
ab,
Peter A.
Wood
b and
Simon
Parsons
*a
aEaStCHEM School of Chemistry and Centre for Science at Extreme Conditions, The University of Edinburgh, King's Buildings, West Mains Road, Edinburgh, Scotland, EH9 3FJ UK. E-mail: S.Parsons@ed.ac.uk; Fax: +0131 650 4743; Tel: +0131 650 5804
bCambridge Crystallographic Data Centre, 12 Union Road, Cambridge, England, CB2 1EZ UK
First published on 23rd September 2015
Abstract
Parameters required to perform PIXEL energy calculations, a semi-empirical method for evaluating intermolecular interactions, have been defined for the transition metals. Using these parameters, lattice energies of thirty-two 1st row, five 2nd row and six 3rd row transition metal complexes have been calculated and compared to experimental values giving correlations of calculated sublimation enthalpies comparable to those obtained for organic crystal structures. Applications of the method are illustrated by analysis of the intermolecular interactions in chromium hexacarbonyl, stacking interactions in bis(acetylacetonato)-oxo-vanadium(IV) and dihydrogen bonding. The results extend the applicability of the PIXEL method from organic materials (ca. 40% of the Cambridge Structural Database (CSD)) to a much wider range of organic and organometallic systems (ca. 85% of the CSD).
1 Introduction
Methods for interpretation of molecular crystal structures have advanced considerably in the past decade. While analysis of intermolecular interactions using graphical tools such as Mercury1 can be achieved in a matter of seconds using fast algorithms based on geometry, it is increasingly common to evaluate intermolecular interaction energies using ab initio methods,2–7 PIXEL calculations,8 symmetry-adapted perturbation theory9 or force fields.10 The results can be used to visualise contributing energy terms using Hirshfeld surfaces11 or energy vectors or frameworks.12–14 This progress has been applied to areas such as polymorphism15 and energy landscapes,16 cocrystals and solvates,17 crystal engineering,18 molecular recognition19 and extreme conditions research.20The techniques listed above have been applied extensively in work on organic materials.21 There is, nevertheless, substantial interest in intermolecular interactions in metal-containing systems.22 Crystal engineering and supramolecular chemistry frequently make use of strategies involving metals. For example, Orpen and co-workers have shown that H-bond acceptors based on metal halides and oxalates can be used to form more reliable and reproducible supramolecular building blocks than those based on purely organic ligands.23,24 Use of diplatinum thiocarboxylate complexes in bottom-up assembly of conductive one-dimensional nanostructures has been suggested on the basis of the strong (~50 kJ mol−1) intermolecular Pt⋯Pt interactions that occur in these systems.25 Density functional theory (DFT) has been used to study the different intermolecular energies of alternative Ru(II) hydrogenation catalysis pathways,26–28 while more recently Li et al. performed calculations to investigate the performance of cobalt and copper analogues of a pre-existing nickel catalyst for olefin purification.29 A great deal of computational effort has been invested in the study of the binding affinities and selectivities of metalloprotein–ligand interactions in these systems,30–32 and the desire to find more efficient methods of drug design means that computational analysis of metal-based pharmaceuticals is an ever expanding field,33 exemplified by the analysis of the interactions of zinc ions with several anti-inflammatory drugs.34 Metal–organic frameworks, large porous structures consisting of metal ions linked by organic ligands, are increasingly being studied as potential gas storage and separation materials, and a variety of computational methods are used to study the adsorption of small molecules in these systems.35–37
The development of the PIXEL method,8,21,38,39 a semi-empirical technique for evaluating intermolecular interactions based on integrations over calculated electron densities of molecules, has allowed energetic analysis in organic crystal structures to be carried out quickly with an accuracy comparable to high level quantum mechanical methods.40,41 PIXEL calculations yield the total lattice energy partitioned into individual molecule–molecule energies, which are themselves partitioned into four terms: coulombic, polarisation, dispersion and repulsion. The separation of contributions allows for the character of individual interactions and overall crystal packing to be inferred from the dominant terms, providing chemical insight. The PIXEL method requires definition of certain atomic parameters, but these are mostly physically measurable quantities such as ionisation potentials, and one of the most appealing features of the method is the transferability of parameters across many different chemical systems. The PIXEL method is thus a potentially valuable addition to established techniques applied to metal-containing systems, such as ab initio methods and calculations based on force fields.
The aims of this paper are (i) to define a validated PIXEL parameter set for use with the d-block metals, and (ii) to illustrate possible applications of the method in interpreting intermolecular interactions in metal-containing structures. This expands the potential applicability of the PIXEL method from organic materials (~40% of the CSD) to the majority of organic and organometallic structures (~85% of the CSD).
2 Parameterisation and validation criteria
2.1 Definition of metal parameters
The four energy terms evaluated during a PIXEL calculation depend on a small number of fundamental atomic parameters. Values of these parameters for atoms common in organic chemistry are embedded in the PIXEL code. In the present work we have defined new values for transition metals and validated them against experimental sublimation enthalpies (ΔHsub) using the convention that lattice energies are approximately equal to –ΔHsub. This procedure assumes that there are no intramolecular structural changes on passing from the solid state to the gas phase. The names used to refer to the parameters in the following sections are those used in the PIXEL program documentation and Gavezzotti's publications, where full details of their definition, use and significance can be found.21,38,39Definition of some parameters is straightforward. ZTOT and ZVAL are the total number of electrons and the number of valence electrons in the neutral atom; POTIO is the first ionisation energy (in atomic units) and WEIGHT is the atomic weight.42 For other parameters a choice among several possibilities needs to be made. Unless otherwise specified, values used for non-metallic elements in the compounds were the program defaults.
Dispersion energies are calculated in a London-type expression in which the ionisation energy of a pixel is used to approximate its ‘oscillator strength’. DIFA (also given the symbol β in Gavezzotti's papers) is a “variable ionisation” parameter which controls the diminution of the ionisation energy of a pixel as the distance from the nucleus increases.21 Gavezzotti's recommended value of 0.4 Å−1 was used throughout. Variation of DIFA(Ti) between 0.1 and 1.0 Å−1 yielded values of the lattice energy of TiCl4 between −46.4 and −57.7 kJ mol−1. A value of −53.0 kJ mol−1 was obtained‡ with the standard value 0.4 Å−1, compared to the experimental sublimation enthalpy of 51.9 kJ mol−1 (calculated from the enthalpies of formation of the gas and solid taken from the NIST Chemistry WebBook, http://webbook.nist.gov/chemistry/).
The covalent radii of atoms, RINTER, are used in PIXEL to check for short internuclear distances, and not used to calculate energies. Values were taken from Emsley's compilation.42RAVDW, the van der Waals radius, is used to assign pixels of electron density to atomic basins. The sets of values reported by Nag et al.43 and Batsanov44 were tested.
The atomic polarisability, POLZE or α in Å3, appears in the calculation of both the polarisation and dispersion terms. For non-metallic species the PIXEL method makes use of the Slater–Kirkwood approximation to estimate α according to eqn (1),
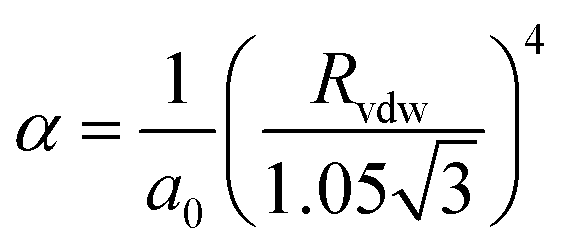 | (1) |
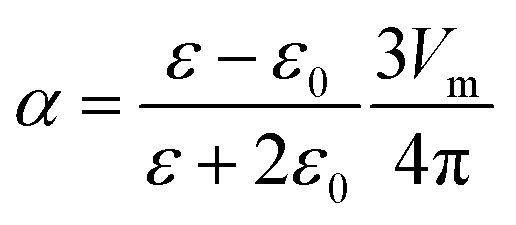 | (2) |
The electronegativity, ELNEG, is used in the calculation of the repulsion energy. Both Pauling and Allred–Rochow values42 were investigated.
2.2 PIXEL calculations and the treatment of ligand parameters
PIXEL calculations can be carried out in one of two ways using the programs PIXELc or PIXELd of Gavezzotti's CLP package of modelling and structure analysis software.45 PIXELd is intended for the calculation of interaction energies of discrete dimers. PIXELc is used for calculations on a complete crystal structure, yielding a lattice energy which is further broken-down into the contributions from individual molecule–molecule pairs out to a specified cluster cut-off distance.PIXELc calculations can be carried out straightforwardly on crystal structures with one or two molecules in the asymmetric unit. More complex cases can be handled with some user intervention. For example, where a molecule lies on a special position the space group symmetry should be lowered and the structure specified using whole molecules. Calculations on disordered structures can be carried out using permutations of PIXELc calculations considering different molecular pairs.46
We thank a referee to this paper for pointing out that some structures may in fact be more readily amenable for processing using a series of PIXELd dimer calculations, while PIXELc can also be used to calculate dimer energies for a user-specified list of symmetry operations if the cluster cut-off distance is set to zero. Although both of these procedures would fail to reflect the many-body nature of the polarisation contribution to the lattice energy, they could be applied to the interpretation of crystal packing. We also note that a correction is applied to PIXELc lattice energies of structures in polar space groups.47 Details of calculations along with an extensive set of tutorial examples are provided with the CLP documentation.
All PIXEL calculations in the present study were carried out using a version of PIXELc48 which had been modified to read a table of transition metal parameters described in section 2.1. For each structure OH, NH and CH distances were normalised to 0.993, 1.015 and 1.089 Å, respectively.49 This procedure involves moving H positions to values typically obtained by neutron diffraction studies and corrects approximately for the effects of asphericity of H-atom electron densities which lead to systematic shortening of distances involving hydrogen atoms when determined by X-ray diffraction. The electron density was obtained in a single-point calculation with a B3LYP functional and a 6-31G** basis-set (Gaussian09)50 for main group elements and first-row transition metals. Second-row transition metal species were treated with the LanL2DZ basis-set, and third-row metals used the LanL2DZ basis-set with pseudopotentials to model the core orbitals of the metal atom. The “cube” format electron density files were then used in PIXEL calculations. Unless otherwise specified, the pixel size for all calculations was 0.16 × 0.16 × 0.16 Å (corresponding to ‘condensation level’ 4), and the cluster cut-off distance was kept at the default value generated by PIXEL for each structure.21
While atomic parameters in PIXEL calculations are intended to be transferable between different compounds, values of atomic polarisabilities may be varied depending on chemical bonding. For instance, three different atomic polarisabilities are used for carbon, depending on whether it is aliphatic, aromatic or ‘bridging aromatic’ as in naphthalene. While such differences might ideally be taken into account for other species (e.g. carbonyl and ether oxygen atoms), the dominance of carbon in organic compounds means that it is much more important to account for variation in its different chemical environments than it is for less abundant atomic species. While in practice only α(C) is usually varied, modification of the atomic polarisabilities of other species has been applied previously by Gavezzotti,51 for example for chloride ions in ionic organic crystals.
Although carbon is a common constituent of many ligands, it may be necessary to consider alternative values of polarisabilities of non-carbon atoms in cases such as homoleptic carbonyl complexes where the molecular surface is composed of exposed oxygen atoms. PIXEL analysis of molecular carbon monoxide using the default parameters in the program yields a lattice energy of −7.9 kJ mol−1. The experimental sublimation enthalpy is 7.9(2) kJ mol−1 (average value from three determinations). However, when carbon monoxide acts as a ligand, PIXEL results were found to be around 20 kJ mol−1 lower than the literature value when the default value of α(O) (0.75 Å3) was used (e.g. Cr(CO)6, literature sublimation enthalpy 69.6 kJ mol−1, calculated lattice energy −47.8 kJ mol−1). Carbonyl oxygen therefore, like carbon, seems to require its own value of α(O) depending on whether the CO is ligating or not. By testing different values of atomic polarisability of O, a value of α(O) = 1.0 Å3 was chosen for this species when carbon monoxide is acting as a ligand, yielding a lattice energy of −70.5 kJ mol−1 for Cr(CO)6. Support for these adjustments was obtained by calculation (AIMAll)52 of atomic polarisabilities in CO and Cr(CO)6 by the Atoms In Molecules method53 as described by Keith52,54 at the B3LYP/6-31G** level, using structures optimised at the same level of theory. The values of α(O) obtained for CO and Cr(CO)6 were 0.57 and 0.86 Å3 respectively, a similar relative increase to the one proposed above. Ligating sulfur atoms were given a polarisability of 3.6 Å3. Program defaults were otherwise used, Table 1 showing the full set of values used for ligand atoms in all calculations.
| Atom | Atomic polarisability |
|---|---|
| H | 0.39 |
| C aliphatic | 1.05 |
| C aromatic | 1.35 |
| C aromatic bridge | 1.90 |
| N | 0.95 |
| O | 0.75 |
| O carbonyl | 1.00 |
| F | 0.40 |
| Cl | 2.30 |
| S | 3.00 |
| S ligating | 3.60 |
2.3 Construction of a validation set of experimental sublimation enthalpies
A compilation of experimental sublimation enthalpies55 was cross-referenced with the Cambridge Structural Database56,57 to obtain a set of transition metal complexes for which both crystal structure and experimental sublimation data are available. Sublimation enthalpies are notoriously difficult to measure, and different determinations may yield wildly disparate results: for example two different measurements of the sublimation enthalpy of ferrocene give values of 64.6 kJ mol−1 and 84.0 kJ mol−1.58,59 For this reason, only compounds with a minimum of two independent sublimation enthalpy determinations were used for validation. The enthalpy values were arithmetically averaged with no weighting after elimination of any egregious outliers. The full validation data set, which contains 43 different compounds, is given in Table 2, and chemical structures are given in the ESI.† Also listed in Table 2 are the CSD refcodes, along with average experimental sublimation enthalpies calculated from data in ref. 55 and PIXEL calculated lattice energies. All complexes investigated had centrosymmetric crystal structures, and no polarisation corrections47 were necessary.| Compound number | CSD refcode | Metal | Experimental sublimation enthalpy | −PIXEL calculated lattice energy | |Difference| (%) |
|---|---|---|---|---|---|
| 1 | ACACCR07 | Cr | 119.5 ± 8.7 | 124.5 | 4.2 |
| 2 | ACACCS | Cr | 120.4 ± 13.3 | 118.1 | 1.9 |
| 3 | ACACCU02 | Cu | 118.7 ± 9.9 | 113.4 | 4.5 |
| 4 | ACACMN21 | Mn | 119.3 ± 6.0 | 126.7 | 6.2 |
| 5 | ACACVO12 | V | 140.6 ± 0.4 | 143.7 | 2.2 |
| 6 | BZCRCO14 | Cr | 94.6 ± 4.7 | 93.8 | 0.8 |
| 7 | CCRTOL01 | Cr | 94.0 ± 1.4 | 97.7 | 3.9 |
| 8 | CDCPTI04 | Ti | 122.5 ± 3.0 | 131.8 | 7.6 |
| 9 | CEHPIO01 | Ti | 94.6 ± 9.0 | 93.5 | 1.2 |
| 10 | COACAC10 | Co | 138.7 ± 4.0 | 136.2 | 1.8 |
| 11 | CPNDYV07 | V | 58.2 ± 1.1 | 76.8 | 32.0 |
| 12 | CUBEAC01 | Cu | 156.6 ± 4.0 | 157.0 | 0.3 |
| 13 | CUQUIN05 | Cu | 166.3 ± 5.3 | 159.6 | 4.0 |
| 14 | DBENCR11 | Cr | 83.6 ± 6.1 | 93.7 | 12.1 |
| 15 | DCYPCO04 | Co | 71.1 ± 1.5 | 75.8 | 6.6 |
| 16 | DERNOD05 | Cu | 124.9 ± 2.9 | 129.5 | 3.7 |
| 17 | DMTCCU | Cu | 150.8 ± 4.6 | 170.9 | 13.3 |
| 18 | DPIMNI | Ni | 128.1 ± 24.2 | 119.2 | 7.5 |
| 19 | DURHEE | Ni | 120.4 ± 11.5 | 99.2 | 17.6 |
| 20 | FEACAC03 | Fe | 121.92 ± 9.0 | 120.0 | 1.6 |
| 21 | FEROCE27 | Fe | 73.6 ± 4.2 | 71.5 | 2.9 |
| 22 | FOHCOU02 | Cr | 69.3 ± 2.6 | 70.5 | 1.7 |
| 23 | IGAGEC | Cr | 117.5 ± 7.8 | 81.6 | 30.6 |
| 24 | IPEZOS | Cu | 129.0 ± 1.9 | 124.0 | 3.9 |
| 25 | IPTCNI10 | Ni | 145.5 ± 3.5 | 152.8 | 5.0 |
| 26 | LIYLIO | Co | 132.6 ± 15.3 | 134.2 | 1.2 |
| 27 | MACACU10 | Cu | 133.0 ± 2.5 | 124.7 | 6.2 |
| 28 | NCKLCN01 | Ni | 71.2 ± 1.0 | 74.1 | 4.1 |
| 29 | NIDCAR06 | Ni | 153.7 ± 2.9 | 151.6 | 1.4 |
| 30 | NISALO01 | Ni | 109.5 ± 3.5 | 115.4 | 5.4 |
| 31 | QQQBWP03 | Cu | 113.5 ± 2.4 | 115.6 | 1.9 |
| 32 | TCBMNI | Ni | 146.0 ± 8.5 | 148.8 | 1.9 |
| 33 | ACACPD01 | Pd | 128.1 ± 4.0 | 138.6 | 8.2 |
| 34 | CYCPRU06 | Ru | 78.8 ± 3.4 | 83.0 | 5.3 |
| 35 | FUBYIK01 | Mo | 72.6 ± 3.5 | 65.7 | 9.5 |
| 36 | HCYPMO02 | Mo | 87.0 ± 7.8 | 82.9 | 4.7 |
| 37 | HQUIPD | Pd | 163.3 ± 6.7 | 154.4 | 5.5 |
| 38 | KOKPEF | Hf | 104.4 ± 4.9 | 94.9 | 9.1 |
| 39 | KOVSOD02 | W | 74.9 ± 2.9 | 55.9 | 25.4 |
| 40 | QQQCXJ02 | Ir | 112.0 ± 21.6 | 115.3 | 2.9 |
| 41 | REGSAY | Hf | 127.2 ± 3.5 | 109.1 | 14.2 |
| 42 | REPKIH | W | 90.4 ± 8.2 | 101.2 | 11.9 |
| 43 | SINWER | Os | 76.2 ± 3.9 | 84.6 | 11.0 |
2.4 Calculations of individual intermolecular interactions
Dimers displaying a variety of intermolecular interaction types were selected to compare the PIXEL results with those calculated using higher level computational methods. A range of interactions was investigated involving chromium hexacarbonyl, vanadyl stacking and metal hydrides participating in dihydrogen bonding.60–62 For each system a combination of Mercury 3.5 and Materials Studio V563 was used to obtain a structural model which was then optimised using Gaussian09. In cases where calculations had been previously reported, we used the same level of theory and basis-set as in the literature study. The optimised structures were then used for PIXELc calculations as described above. Further computational details are given in the relevant sections below, but Hirshfeld surface analysis was carried out with CrystalExplorer 3.1,64 where the required wavefunctions were calculated with the program TONTO 3.2 rev. 4048,65 while analysis of PIXEL results was accomplished using processPIXEL.123 Results
3.1 Parameter set selection and reproduction of experimental sublimation enthalpy data
Five different parameter sets were constructed using different combinations of methods for estimation of the van der Waals radii, polarisability and electronegativity, as defined in section 2.1. The different combinations and values for each parameter set are given in Tables S2 and S3 in the ESI.† Following Gavezzotti,40 the performance of the sets was quantified using the gradients and correlation coefficients of straight-line fits of the experimental sublimation enthalpies of Table 2 to the calculated lattice energies. The straight-line fitting statistics are listed in Table S2;† the fits used unit weights and were constrained to intercept at the origin.The data in Table S2† show that there is little difference between the performance of the different parameter sets, demonstrating the robustness of the PIXEL method to different choices of reasonable parameters. We recommend parameter set 5 (Table 3) because both the volume (and therefore the polarisability) of an elemental metal and its Allred–Rochow electronegativity are more unambiguously defined in terms of readily accessible experimental data than the quantities used in other parameter sets. Furthermore, the gradient for 2nd and 3rd row transition metals for set 5 is nearer unity than the other sets.
| Atom | DIFA | RAVDW | RINTER | ZTOT | ZVAL | POLZE | ELNEG | POTIO | WEIGHT |
|---|---|---|---|---|---|---|---|---|---|
| Ti | 0.4 | 2.15 | 1.32 | 22 | 4 | 4.18 | 1.32 | 0.251 | 47.88 |
| V | 0.4 | 2.05 | 1.22 | 23 | 5 | 3.31 | 1.45 | 0.248 | 50.94 |
| Cr | 0.4 | 2.05 | 1.17 | 24 | 6 | 2.86 | 1.56 | 0.249 | 52.00 |
| Mn | 0.4 | 2.05 | 1.17 | 25 | 7 | 2.93 | 1.60 | 0.273 | 54.94 |
| Fe | 0.4 | 2.05 | 1.16 | 26 | 8 | 2.81 | 1.64 | 0.289 | 55.85 |
| Co | 0.4 | 2.00 | 1.16 | 27 | 9 | 2.62 | 1.70 | 0.289 | 58.93 |
| Ni | 0.4 | 2.00 | 1.15 | 28 | 10 | 2.61 | 1.75 | 0.281 | 58.69 |
| Cu | 0.4 | 2.00 | 1.35 | 29 | 11 | 2.81 | 1.75 | 0.284 | 63.55 |
| Zn | 0.4 | 2.10 | 1.31 | 30 | 12 | 3.63 | 1.66 | 0.345 | 65.38 |
| Zr | 0.4 | 2.30 | 1.45 | 40 | 12 | 5.56 | 1.33 | 0.251 | 91.22 |
| Nb | 0.4 | 2.15 | 1.34 | 41 | 13 | 4.30 | 1.60 | 0.253 | 92.91 |
| Mo | 0.4 | 2.10 | 1.29 | 42 | 14 | 3.72 | 2.16 | 0.261 | 95.94 |
| Tc | 0.4 | 2.05 | 1.23 | 43 | 15 | 3.41 | 1.90 | 0.267 | 98.91 |
| Ru | 0.4 | 2.05 | 1.24 | 44 | 16 | 3.23 | 2.20 | 0.271 | 101.07 |
| Rh | 0.4 | 2.00 | 1.25 | 45 | 17 | 3.29 | 2.28 | 0.274 | 102.91 |
| Pd | 0.4 | 2.05 | 1.28 | 46 | 18 | 3.51 | 2.20 | 0.307 | 106.42 |
| Ag | 0.4 | 2.10 | 1.34 | 47 | 19 | 4.07 | 1.93 | 0.278 | 107.87 |
| Cd | 0.4 | 2.20 | 1.41 | 48 | 20 | 5.15 | 1.69 | 0.330 | 112.41 |
| Hf | 0.4 | 2.35 | 1.44 | 72 | 12 | 5.32 | 1.23 | 0.251 | 178.49 |
| Ta | 0.4 | 2.20 | 1.34 | 73 | 13 | 4.31 | 1.33 | 0.277 | 180.95 |
| W | 0.4 | 2.10 | 1.30 | 74 | 14 | 3.78 | 1.40 | 0.289 | 183.85 |
| Re | 0.4 | 2.05 | 1.28 | 75 | 15 | 3.51 | 1.46 | 0.288 | 186.21 |
| Os | 0.4 | 2.00 | 1.26 | 76 | 16 | 3.34 | 1.52 | 0.310 | 190.20 |
| Ir | 0.4 | 2.00 | 1.26 | 77 | 17 | 3.40 | 1.55 | 0.330 | 192.22 |
| Pt | 0.4 | 2.05 | 1.29 | 78 | 18 | 3.61 | 1.44 | 0.329 | 195.08 |
| Au | 0.4 | 2.10 | 1.34 | 79 | 19 | 4.04 | 1.42 | 0.339 | 196.97 |
| Hg | 0.4 | 2.05 | 1.44 | 80 | 20 | 5.87 | 1.44 | 0.384 | 200.59 |
For parameter set 5 the overall gradient of the straight line fit is 0.99(1) with a correlation coefficient of 0.92 (Fig. 1). These data compare favourably with respective values of 0.96 and 0.89 obtained by Gavezzotti for 172 organic crystal structures.21 The apparent improvement over organic materials obtained for the metal complexes is probably ascribable to limitation of the validation data-set to compounds for which multiple sublimation enthalpy determinations are available. If all available data (105 complexes containing first, second and third row metals) are used the gradient and correlation coefficient are 0.96(2) and 0.69, respectively (Fig. 2).
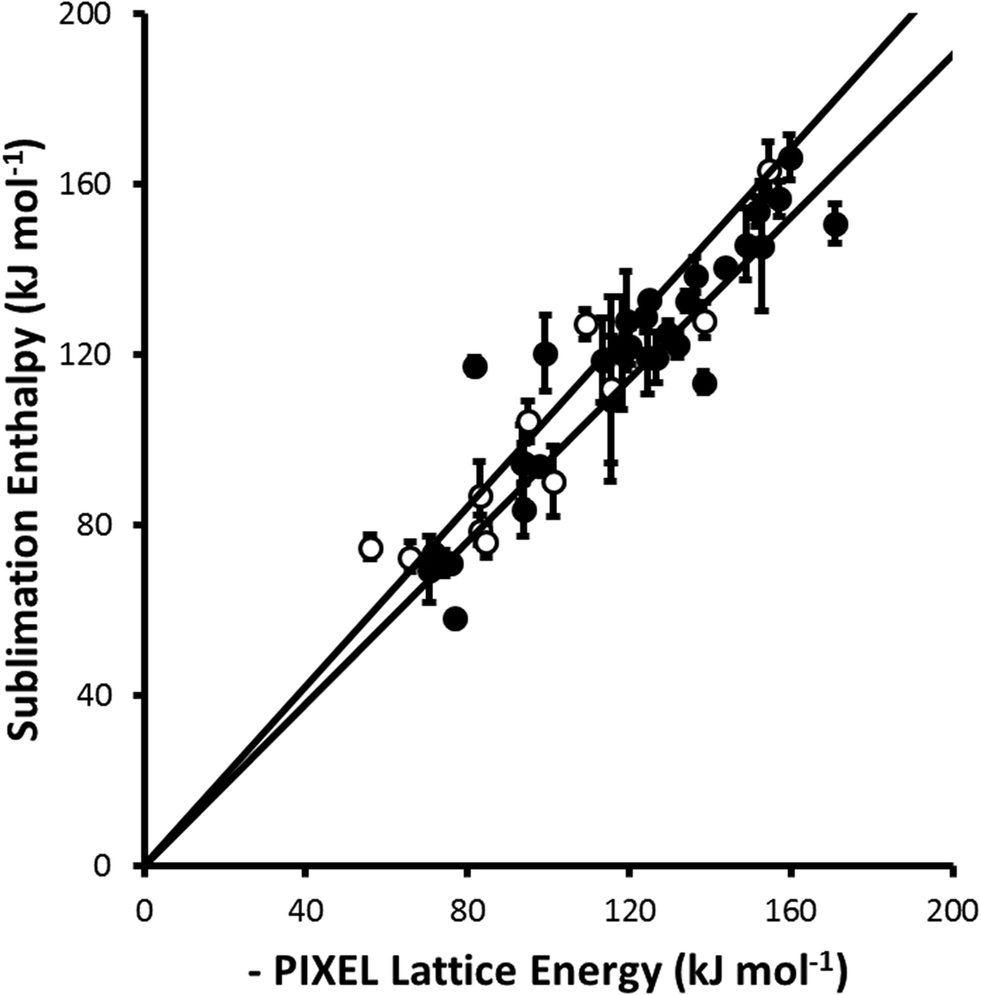 | ||
| Fig. 1 Comparison of experimental sublimation enthalpies with values calculated by PIXEL (parameter set 5) for 1st row (closed circles) and 2nd and 3rd row (open circles) transition metal complexes for which the sublimation enthalpy has been determined multiple times. The black lines are ±5% of the experimental sublimation enthalpy. The least-squares straight line through the data points is y = 0.99(1)x (R = 0.92). | ||
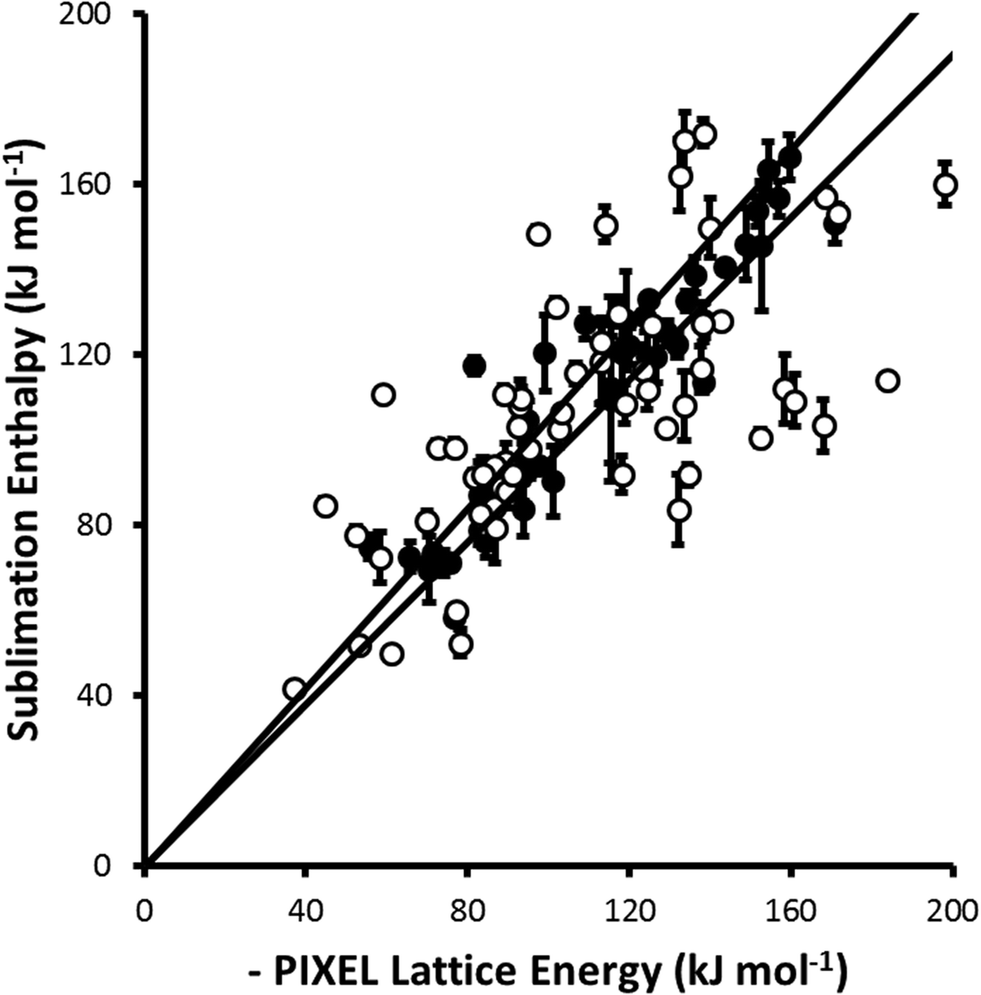 | ||
| Fig. 2 A similar comparison to that shown in Fig. 1 but including complexes (shown as open circles) for which the sublimation enthalpy has been determined only once. All points from Fig. 1 are shown as closed circles. The least-squares straight line through the data points is y = 0.96(2)x (R = 0.69). | ||
The data presented in Fig. 1 are dominated by first-row transition metal complexes, these data alone yielding a gradient and correlation coefficient equal to 0.99(2) and 0.92, respectively. The data for second and third row complexes are more limited, and the fitting statistics are 1.02(3) and 0.92. Some elements, such as technetium and gold, are not represented at all.
Overall, while the data presented above indicate that the PIXEL method can be applied with some confidence to first-row metal complexes, more data are needed to establish this for compounds containing heavier metals.
It is important to note that the assumption made in calculating a lattice energy by the PIXEL method is that no change in molecular structure occurs on passing from the crystalline state to the gas phase. PIXEL calculations of the lattice energies of amino acids, for example, are in poor agreement with experimental values because amino acids exist as zwitterions in the solid state but as neutral molecules in the gas phase. The energy associated with the transfer of a proton from the ammonium to the carboxylate group does not form part of the PIXEL analysis, and would need to be calculated separately in any calculation aiming to reproduce the experimental sublimation enthalpy. This consideration may account for some of the differences between observed and calculated data presented in Table 2.
The experimental sublimation enthalpies of the bis-cyclopentadienyl complexes of Cr, Fe, Ni, Ru and Os all lie between 70 and 80 kJ mol−1, and are reproduced to within 10 kJ mol−1 in the PIXEL calculations. By contrast the relative difference between the experimental and PIXEL values for Cp2V is the poorest in Table 2 (entry 11), but it is notable that the experimental value seems anomalously low at 58.2(11) kJ mol−1. The lattice energy of tris(hexafluoroacetylacetonato-O,O′)-chromium(III) (entry 23) is under-estimated [calc. 81.6, expt. 117.5(78) kJ mol−1], a finding consistent with previous PIXEL calculations on fluorinated polycyclic hydrocarbons.66 Decreasing the damping parameter used to calculate dispersion energies from 3.0 to 2.4 together with a small increase in α(F) from 0.4 to 0.5 Å3 to account for the effect of negative charge distribution in the (anionic) ligand yields a lattice energy of 113.4 kJ mol−1, illustrating the sensitivity of the calculation to the parameterisation used for fluorine.
4 Examples
4.1 Chromium hexacarbonyl
Chromium hexacarbonyl (CSD refcode FOHCOU02)67 crystallises in space group Pnma with the molecules forming layers in the mirror planes at y = 1/4 and 3/4. The sublimation enthalpy is 69.3(26) kJ mol−1 (an average of twelve determinations),55 and the lattice energy calculated by PIXEL is −70.5 kJ mol−1 (see Table 2). The three principal intermolecular interactions are to the molecules labelled A, B and C in Fig. 3; Table 4 shows the breakdown of the component energy terms and the Cr⋯Cr distance for each interaction. | ||
| Fig. 3 Energy vectors (obtained using processPIXEL) for the total intermolecular energies of chromium hexacarbonyl as viewed down the crystallographic c-axis. The energy vectors show the interaction energies scaled to that of the strongest interaction. The reference molecule is shown in green, and the strongest interaction is between this and molecule A. The interaction to molecule B is the second strongest, and that to molecule C is the weakest. | ||
| Interacting molecule | E coul | E disp | E pol | E rep | E tot | Cr⋯Cr (Å) |
|---|---|---|---|---|---|---|
| A | −5.8 | −18.7 | −1.8 | 13.0 | −13.3 | 6.236 |
| B | −2.6 | −16.9 | −1.2 | 8.9 | −11.9 | 6.213 |
| C | −4.3 | −11.2 | −1.1 | 7.4 | −9.1 | 6.888 |
The data in Table 4 confirm that the interactions are predominantly dispersion based. The influence of the Cr-atom on the dispersion term in interaction A can be demonstrated by setting its polarizability close to zero§ (0.0001 Å3), which changes the dispersion energy from −18.7 to −16.6 kJ mol−1. Setting both α(Cr) and α(O) to zero gives Edisp = −3.5 kJ mol−1 showing that the bulk of the interaction energy derives from the oxygen atoms. The role of electrostatics in the three interactions is illustrated in Fig. 4, where the electrostatic potential of each molecule is mapped onto its Hirshfeld surface.11,68 Favourable overlap occurs when the negatively-charged (red) regions of one molecule are in contact with the positively-charged (blue) regions of a neighbouring molecule, and the white lines separating these regions are contiguous. This arrangement is seen for interactions A and C, but in the interaction with molecule B there is some overlap between negative regions. This is the source of the more negative (stabilising) coulombic terms for interactions A and C, which is not apparent from geometric analysis alone.
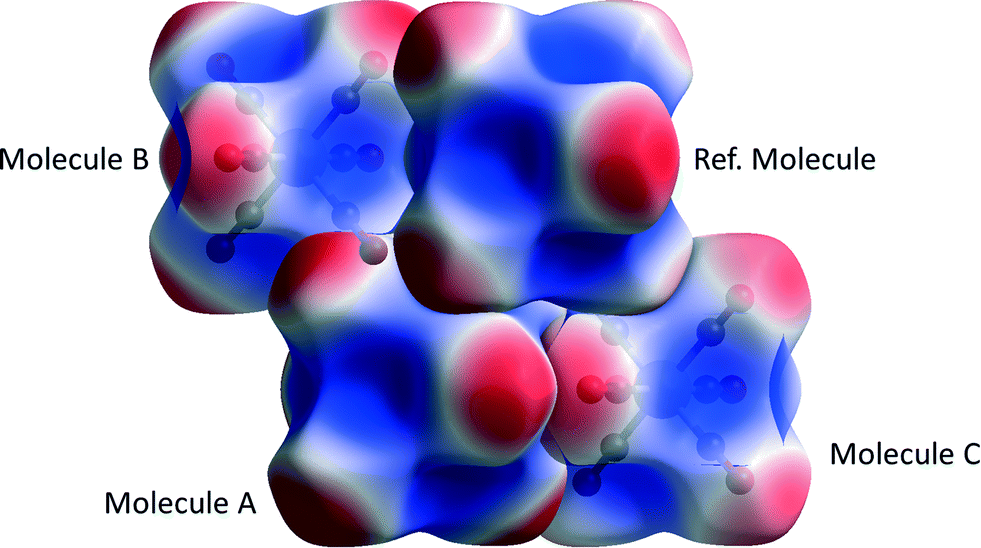 | ||
| Fig. 4 Coulombic interactions in Cr(CO)6 visualised using the electrostatic potential (ESP) mapped onto Hirshfeld surfaces. The surface is mapped from −0.036 au (red) to 0.047 au (blue). Wave functions were obtained at the HF/STO-3G level. | ||
4.2 Bis(acetylacetonato)-oxo-vanadium(IV)
In the crystal structure of bis(acetylacetonato)-oxo-vanadium(IV) (VO(acac)2, CSD refcode ACACVO12),69 the molecules are arranged in offset stacks disposed about inversion centres. The spacing between the two planes defined by the four ligating acac oxygen atoms in each molecule is 3.46 Å. A similar motif occurs in the crystal structures of other vanadyl complexes.70–72 The sublimation enthalpy is 140.6(4) kJ mol−1 (the average of two measurements),55 and the lattice energy estimated by PIXEL is −143.7 kJ mol−1. Based on the PIXEL results, the stacking interaction is the strongest intermolecular contact with an energy of −65.0 kJ mol−1, which is an order of magnitude higher than in typical π-stacking interactions involving phenyl groups,41 and similar to a strong hydrogen bond. A series of single-point ab initio calculations¶ performed at the crystal structure geometry with ‘normalised’ H-atom positions confirms the value obtained by PIXEL: B3LYP-D/6-31G*: −65.9 kJ mol−1; B2PLYP-D/6-31G*: −52.9 kJ mol−1; MO5-2X/6-311++G**: −53.5 kJ mol−1; SCS MP2: −67.7 kJ mol−1.The component terms (in kJ mol−1) of the stacking energy are: Ecoul = −28.8, Edisp = −67.9, Epol = −9.8 and Erep = +41.5, showing that, though the dispersion term is the largest, there is also substantial electrostatic character (Fig. 5). The contribution of the metal atom to the dispersion energy can be estimated by running the PIXEL calculation with α(V) set to approximately zero. This procedure reduces the magnitude of the interaction energy by 16.4 kJ mol−1 to −51.5 kJ mol−1, showing that the relatively high polarizability of vanadium (3.31 Å3) has an important influence on the intermolecular interaction energies. The magnitude of the coulombic term results from the efficient overlap of positive and negative regions of the electrostatic potentials of the two molecules (Fig. 6).
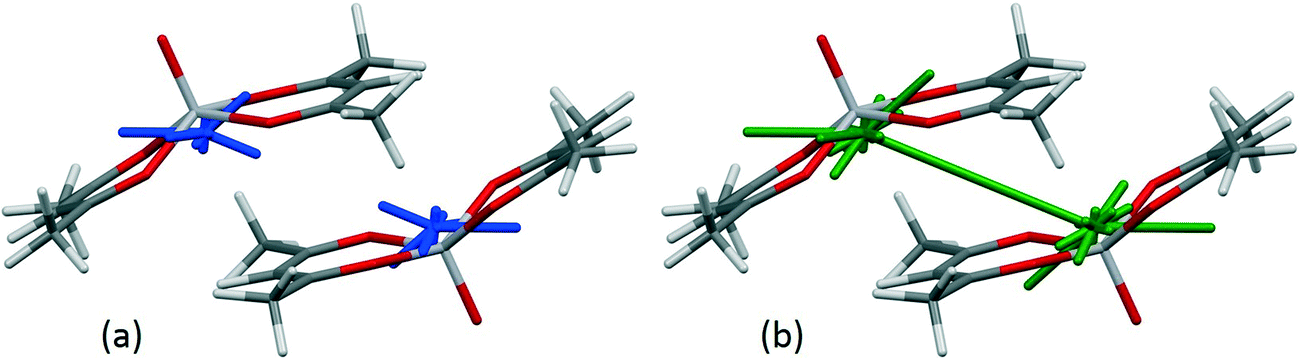 | ||
| Fig. 5 Energy vectors for the coulombic (a) and dispersion (b) components of the stacking interaction between bis(acetylacetonato)-oxo-vanadium(IV) molecules. | ||
 | ||
| Fig. 6 The stacking interaction in bis(acetylacetonato)-oxo-vanadium(IV) shown using the electrostatic potential mapped onto Hirshfeld surfaces. The surface is mapped from −0.005 au (red) to +0.005 au (blue). Wavefunctions were obtained using DFT with the Becke88 exchange potential and LYP correlation potential, and the 6-31G* basis-set. | ||
4.3 Dihydrogen bonding
A metal-bound hydride may be sufficiently negatively charged such that it can act as a hydrogen bond acceptor, forming a dihydrogen bond.74 Theoretical analysis of such interactions has shown that they are attractive and are predominantly electrostatic in character.75 Interaction energies for a series of dihydrogen-bonded dimers comprising a Ru-hydride complex and carboxylic acids or alcohols with different levels of fluorination have been evaluated using either DFT (B3LYP) or Hartree–Fock (HF) calculations with the LanL2DZ basis-set.60–62 PIXEL calculations were carried out using electron densities calculated with the same geometries, functionals and basis-sets as reported for the DFT calculations, and the results are compared in Fig. 7 and Table 5.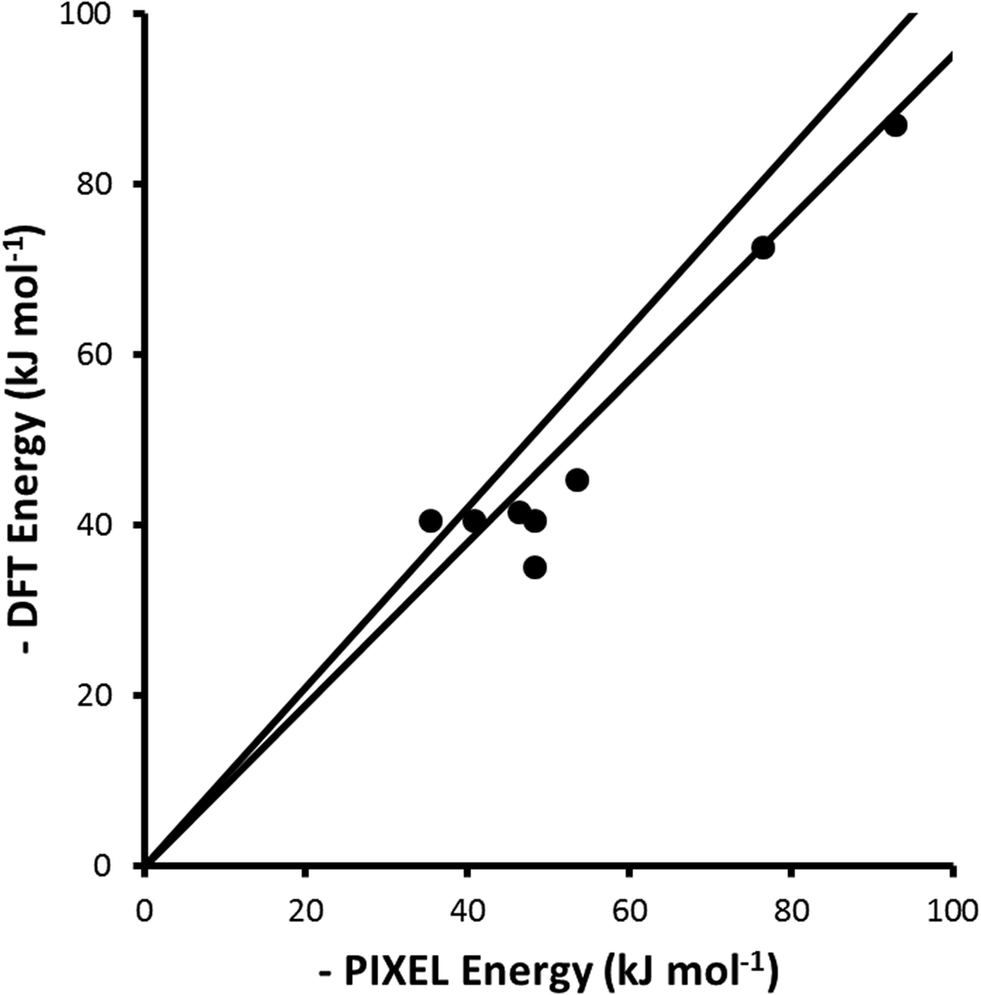 | ||
| Fig. 7 Comparison of PIXEL calculated energies with DFT reference energies for a selection of dihydrogen bonded complexes. The black lines are ±5% of the DFT calculated interaction energy. The least-squares straight line through the data points is y = 0.91x (R = 0.96). | ||
| Complex | Coul | Disp | Rep | Pol | Tot | DFT | Ref. |
|---|---|---|---|---|---|---|---|
| CpRu(CO)(PH3)H⋯HOOCCF3 | −53.0 | −22.4 | 63.6 | −36.6 | −48.4 | −35.1 | 60 |
| CpRu(CO)(PH3)H⋯HOC(CF3)3 | −50.7 | −27.3 | 65.0 | −35.4 | −48.4 | −40.6 | 60 |
| CpMo(dpe)H2H⋯HOCH(CF3)2 | −62.3 | −35.1 | 81.3 | −30.3 | −46.4 | −41.6 | 61 |
| CpW(dpe)H2H⋯HOCH(CF3)2 | −69.6 | −36.6 | 85.6 | −32.9 | −53.5 | −45.3 | 61 |
| PP3RuHHax⋯HOCH3 | −54.8 | −15.4 | 57.1 | −22.3 | −35.4 | −40.6 | 62 |
| PP3RuHHeq⋯HOCH3 | −51.8 | −18.8 | 52.8 | −23.1 | −40.9 | −40.5 | 62 |
| PP3RuHHax⋯HOCF3 | −103.6 | −20.9 | 92.6 | −61.0 | −92.9 | −86.9 | 62 |
| PP3RuHHeq⋯HOCF3 | −92.6 | −23.7 | 117.1 | −77.4 | −76.6 | −72.6 | 62 |
The level of agreement between the PIXEL and DFT results is similar to that found for the sublimation enthalpies: the gradient of the straight line in Fig. 7 is 0.91 and the correlation coefficient 0.96; differences are of the order of 5 kJ mol−1. The data in Table 5 show that the interactions are predominantly electrostatic in character, in accordance to observations made by Liu and Hoffmann, and that the coulombic term increases with increasing fluorination of the donor as expected on the basis of induction effects. The increase in the dispersion interaction which also occurs on fluorination is a result of a change in the orientation of the alcohol (Fig. 8) which enables formation of a secondary dispersion interaction between the fluorine atoms on the donor and the hydrogen atoms of the Ru complex.76
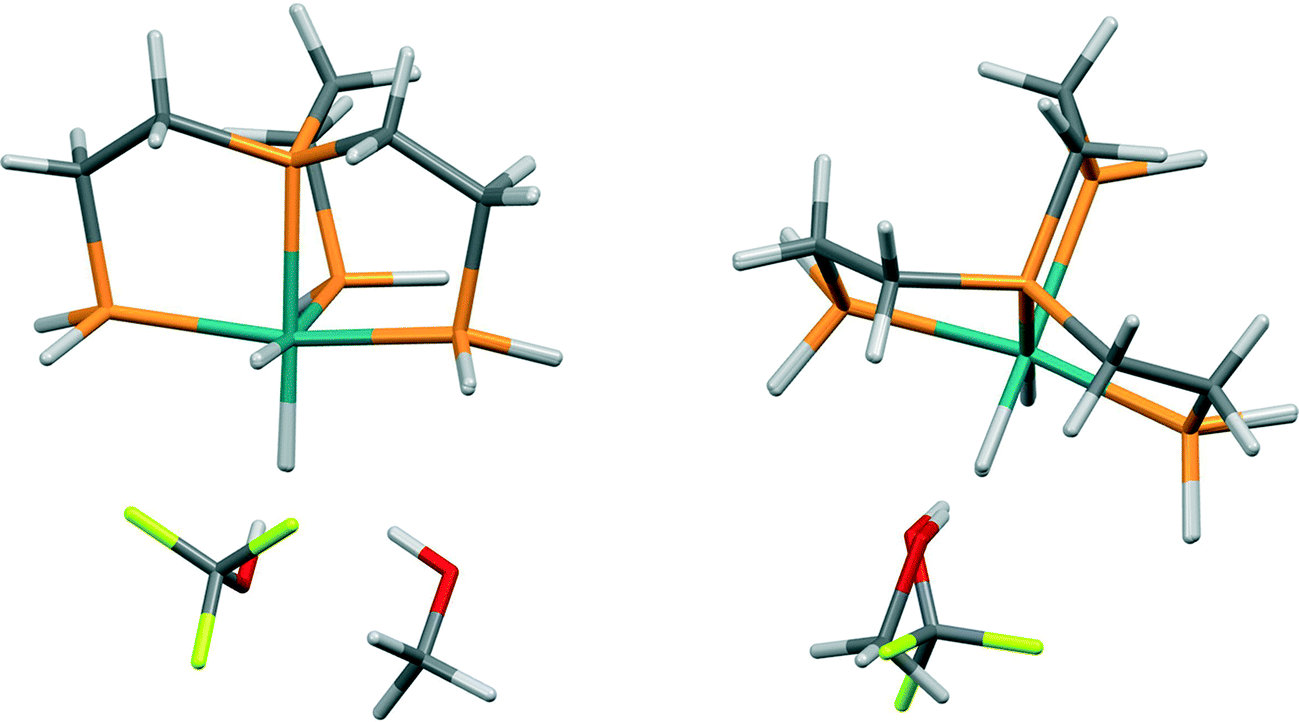 | ||
| Fig. 8 Binding geometries to the axial hydride (left) and equatorial hydride (right) in PP3RuHH where PP3 is P(CH2CH2PH2)3. Changes in the disposition of the alcohol upon fluorination are shown by an overlay in each case. | ||
5 Conclusions
A straight-line fit of experimental sublimation enthalpies of a series of first-row transition metal complexes to the lattice energy magnitudes calculated using the PIXEL method has a gradient of 0.99(2) and a correlation coefficient of 0.92. These figures indicate that the performance of the PIXEL parameterisation for molecules containing first-row metals is similar to that described by Gavezzotti for organic materials.The use of the method has been illustrated using interactions in the crystal structures of chromium hexacarbonyl and bis(acetylacetonato)-oxo-vanadium(IV). Intermolecular interactions in Cr(CO)6 are dominated by dispersion, as expected, but the shortest interaction is not the strongest on account of the second shortest interaction having a more complementary match of electrostatic potentials. Stacking interactions in VO(acac)2 have an energy in the region of 60 kJ mol−1, a figure confirmed by a variety of flavours of DFT calculation. This energy is similar to a strong acid–base hydrogen bond such as benozoic acid–imidazole,21 and explains why stacking interactions are a recurring feature of vanadyl crystal structures. Application to guest-binding in metal–organic frameworks will be described in a future paper.
The gradient and correlation coefficient of the fit between sublimation enthalpies and PIXEL lattice energies for second and third-row complexes are 1.02(3) and 0.92, respectively, but these figures are based on a much less extensive set of data than those for the first-row systems. The parameters for 2nd and 3rd row transition metals should be used with caution, but they are consistent with DFT results for dihydrogen bonds involving ruthenium hydride complexes and intermolecular embraces.
The PIXEL method assumes that an interaction is truly non-covalent, and it cannot be applied to interactions involving, for example, partial bond formation or other electron-sharing regimes for which full quantum mechanical treatments are necessary. Distinction between non-covalent and more complex interactions is often quite intuitive in organic chemistry, but this may not be the case in transition metal chemistry, and so caution is also necessary in this context. It would not be appropriate, for example, to apply PIXEL calculations to modelling the transition between long and short Jahn–Teller distortions or to aurophilic interactions, which both demand high-level quantum mechanical treatments even though the interatomic distances which characterise them extend to 3 Å and beyond. This said, comparison of PIXEL and quantum mechanical treatments for such systems might be used to detect the presence of complex behaviour.
The accurate and efficient quantification of crystal-packing and intermolecular energies is having a transformative effect on solid-state organic chemistry. It is no longer necessary to base the interpretation of a structure or a crystal engineering strategy on the assumption that short interactions are strong interactions.77 Systematic quantification of molecule–molecule energies further reveals important interactions that are easily missed on the basis of analysis of distances alone, a particular issue with electrostatic and van der Waals interactions which lack characteristic geometric signatures.78 While the PIXEL calculations do not replace quantum mechanical methods, they have proved to be an extremely valuable tool for interpretation of the thermodynamic stability of crystalline organic phases. The parameterisation described here should enable these advantages to be extended to systems containing metals, an extension which dramatically increases the domain of applicability for the PIXEL method.
5 Code availability
A version of the CLP package incorporating the PIXEL parameterisation given here for the first-row metals is available from Professor Gavezzotti's web-site, http://users.unimi.it/gavezzot. PIXEL calculations using parameters for the second and third row metals can be carried out by editing the symbol, wei, ravrg, ravdw, zeta, zval, polat, elneg, potio and difa data statements in the file alldat.for and re-compiling the code. These arrays respectively correspond to the Atom, WEIGHT, RINTER, RAVDW, ZTOT, ZVAL, POLZE, ELNEG, POTIO and DIFA entries in Table 3.Acknowledgements
We thank the Cambridge Crystallographic Data Centre and the EPSRC for studentship funding to AGPM. This work has made use of the resources provided by the EaStCHEM Research Computing Facility (http://www.eastchem.ac.uk/facilities), which is partially supported by the eDIKT initiative. (http://www.edikt.org.uk). We also thank Professor Nik Kaltsoyannis (University College London) for helpful advice, Tanya van Mourik (University of St Andrews) for assistance with SCS MP2 calculations, and Professor Andrew Bond for his assistance in the use of processPIXEL with transition metal complexes. We also thank Dr Neil Feeder and Dr Colin Groom (CCDC) for their comments on the manuscript and Professor Angelo Gavezzotti for his advice, insight and interest over many years.References
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- G. J. O. Beran, Angew. Chem., Int. Ed., 2015, 54, 396–398 CAS.
- R. Centore, M. Causa, S. Fusco and A. Carella, Cryst. Growth Des., 2013, 13, 3255–3260 Search PubMed.
- H. C. S. Chan, J. Kendrick, M. A. Neumann and F. J. J. Leusen, CrystEngComm, 2013, 15, 3799–3807 RSC.
- J. A. Chisholm, S. Motherwell, P. R. Tulip, S. Parsons and S. J. Clark, Cryst. Growth Des., 2005, 5, 1437–1442 Search PubMed.
- O. V. Shishkin, S. V. Shishkina, A. V. Maleev, R. I. Zubatyuk, V. Vasylyeva and K. Merz, ChemPhysChem, 2013, 14, 847–856 CrossRef CAS PubMed.
- J. Yang, W. Hu, D. Usvyat, D. Matthews, M. Schuetz and G. K.-L. Chan, Science, 2014, 345, 640–643 CrossRef CAS PubMed.
- A. Gavezzotti, Z. Kristallogr., 2005, 220, 499–510 CAS.
- E. G. Hohenstein and C. D. Sherrill, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 304–326 CAS.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- A. D. Bond, J. Appl. Crystallogr., 2014, 47, 1777–1780 CrossRef CAS.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735–3738 RSC.
- O. V. Shishkin, V. V. Medvediev, R. I. Zubatyuk, O. O. Shyshkina, N. V. Kovalenkoc and J. K. Volovenko, CrystEngComm, 2012, 14, 8698–8707 RSC.
- F. P. A. Fabbiani, C. R. Pulham and J. E. Warren, Z. Kristallogr., 2014, 229, 667–675 CAS.
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. F. Johnston and A. J. Florence, Cryst. Growth Des., 2013, 13, 1602–1617 Search PubMed.
- A. O. Surov, K. A. Solanko, A. D. Bond, A. Bauer-Brandl and G. L. Perlovich, CrystEngComm, 2015, 17, 4089–4097 RSC.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 Search PubMed.
- A. Gavezzotti, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 905–908 CrossRef CAS PubMed.
- R. D. L. Johnstone, D. Francis, A. R. Lennie, W. G. Marshall, S. A. Moggach, S. Parsons, E. Pidcock and J. E. Warren, CrystEngComm, 2008, 10, 1758–1769 RSC.
- A. Gavezzotti, Molecular Aggregation - Structure Analysis and Molecular Simulation of Crystals and Liquids, Oxford University Press, New York, 1st edn, 2007 Search PubMed.
- I. Dance, CrystEngComm, 2003, 5, 208–221 RSC.
- C. J. Adams, A. Angeloni, A. G. Orpen, T. J. Podesta and B. Shore, Cryst. Growth Des., 2006, 6, 411–422 Search PubMed.
- C. J. Adams, P. C. Crawford, A. G. Orpen and T. J. Podesta, Dalton Trans., 2006, 4078–4092 RSC.
- A. Perez Paz, L. A. Espinosa Leal, M.-R. Azani, A. Guijarro, P. J. Sanz Miguel, G. Givaja, O. Castillo, R. Mas-Balleste, F. Zamora and A. Rubio, Chem. – Eur. J., 2012, 18, 13787–13799 CrossRef CAS PubMed.
- D. Di Tommaso, S. A. French and C. R. A. Catlow, J. Mol. Struct.: THEOCHEM, 2007, 812, 39–49 CrossRef CAS.
- D. Di Tommaso, S. A. French, A. Zanotti-Gerosa, F. Hancock, E. J. Palin and C. R. A. Catlow, Inorg. Chem., 2008, 47, 2674–2687 CrossRef CAS PubMed.
- S. A. French, D. Di Tommaso, A. Zanotti-Gerosa, F. Hancock and C. R. A. Catlow, Chem. Commun., 2007, 2381–2383 RSC.
- H. Li, E. N. Brothers and M. B. Hall, Inorg. Chem., 2014, 53, 9679–9691 CrossRef CAS PubMed.
- T. Dudev and C. Lim, Annu. Rev. Biophys., 2008, 37, 97–116 CrossRef CAS PubMed.
- D. P. Martin, Z. S. Hann and S. M. Cohen, Inorg. Chem., 2013, 52, 12207–12215 CrossRef CAS PubMed.
- I. Yruela, Metallomics, 2013, 5, 1090–1109 RSC.
- A. De Almeida, B. L. Oliveira, J. D. G. Correia, G. Soveral and A. Casini, Coord. Chem. Rev., 2013, 257, 2689–2704 CrossRef CAS.
- H. Chiniforoshan, L. Tabrizi, M. Hadizade, M. R. Sabzalian, A. N. Chermahini and M. Rezapour, Spectrochim. Acta, Part A, 2014, 128, 183–190 CrossRef CAS PubMed.
- R. Q. Snurr, A. O. Yazaydin, D. Dubbeldam and H. Frost, in Metal-Organic Frameworks: Design and Application, ed. L. R. MacGillivray, John Wiley & Sons, Inc., Hoboken, New Jersey, 2010, ch. 11, pp. 313–339 Search PubMed.
- S. Keskin, J. Liu, R. B. Rankin, J. K. Johnson and D. S. Sholl, Ind. Eng. Chem. Res., 2009, 48, 2355–2371 CrossRef CAS.
- T. Düren, Y.-S. Bae and R. Q. Snurr, Chem. Soc. Rev., 2009, 38, 1237–1247 RSC.
- A. Gavezzotti, J. Phys. Chem. B, 2002, 106, 4145–4154 CrossRef CAS.
- A. Gavezzotti, J. Phys. Chem. B, 2003, 107, 2344–2353 CrossRef CAS.
- L. Maschio, B. Civalleri, P. Ugliengo and A. Gavezzotti, J. Phys. Chem. A, 2011, 115, 11179–11186 CrossRef CAS PubMed.
- W. B. Schweizer and J. D. Dunitz, J. Chem. Theory Comput., 2006, 2, 288–291 CrossRef CAS PubMed.
- J. Emsley, The Elements, Oxford University Press, New York, 1989 Search PubMed.
- S. Nag, K. Banerjee and D. Datta, New J. Chem., 2007, 31, 832–834 RSC.
- S. S. Batsanov, Inorg. Mater., 2001, 37, 871–885 CrossRef CAS.
- A. Gavezzotti, New J. Chem., 2011, 35, 1360–1368 RSC.
- K. A. Solanko and A. D. Bond, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 437–445 CrossRef CAS PubMed.
- B. P. van Eijck and J. Kroon, J. Phys. Chem. B, 1997, 101, 1096–1100 CrossRef CAS.
- A. Gavezzotti, A computer program package for the calculation of intermolecular interactions and crystal energies, University of Milan, Italy, 2003 Search PubMed.
- F. H. Allen and I. J. Bruno, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 380–386 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gasussian, Inc., 2009 Search PubMed.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 396–406 CrossRef CAS PubMed.
- T. A. Keith, A software package for performing quantitative and visual QTAIM (Quantum Theory of Atoms in Molecules) analyses of molecular systems, TK Gristmill Software, Overland Park, KS, USA, 12.06.03 edn, 2012 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- T. A. Keith, in The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, ed. C. F. Matta and R. J. Boyd, WILEY-VCH, Weinham, 2007, ch. 3, pp. 61–95 Search PubMed.
- J. S. Chickos and W. E. Acree, J. Phys. Chem. Ref. Data, 2002, 31, 537–698 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- R. M. Stephenson and S. Malanowski, Handbook of the Thermodynamics of Organic Compounds, Elsevier, New York, 1987 Search PubMed.
- G. Beech and R. Lintonbon, Thermochim. Acta, 1971, 2, 86–88 CrossRef CAS.
- N. V. Belkova, M. Besora, L. M. Epstein, A. Lledos, F. Maseras and E. S. Shubina, J. Am. Chem. Soc., 2003, 125, 7715–7725 CrossRef CAS PubMed.
- I. Andrieu, N. V. Belkova, M. Besora, E. Collange, L. M. Epstein, A. Lledos, R. Poli, P. O. Revin, E. S. Shubina and A. V. Vorontsov, Russ. Chem. Bull., 2003, 52, 2679–2682 CrossRef.
- E. I. Gutsul, N. V. Belkova, M. S. Sverdlov, L. M. Epstein, E. S. Shubina, V. I. Bakhmutov, T. N. Gribanova, R. M. Minyaev, C. Bianchini, M. Peruzzini and F. Zanobini, Chem. – Eur. J., 2003, 9, 2219–2228 CrossRef CAS PubMed.
- Accelrys, Accelrys Software Inc., Cambridge, UK, Release 5.5.3 edn, 2010 Search PubMed.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer 3.1, University of Western Australia, 2012 Search PubMed.
- D. Jayatilaka and D. J. Grimwood, in Computational Science - ICCS 2003, Pt IV, Proceedings, ed. P. M. A. Sloot, D. Abramson, A. V. Bogdanov, J. J. Dongarra, A. Y. Zomaya and Y. E. Gorbachev, 2003, vol. 2660, pp. 142–151 Search PubMed.
- F. Cozzi, S. Bacchi, G. Filippini, T. Pilati and A. Gavezzotti, Chem. – Eur. J., 2007, 13, 7177–7184 CrossRef CAS PubMed.
- A. Whitaker and J. W. Jeffery, Acta Crystallogr., 1967, 23, 977–984 CrossRef CAS.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, CrystEngComm, 2008, 10, 377–388 CAS.
- M. Hoshino, A. Sekine, H. Uekusa and Y. Ohashi, Chem. Lett., 2005, 34, 1228–1229 CrossRef CAS.
- D. I. Arnold, F. A. Cotton, J. H. Matonic and C. A. Murillo, Chem. Commun., 1996, 2113–2114 RSC.
- P. K. Hon, R. L. Belford and C. E. Pfluger, J. Chem. Phys., 1965, 43, 3111–3115 CrossRef CAS.
- U. Schilde, W. Bansse, E. Ludwig and E. Uhlemann, Z. Kristallogr., 1995, 210, 627–628 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- V. I. Bakhmutov, Dihydrogen Bonds: Principles, Experiments and Applications, John Wiley & Sons, Inc., Hoboken, NJ, 2008 Search PubMed.
- Q. Liu and R. Hoffmann, J. Am. Chem. Soc., 1995, 117, 10108–10112 CrossRef CAS.
- H. Jacobsen, Phys. Chem. Chem. Phys., 2009, 11, 7231–7240 RSC.
- J. Dunitz, IUCrJ, 2015, 2, 157–158 CrossRef CAS PubMed.
- A. Gavezzotti, CrystEngComm, 2013, 15, 4027–4035 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c5ce01522g |
| ‡ Test calculations were performed using parameter set 5 (see below). |
| § Setting the value to exactly zero causes the PIXELc program to use a default value of α. |
| ¶ MO5-2X performed with Gaussian09, B3LYP-D, B2PLYP-D and SCS MP2 performed with ORCA.73 |
| This journal is © The Royal Society of Chemistry 2015 |