
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Contrasting crystal packing arrangements in triiodide salts of radical cations of chiral bis(pyrrolo[3,4-d])tetrathiafulvalenes†
Lee
Martin
*a,
Songjie
Yang
a,
Andrew C.
Brooks
ab,
Peter N.
Horton
c,
Louise
Male
c,
Ourida
Moulfi
a,
Lydie
Harmand
a,
Peter
Day
b,
William
Clegg
d,
Ross W.
Harrington
d and
John D.
Wallis
*a
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: lee.martin@ntu.ac.uk
bUniversity College London, Department of Chemistry, 20 Gordon Street, London WC1H 0AJ, UK
cDepartment of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
dSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
First published on 20th August 2015
Abstract
Crystal structures of six 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 triiodide salts of a series of enantiopure bis(pyrrolo[3,4-d])TTF derivatives, the first structures of radical cation salts reported for this bis(pyrrolo) donor system, show three different arrangements of triiodide ions, organised either in head-to-tail pairs, in infinite lines, or in a castellated arrangement. The complex crystal structures, obtained by electrocrystallisation, are influenced by the presence of solvent, for example changing an ABCABC packing arrangement to ABAB with inclusion of THF, as well as by the size of the chiral side chain.
:1 triiodide salts of a series of enantiopure bis(pyrrolo[3,4-d])TTF derivatives, the first structures of radical cation salts reported for this bis(pyrrolo) donor system, show three different arrangements of triiodide ions, organised either in head-to-tail pairs, in infinite lines, or in a castellated arrangement. The complex crystal structures, obtained by electrocrystallisation, are influenced by the presence of solvent, for example changing an ABCABC packing arrangement to ABAB with inclusion of THF, as well as by the size of the chiral side chain.
Introduction
The electrical properties of the radical cation salts of donors such as TTF 1 and BEDT-TTF 2 have attracted a lot of attention. Over 30 years ago Dunitz raised the question as to how the chirality of a material might affect its electrical properties, and prepared the enantiopure (S,S,S,S)-tetramethyl(BEDT-TTF) donor 3 and a family of its semi-conducting radical cation salts.1 In subsequent studies several series of salts from both enantiomers of 3 and its racemate were prepared. For the 2
 :1 salts with AsF6− or SbF6− the conductivities and activation energies were similar, while for the 1:1 salts with triiodide the racemate salt had lower conductivity and higher activation energy than the enantiomerically pure salts.2 Very recently, this series of stereoisomers has been electrocrystallised with the tris(chloranilato)ferrate(III) ion to provide materials combining chirality, electrical conductivity and magnetic properties.3 In addition donor 3 and a mixed metal oxalate network form a chiral ferromagnetic material.4
:1 salts with AsF6− or SbF6− the conductivities and activation energies were similar, while for the 1:1 salts with triiodide the racemate salt had lower conductivity and higher activation energy than the enantiomerically pure salts.2 Very recently, this series of stereoisomers has been electrocrystallised with the tris(chloranilato)ferrate(III) ion to provide materials combining chirality, electrical conductivity and magnetic properties.3 In addition donor 3 and a mixed metal oxalate network form a chiral ferromagnetic material.4
Rikken has shown that, for a chiral conducting material in a coaxial magnetic field, the size of the electrical resistance has a dependence on the handedness of the material, a property termed magnetochiral anisotropy, with measurements made on bismuth helices and carbon nanotubes.5 To provide further materials for investigation we and others have prepared a range of enantiopure organosulfur donors, e.g.4–106–12 and have reported some of their radical cation salts.7,8,10,11 In particular, Avarvari, Rikken et al. have shown that the metallic 2:1 salts of R,R or S,S-8 with ClO4− crystallise in enantiomorphous hexagonal space groups and indeed show magnetochiral anisotropy.13
An alternative approach is to form salts of an achiral donor with a chiral anion, e.g. a tris(oxalate)metallate in (BEDT-TTF)4[(H3O)Cr(C2O4)3]·C6H5CN where one polymorph has the enantiomers partitioned into separate layers.14 By using R-(−)-carvone as solvent, the first BEDT-TTF salts with a single enantiomer of the tris(oxalate)metallate have been obtained.15 Further BEDT-TTF salts have been made with the enantiopure Sb2(L-tartrate)2 anion16 as well as with racemic anions such as SF5CHFCF2SO3− and Fe(III)(croconate)3, and the salt of enantiopure tetramethyl-BEDT-TTF with the racemic TRISPHAT anion has been prepared.17–19 Chirality has also been introduced using a solvent e.g. (S)-sec-phenylethyl alcohol which is included in crystals of BEDT-TTF radical cation salts.20 The various strategies for preparing chiral molecular conductors have been reviewed.21
We recently reported the preparation and crystal packing arrangements of four new chiral bis(pyrrolo)[3,4-d]tetrathiafulvalene derivatives 11–14 in which the two stereogenic centres carry a hydrogen, a methyl and either a phenyl or 1-naphthyl group, and the TTF is fused to pyrrole or pyrroline rings.22 Most notable was that donors 11 and 12, which have fully unsaturated heterocyclic systems, adopt crystal packings with 43 or near 43 chiral axes relating successive layers of donors. We now report the first radical cation salts obtained with these materials. Note that for 11 and 13 the stereogenic centres have the S configuration, and for 12 and 14 they have the R configuration.
Results and discussion
Cyclic voltammetry measurements demonstrated that all four donors showed two oxidation processes, with the pyrrolo-fused donors having slightly lower oxidation potentials than the dihydropyrrolo-fused systems (Table 1). Electrocrystallisations with tetrabutylammonium triiodide have produced crystalline salts from three of these donors whose structures have been determined by single-crystal X-ray crystallography: 11·I3 and 11·I3·0.5THF; a series of three related salts from 12, viz.12·I3·solvent (THF, chlorobenzene or DCM); and 13·I3 (Tables 2 and 3). These are the first radical cation salts reported from the bis(pyrrolo)TTF system, though this molecular unit has been incorporated in catenanes and rotaxanes in which the TTF system forms charge transfer interactions with species such as 4,4′-bipyridinium dications in cyclo-bis(paraquat-p-phenylene).23 For salts of donors 11 and 12, the cations show lengthening of the central double bond from 1.352–1.356 Å to ca. 1.40 Å, and shortening of the C–S bonds, especially those at the centre of the molecule (from 1.744–1.783 Å to ca. 1.73 Å), but only minor changes in the pyrrole rings.| Donor | E 1 (V) | E 2 (V) |
|---|---|---|
| a Measured relative to Ag/AgCl at a platinum electrode in dichloromethane containing 0.1 M n-Bu4NPF6 as charge carrier and using a 100 mV s−1 scan. | ||
| 11 | 0.36 | 0.86 |
| 12 | 0.36 | 0.86 |
| 13 | 0.40 | 0.90 |
| 14 | 0.41 | 0.88 |
| 11·I3·0.5THF | 11·I3 | 12·I3·THF | 12·I3·C6H5Cl | 12·I3·DCM | 13·I3 | |
|---|---|---|---|---|---|---|
| Formula | C26H22N2S4·I3·0.5C4H8O | C26H22N2S4·I3 | C34H26N2S4·I3·C4H8O | C34H26N2S4·I3·C6H5Cl | C34H26N2S4·I3·CH2Cl2 | C26H26N2S4·I3 |
| M r | 907.45 | 871.39 | 1043.66 | 1084.06 | 1056.43 | 875.43 |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| a (Å) | 9.446(6) | 9.4562(16) | 25.5744(5) | 26.3842(14) | 25.618(4) | 9.484(10) |
| b (Å) | 17.339(11) | 9.5572(14) | 8.9609(2) | 9.0879(2) | 8.9466(5) | 19.551(14) |
| c (Å) | 37.04(2) | 53.944(9) | 19.6079(5) | 19.4801(9) | 19.623(2) | 30.639(3) |
| α (°) | 90 | 89.206(5) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 87.608(3) | 123.5900(10) | 123.371(7) | 123.406(18) | 90 |
| γ (°) | 90 | 60.661(5) | 90 | 90 | 90 | 90 |
| V (Å3) | 6067(6) | 4246.0(12) | 3743.19(15) | 3900.8(3) | 3754.5(7) | 5681(7) |
| Z | 8 | 6 | 4 | 4 | 4 | 8 |
| Space group | P212121 | P1 | C2 | C2 | C2 | P212121 |
| T (K) | 120 | 120 | 120 | 120 | 120 | 120 |
| D c (g cm−3) | 1.99 | 2.04 | 1.85 | 1.85 | 1.87 | 2.04 |
| λ (Å) | 0.6893 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| μ (mm−1) | 2.91 | 3.63 | 2.75 | 2.72 | 2.91 | 3.62 |
| No. of unique reflections | 8693 | 19478 |
8218 | 6467 | 6390 | 10456 |
| No. of unique reflections with F > 4σ(F) | 6035 | 10434 |
7638 | 5804 | 5896 | 4940 |
| No. of refined parameters | 682 | 1891 | 419 | 454 | 427 | 356 |
| Flack parameter | 0.10(6) | 0.12(3) | 0.09(2) | −0.02(2) | 0.13(3) | 0.12(6) |
| R 1, wR2 for reflections with F > 4σ(F) | 0.076, 0.161 | 0.084, 0.187 | 0.036, 0.081 | 0.028, 0.067 | 0.049, 0.133 | 0.159, 0.256 |
| Salt | Solvent | Current (μA) | Time (days) | Appearance of crystals |
|---|---|---|---|---|
| 11·I3·0.5THF | THF | 1.0 | 26 | Thin black needles |
| 11·I3 | Dichloromethane | 1.0 | 26 | Black diamond plates |
| 12·I3·THF | THF (20 ml) | 0.5 | 28 | Black plates |
| 12·I3·C6H5Cl | Chlorobenzene | 5.0 | 28 | Black plates |
| 12·I3·DCM | Dichloromethane | 5.0 | 28 | Black plates |
| 13·I3 | 1,1,1,-Trichloroethane | 0.5 | 20 | Black plates |
The six triiodide salts are all of 1:1 stoichiometry but show a variety of packing modes, with inclusion of solvent in some cases, in which the triiodides are packed alongside the donors either in head-to-tail pairs, or in continuous lines, or even in a castellated array. The donor cations are packed in face-to-face pairs. Incorporation of iodides parallel to the TTF unit has been observed in triiodide salts of TTF itself and its derivatives bearing two fused cyclopentene or cyclohexene rings,24 as well as in bis(2-hydroxypropylthio)ethylenedithio-TTF.25 The salts were found to be electrically insulating; data for salt 12·I3·THF could not be measured.
Salts 11·I3 and 11·I3·0.5THF
Two 1:1 triiodide salts of donor 11 have been characterized structurally: a triclinic phase grown as thin black diamonds from dichloromethane, and an orthorhombic THF hemisolvate phase obtained as thin black needles. They both contain pairs of triiodide ions lying head-to-tail which lie side-by-side with pairs of donor cations which are oriented face-to-face. The bonding geometry at each pyrrole nitrogen atom is planar.
The asymmetric unit of the THF solvate phase contains two donor cations and two triodide anions along with one THF molecule, which in the space group P212121 gives eight cations, eight anions and four THF molecules per unit cell. Two important structural motifs can be identified: a pair of crystallographically independent donor cations packed face-to-face but with their TTF units offset in the direction of the longest molecular axis (Fig. 1), and a unit formed by two triiodides which are organized “head-to-tail” (I⋯I: 3.578(2) Å, with I–I⋯I angles 170.1 and 173.1°). These motifs are packed alternately along the b axis, and the 21 axis along c produces a herringbone pattern of these units in the bc plane (Fig. 2). Successive planes in the a-axis direction are related by the 21 axes along a (Fig. 2). The THF solvent molecules are surrounded principally by the chiral side chains of the donor cations. An alternative way of considering the packing arrangement is that the two structural motifs are packed into blocks extending half a unit cell length along the c axis, and the 21 axes along b and c produce the herringbone arrangement between successive blocks. The blocks interface via the phenylethyl sidechains of the donor cations and the THF molecules lie in this region.
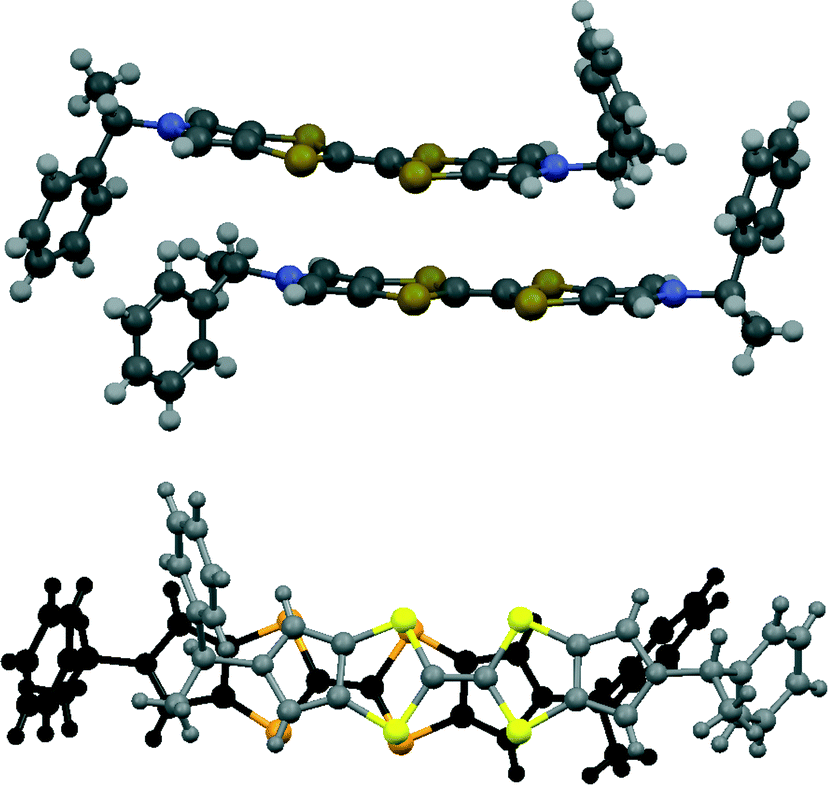 | ||
| Fig. 1 Pair of donor cations in 11·I3·0.5THF showing their offset, face-to-face, with orientation viewed side on (above) and perpendicular to the heterocyclic system (below). | ||
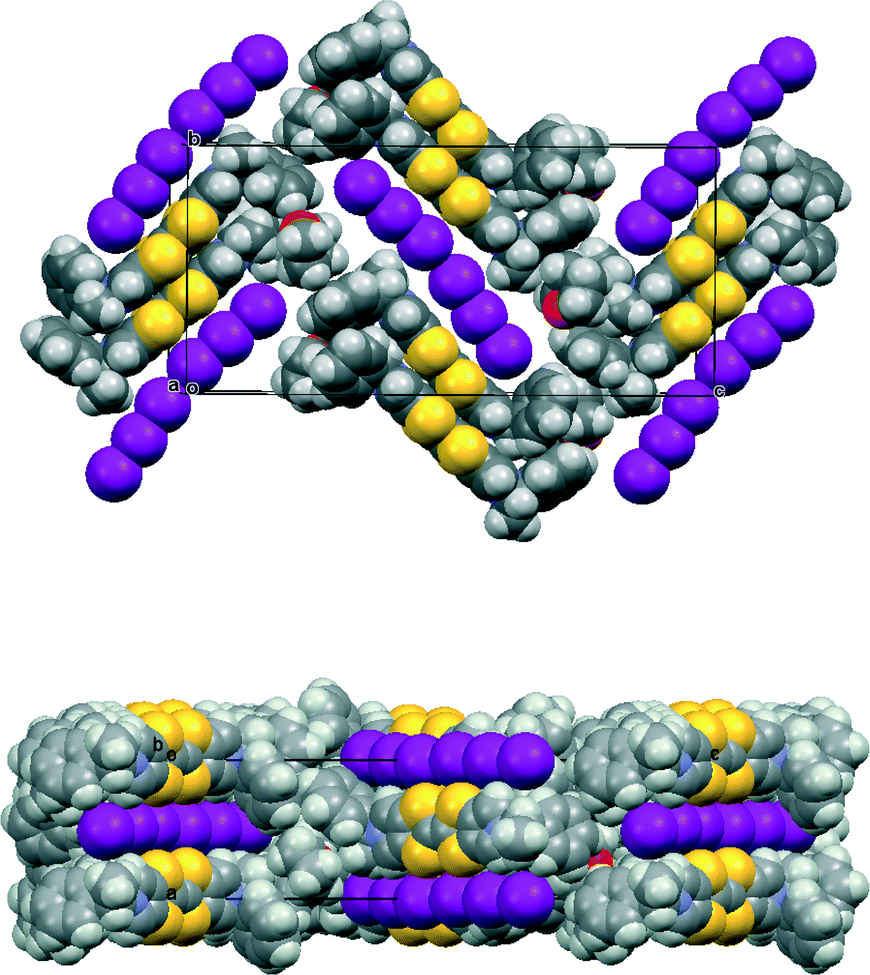 | ||
| Fig. 2 Crystal packing of 11·I3·0.5THF viewed down the a axis (above) and down the b axis (below). | ||
Each (I3−)2 unit is surrounded by four donor cation pairs and vice-versa. Thus two cation pairs lie edge-on to the (I3−)2 unit, and two others present a donor face. The I⋯S contacts are shorter to the edge-on donor cation pair with shortest contacts of 3.746(6) and 3.750(7) Å, while the shortest I⋯S contacts to the faces of the other two donor cations are >4.1 Å. There are also quite short S⋯S contacts between the adjacent donor cation pairs surrounding an (I3−)2 unit, notably 3.406(10), 3.412(10), 3.496(10) and 3.557(11) Å. The 1:1 salts of the enantiomers of tetramethyl-BEDT-TTF 3 with triiodide also contain head-to-tail pairs of triiodide,3 a motif which has a similar length to the donor molecules.
The donor cation pairs lie with their bis(pyrrolo)TTF planes at 5.0° to each other, but slipped so that the central double bond for each donor overlaps with a dithiolo-pyrrole group of the other one (Fig. 1). One donor cation plane is perfectly planar and the other is slightly bowed. There are two very short S⋯S contacts between these cations of 3.282(10) and 3.297(9) Å, which are ca. 0.6 Å shorter than the sum of two van der Waals radii for the asymmetrically shaped bonded S atom.26 The conformations about the exocyclic N–C bonds at the ends of the donor cations are different. The phenyl group at one stereogenic centre and the hydrogen atom at the other are oriented roughly perpendicular to the heterocyclic plane and towards the other donor cation in the pair. The pair of donors fit together with the “perpendicular” phenyl group of each donor wrapping over the end of the other donor (Fig. 1).
The unsolvated triclinic phase in space group P1 is similar, but there are six independent donor cations, packed in three pairs, and three independent linear (I3−)2 units with I⋯I separations of 3.498(3), 3.553(4) and 3.609(4) Å. A pair of donor cations is packed with a linear arrangement of two triodide ions to form blocks perpendicular to the c axis (Fig. 3), similar to those in the orthorhombic phase. The difference is that in the triclinic phase there are three symmetry-unrelated blocks of this type sequentially along the c axis, which is over 50 Å long, with interfaces between the blocks formed by the phenylethyl sidechains (Fig. 3). In the orthorhombic phase there are just two, symmetry-related, blocks along the c axis. The modes of overlap between the members of the three independent pairs of donor cations are also similar to that in the orthorhombic phase, and within each pair there are two close S⋯S contacts in the range 3.341(14)–3.388(14) Å. Furthermore, there are S⋯S contacts between each set of donor cation pair neighbours surrounding each (I3−)2 unit in the range 3.503(13)–3.522(12) Å.
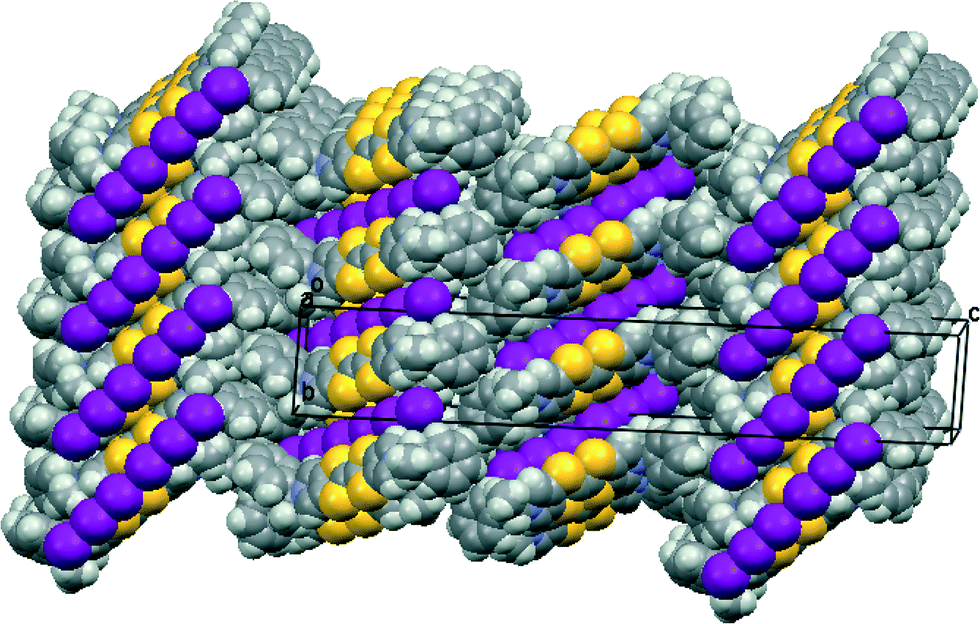 | ||
| Fig. 3 Crystal packing of 11·I3 showing one block of donor cation pairs and (I3−)2 units extending one third the way along the c axis and the arrangement of successive blocks along the c axis, viewed down the a axis. | ||
The state of oxidation of TTF and derived donors can be detected by observation of particular stretches in the Raman spectrum.27 While donor 11 showed stretches at 1485 and 1549 cm−1, in the two salts of this donor, the corresponding stretches occurred at lower wavenumber, 1412–1414 and 1492–1496 cm−1 (Table 4). These are the first measurements on the bis(pyrrolo)TTF system, but the results are reasonably consistent with a +1 oxidation state by comparison with results from TTF itself.
| Donor/salt | Raman frequencies (cm−1) |
|---|---|
| 11 | 1485, 1549 |
| 11·I3 | 1414, 1496 |
| 11·I3·0.5THF | 1412, 1492 |
| 13 | 1471, 1538 |
| 13·I3 | 1412, 1499 |
Three 12·I3 solvates with THF, chlorobenzene and DCM
Donor 12 formed a series of 12·I3·solvent radical cation salts by electrocrystallisation with tetrabutylammonium triiodide in THF, chlorobenzene or dichloromethane. Single crystal diffraction studies on crystals of all three substances showed that their monoclinic unit cells are very similar in dimensions. For all three cases the space group is C2, with one donor cation, a solvent molecule and two halves of two triiodide anions in the asymmetric unit; the central iodine atom of each triiodide ion lies on a crystallographic 2-fold axis. Aspects of the crystal structure of the THF solvate are shown in Fig. 4 and 5. The THF molecule is disordered between two orientations. The triiodide ions lie head-to-tail forming infinite rows along the c direction, with an I⋯I separation of 3.9363(5) Å and I–I⋯I angles of 174.22(3) and 177.14(2)°. The I⋯I separation is ca. 0.33 Å longer than the longest I⋯I separation in the triiodide salts of 11. The donor cations occur in pairs related by a two-fold rotation axis with their TTF groups directly facing each other, and with S⋯S contacts between them of 3.272(4) and 3.306(4) Å; rows of these pairs lie between the rows of triiodides. The donor cations adopt a conformation with both naphthyl groups directed out perpendicular from the donor cation pair. The bulkier naphthyl side chains in donor 12, rather than phenyl groups in donor 11, prevent the formation of such compact donor cation pairs as in the salts of 11. The donor cation retains the almost planar bonding geometry at nitrogen. The heterocyclic system of the donor cation species is very slightly bowed due to steric effects between donor cation side chains. One of the two crystallographically unique triiodides lies side-by-side with the four TTF units of two donor cation pairs, so that there are sixteen S⋯I contacts from as short as 3.586(2) and 3.592(2) Å up to 3.856(2) Å. The second triiodide lies among the chiral hydrocarbon sidechains of donor cations and is not involved in any short contacts. Donor cation pairs form rows, parallel to the triiodide chains, and interface to each other by roughly face-to-face π/π interactions between naphthyl groups whose mean planes lie at 16.1° to each other. The methyl group at the stereogenic centre prevents very close packing of the aromatic planes, though the outer edges of the rings do make C⋯C contacts in the range 3.40–3.46 Å. Naphthyl groups of donor cations in neighbouring rows interface in an edge-to-face manner, though the H⋯C contacts are not particularly short (2.9–2.97 Å). The THF molecules lie in cavities formed by several naphthalene rings and a TTF group, which are positioned just to one side of the point where two triiodide ions interface end-to-end.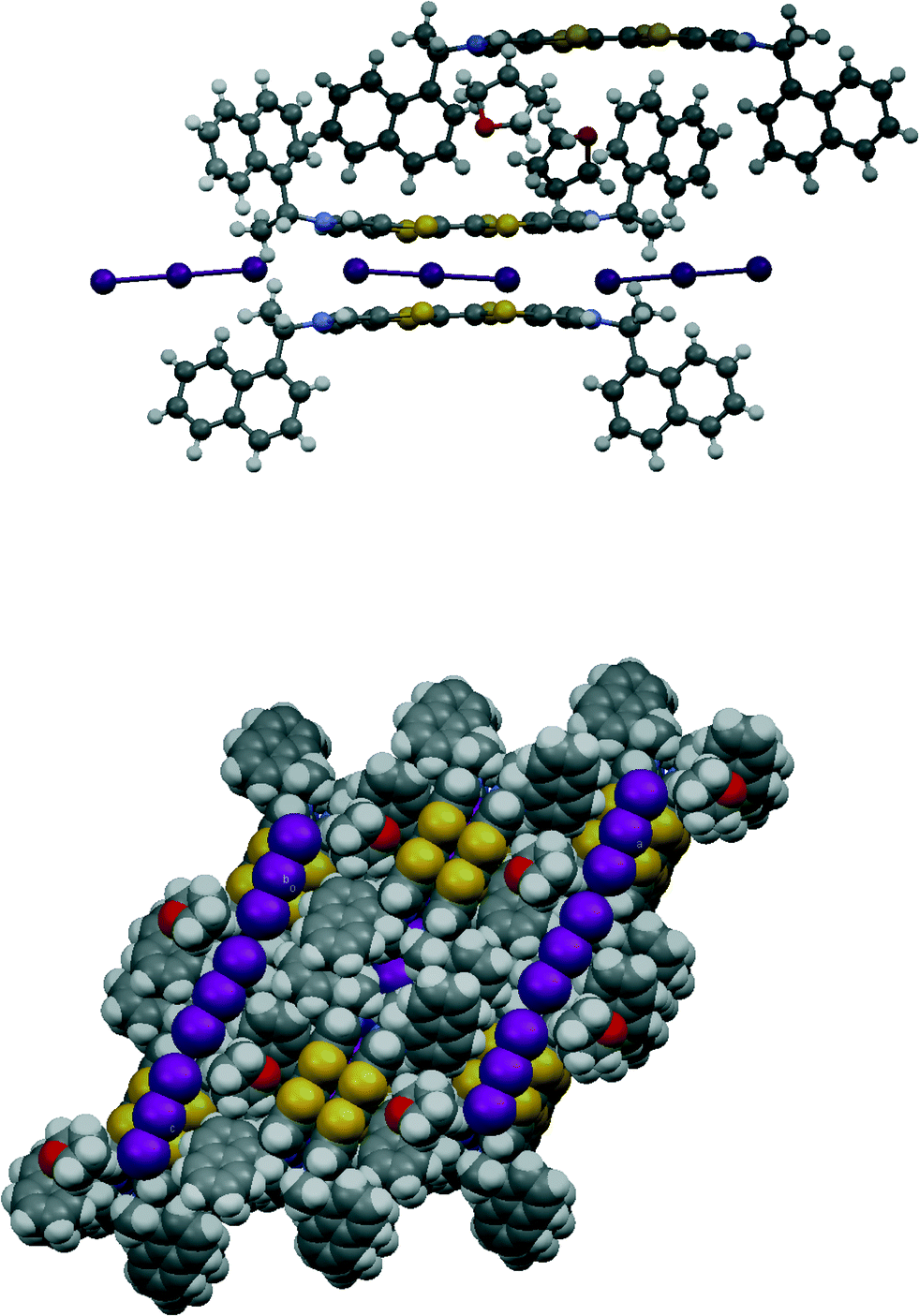 | ||
| Fig. 4 Section of crystal packing in 12·I3·THF showing the conformation of the donor and the relative location of the lines of triiodides and the THF molecules (above), view of crystal packing looking down the b axis (below). | ||
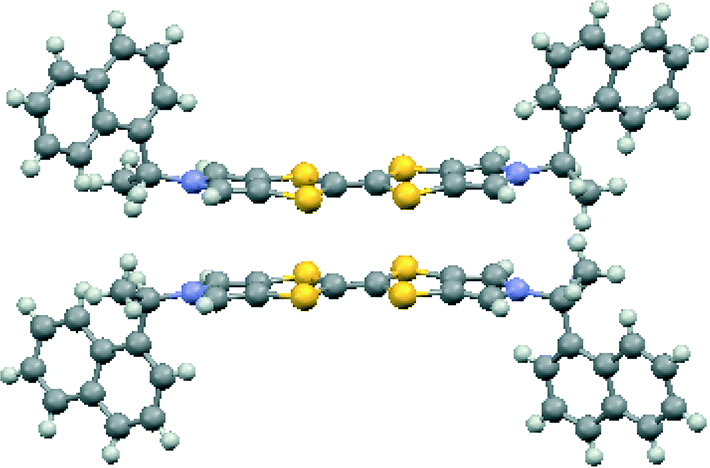 | ||
| Fig. 5 The donor pair in 12·I3·THF. | ||
The corresponding 12·I3 chlorobenzene solvate has a similar crystal structure (Fig. 6 and 7), but with chlorobenzene molecules filling the voids between the donor cation pairs. Indeed, there are Cl⋯benzene contacts between adjacent solvent molecules, Cl⋯centroid(benzene) = 3.429 Å, with the Cl⋯centroid vector roughly perpendicular to the benzene ring and a C–Cl⋯centroid angle of 148.9°, which link the solvent molecules in the b direction throughout the structure, perpendicular to the rows of triiodides and donor cation pairs. These chlorobenzene rows pass to one side of the perpendicular triiodide rows at just the point where the triiodide ions interface end-to-end, so that there are contacts from the two terminal iodine atoms to a meta-hydrogen of chlorobenzene of 3.23 and 3.35 Å. The unit cell is ca. 50 Å3 larger than for the THF solvate, expanding along the a and b axes but contracting slightly along the c axis. The packing of the donor cations and triiodide anions has expanded somewhat, thus there are fourteen S⋯I contacts in the range 3.6390(15)–3.8517(15) Å compared to sixteen in the range 3.586–3.856 Å. However, the S⋯S contacts within a donor cation pair of 3.297(3) and 3.307(3) Å are very similar to those in the THF solvate (3.272 and 3.306 Å). Along the chain of triiodides the I⋯I separations are slightly longer (3.9887(5) cf. 3.9363 Å) and the I–I⋯I angles notably smaller (166.55(2) and 169.75(2) cf. 174.22 and 177.14°) than in the THF solvate, so the relative orientation of triiodide ions is further away from linear compared to the THF solvate, and thus compatible with the slightly shorter c axis.
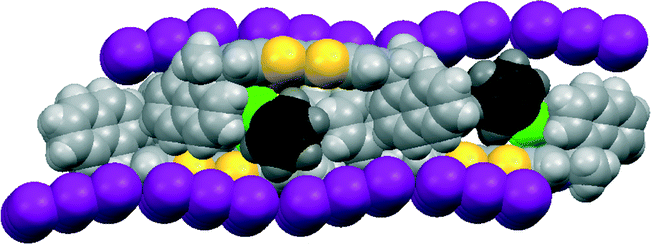 | ||
| Fig. 6 View of the structure of 12·I3·chlorobenzene, showing how the solvent molecules lie close to the interface between triiodide ions. | ||
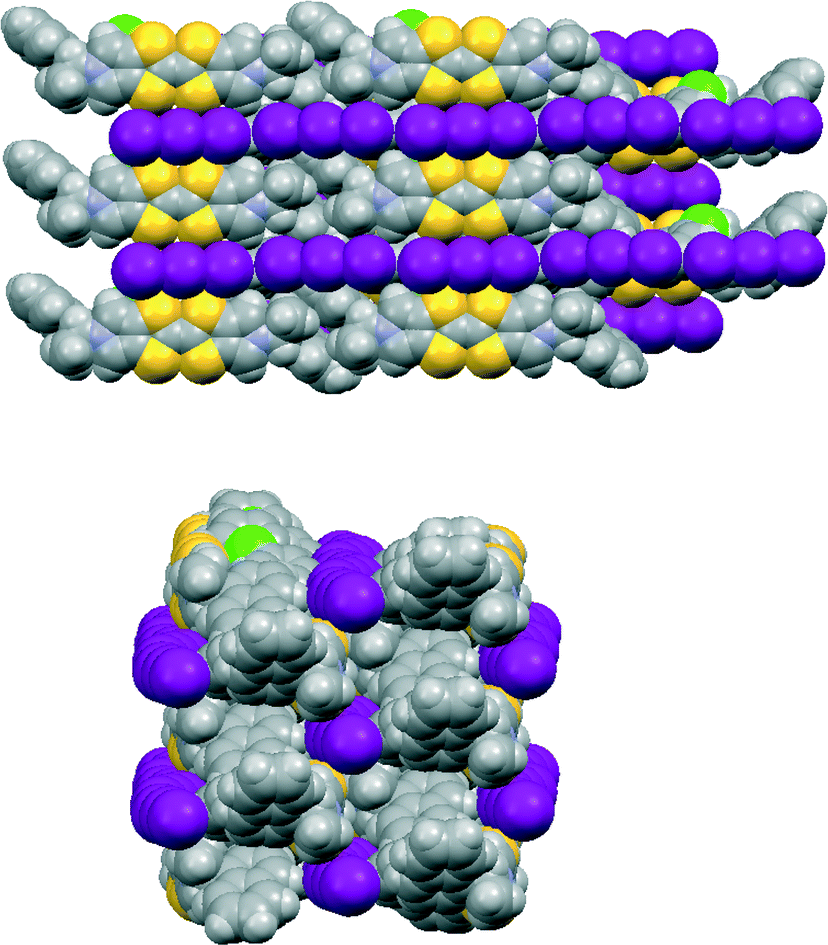 | ||
| Fig. 7 Crystal packing in 12·I3·chlorobenzene viewed on the bc plane (above) and viewed along the c axis (below). | ||
The crystal of the dichloromethane solvate was twinned, but a reasonable model was refined (Fig. 8). The volume of the unit cell is very similar to that of the THF solvate. The overall organization of the crystal structure is very similar to the other solvates, however the smaller size of the solvent has provided the opportunity for one triodide ion to adopt two different orientations, in a 3:1 ratio, which are coupled with two orientations of the dichloromethane molecule which can be accommodated in the ample solvent pocket. The components of the disordered triiodide share a common location for the central iodine atom, and the two orientations lie at ca. 9.8°.
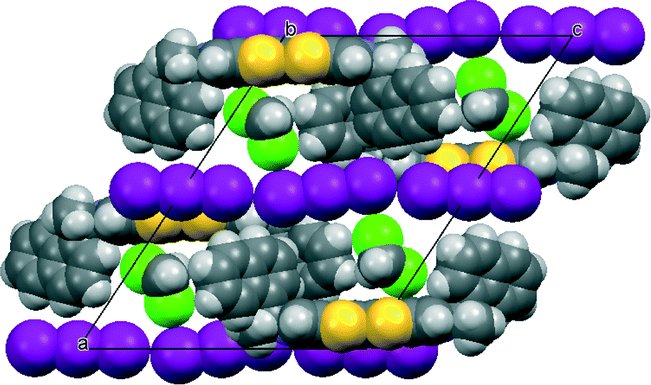 | ||
| Fig. 8 Crystal packing of 12·I3·CH2Cl2 viewed down the b axis with one orientation for the triiodide and associated dichloromethane molecule. | ||
Salt 13·I3
The salt of donor 13 with I3− provides an unusual packing arrangement; the structure is not as well defined as for the other salts (R = 0.159) even though a number of crystals were investigated with different X-ray sources, and so only the main features will be outlined. The cations and anions are packed together in layers perpendicular to the a axis with two layers per unit cell. Running through the layers in the b direction are castellated packing arrangements of triiodide ions with I⋯I contacts of 3.812(7) and 4.000(9) Å and I–I⋯I angles at the terminal I atoms of 92.2(2) and 111.8(2)° (Fig. 9). Two crystallographically independent donor cations form a face-to-face pair with their TTF planes lying perpendicular to the packing plane. Pairs are packed into bands extending along the b axis. There is considerable residual difference electron density concentrated around the triiodides suggesting extensive positional disorder. The Raman spectra for donor 13 and this salt (Table 4) are consistent with the +1 oxidation state.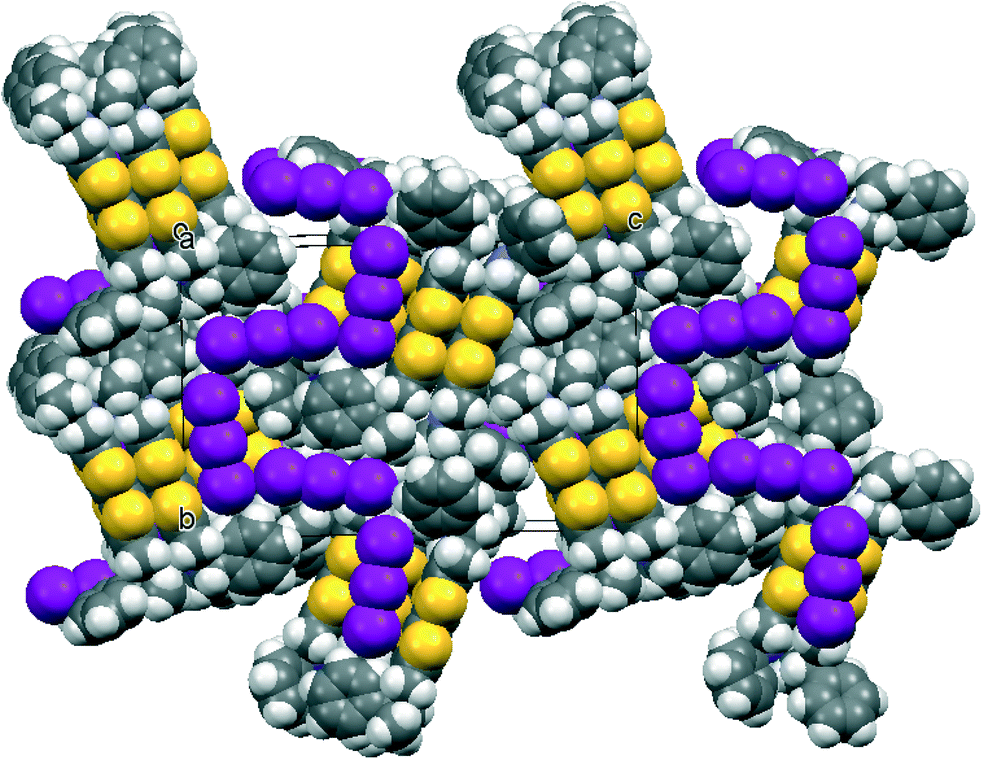 | ||
| Fig. 9 Packing arrangement of 13·I3 viewed in the bc plane showing the castellated arrangement of triiodide ions. Note that the apparent space between the castellated triodides and phenyl groups is filled by phenyl rings from the next layer above. | ||
Conclusions
The crystal structures of 1:1 triiodide salts of enantiopure donors 11–13 show a range of packing arrangements involving donor cation pairs with triiodides which are arranged in head-to-tail pairs, continuous lines, and a castellated arrangement, but do not show the helical packing observed in the neutral donors 11 and 12. Future work will explore the incorporation of alternative non-linear ions. It may also be important to pay attention to the exact conditions for preparation of the salts. For enantiopure tetramethyl-BEDT-TTF, this has formed two phases with triiodide, a 1:1 phase with radical cation pairs separated by triiodides,3 as for 11–13, but also a 2:~0.71 salt which, by comparison of its cell parameters with other salts, probably contains stacked donors.2 If larger side chains are to be used to confer chirality it is likely to be preferable to prepare donors which have such features at only one end of the molecule.
Experimental
Preparation of radical cation salts
Two-compartment H-shaped cells with a volume of 40 ml were used to grow crystals by electrocrystallisation. Each cell had two platinum electrodes, separated by two porous glass frits to prevent contamination by reduction products. The electrodes were cleaned by washing in concentrated HNO3 followed by applying a voltage across the electrodes in 1 M H2SO4 in each direction until the evolution of H2 and O2 was observed at the electrodes. Finally the electrodes were washed in distilled water and thoroughly dried. The cells were fixed inside sand-filled compartments to minimise vibration and were kept at ambient temperature. The donor (10 mg) was placed in one half of the cell and the cell was filled with a solution of tetrabutylammonium triiodide (120 mg) in 40 ml of solvent unless otherwise stated, and a constant current source was used to control the crystal growth rate.Conductivity measurements
Four-probe DC transport measurements were made on crystals of all triiodide salts, with the exception of 12·I3·THF, at room temperature using an Oxford Instruments Maglab System 2000. Gold wires (0.0025 mm diam.) were attached to the crystal, and the attached wires were connected to an eight-pin integrated circuit plug with carbon conductive cement (RS components). The resistance of the contacts was assumed to be negligible since the crystal had a high intrinsic resistance. Conductivity measurements on all five salts showed they were insulators.Raman spectroscopy
Raman spectra were recorded at room temperature with a Renishaw InVia Raman microscope equipped with a diode laser (785 nm) and a 1200 l mm−1 grating using a laser power of ca. 1 mW. The recorded spectra are the result of 20 accumulations of 5 seconds each from 1200 to 1650 cm−1 to observe the TTF ν3 and ν4 vibrations.X-Ray crystallography
Datasets for 11·I3, 12·I3·THF and 13·I3 were measured at the EPSRC National Crystallography Service, University of Southampton,28 using a Bruker-Nonius FR951 rotating anode, with collimation by 10 cm confocal mirrors, or in the case of 11·I3 a graphite monochromator, and a Bruker-Nonius diffractometer APEX II diffractometer equipped with a CCD detector at 120 K. The dataset for 11·I3·0.5THF was measured with synchrotron X-radiation (λ = 0.6893 Å) at Daresbury Laboratory SRS station 9.8, equipped with a silicon 111 monochromator and using a Bruker APEX2 CCD diffractometer at 120 K.29 Datasets for 12·I3·chlorobenzene and 12·I3·DCM were measured at NTU on an Agilent Excalibur System equipped with Sapphire detector at 120 K. The crystal of 12·I3·DCM was twinned and the two components are related by the law: 1 0 1.438/0 −1 0/0 0 −1. Because of the difficulties in obtaining a satisfactory structure refinement, several crystals of 13·I3 were investigated, some of them with synchrotron facilities at both SRS and Diamond Light Source beamline I19; these repeated attempts produced no improvement on the structure reported here, but did confirm the choice of crystal system and space group in preference to a twinned structure of lower symmetry. Structures were solved with SHELXS-97 and refined with SHELXL-9730 using the X-SEED interface31 and CRYSTALS.32 Structures were refined with anisotropic atomic displacement parameters for non-hydrogen atoms, with the exception of 13·I3, where only iodine and sulfur atoms were treated anisotropically. Hydrogen atoms were placed geometrically and treated with a riding model. Geometric analyses were made with PLATON33 and illustrations with MERCURY 3.3.34Acknowledgements
We thank the EPSRC for support (EP/C510488/1) and for funding the National Crystallography Service at Southampton and Newcastle, and University College, London and Nottingham Trent University for a studentship (ACB). LM thanks the Royal Society for Research Grants (RG100853 and RG081209), International Exchange Scheme (IE130367), and International Joint Project (JP0869972). LM thanks the RSC for a Journals Grant for International Authors. We thank Mr. Brian O'Neill and Mr. Peter Moore for the construction of constant current sources, and STFC and Diamond Light Source for access to synchrotron facilities.References
- J. D. Dunitz, A. Karrer and J. D. Wallis, Helv. Chim. Acta, 1986, 69, 69–70 CrossRef CAS PubMed; A. Karrer, J. D. Wallis, J. D. Dunitz, B. Hilti, C. W. Mayer, M. Bürkle and J. Pfeiffer, Helv. Chim. Acta, 1987, 70, 942–953 CrossRef PubMed.
- F. Pop, S. Laroussi, T. Cauchy, C. J. Gomez-Garcia, J. D. Wallis and N. Avarvari, Chirality, 2013, 25, 466–474 CrossRef CAS PubMed; S. Yang, F. Pop, C. Melan, A. C. Brooks, L. Martin, P. Horton, P. Auban-Senzier, G. L. J. A. Rikken, N. Avarvari and J. D. Wallis, CrystEngComm, 2014, 16, 3906–3916 RSC.
- M. Atzori, F. Pop, P. Auban-Senzier, R. Clérac, E. Canadell, M. L. Mercuri and N. Avarvari, Inorg. Chem., 2015, 54, 3643–3653 CrossRef CAS PubMed.
- E. Coronado, J. R. Galán-Mascarós, A. I. Coldea, P. Goddard, J. Singleton, J. D. Wallis, S. J. Coles and A. Alberola, J. Am. Chem. Soc., 2010, 132, 9271–9273 CrossRef PubMed.
- G. L. J. A. Rikken, J. Fölling and P. Wyder, Phys. Rev. Lett., 2001, 87, 236602 CrossRef CAS; G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493–494 CrossRef; V. Krstić, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315–11319 CrossRef PubMed.
- F. Leurquin, T. Ozturk, M. Pilkington and J. D. Wallis, J. Chem. Soc., Perkin Trans. 1, 1997, 3173–3178 RSC.
- S. Yang, A. C. Brooks, L. Martin, P. Day, M. Pilkington, W. Clegg, R. W. Harrington, L. Russo and J. D. Wallis, Tetrahedron, 2010, 66, 6977–6989 CrossRef CAS PubMed.
- I. Awheda, S. J. Krivickas, S. Yang, L. Martin, M. A. Guziak, A. C. Brooks, F. Pelletier, M. Le Kerneau, P. Day, P. N. Horton, H. Akutsu and J. D. Wallis, Tetrahedron, 2013, 69, 8738–8750 CrossRef CAS PubMed; L. Martin, J. D. Wallis, M. A. Guziak, J. Oxspring, J. R. Lopez, S. Nakatsuji, J. Yamada and H. Akutsu, CrystEngComm, 2014, 16, 5424–5429 RSC.
- R. J. Brown, A. C. Brooks, J.-P. Griffiths, B. Vital, P. Day and J. D. Wallis, Org. Biomol. Chem., 2007, 3172–3182 CAS.
- F. Pop, P. Auban-Senzier, A. Frackowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed.
- C. Réthoré, N. Avarvari, E. Canadell, P. Auban-Senzier and M. Fourmigué, J. Am. Chem. Soc., 2005, 127, 5748–5749 CrossRef CAS PubMed; A. M. Madalan, C. Réthoré, M. Fourmigué, E. Canadell, E. B. Lopes, M. Almeida, P. Auban-Senzier and N. Avarvari, Chem. – Eur. J., 2010, 16, 528–537 CrossRef PubMed.
- M. Chas, M. Lemarié, M. Gulea and N. Avarvari, Chem. Commun., 2008, 220–222 RSC; M. Chas, F. Riobé, R. Sancho, C. Minguillon and N. Avarvari, Chirality, 2009, 21, 818–825 CrossRef CAS PubMed.
- F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 3757 Search PubMed.
- L. Martin, S. S. Turner, P. Day, K. M. A. Malik, S. J. Coles and M. B. Hursthouse, Chem. Commun., 1999, 513 RSC; L. Martin, S. S. Turner, P. Day, P. Guionneau, J. A. K. Howard, K. M. A. Malik, M. B. Hursthouse, M. Uriuchi and K. Yakushi, Inorg. Chem., 2001, 40, 1363–1371 CrossRef CAS PubMed.
- L. Martin, P. Day, P. Horton, S. Nakatsuji, J. Yamada and H. Akutsu, J. Mater. Chem., 2010, 20, 2738–2742 RSC; L. Martin, P. Day, S. Nakatsuji, J. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369–1372 RSC.
- E. Coronado, J. R. Galan-Mascaros, C. J. Gomez-Garcia, A. Murcia-Martinez and E. Canadell, Inorg. Chem., 2004, 43, 8072–8077 CrossRef CAS PubMed.
- J. A. Schlueter, B. H. Ward, U. Geiser, H. H. Wang, A. M. Kini, J. Parakka, E. Morales, H. J. Koo, M. H. Whangbo, R. W. Winter, J. Mohtasham and G. L. Gard, J. Mater. Chem., 2001, 11, 2008–2013 RSC.
- E. Coronado, S. Curreli, C. Giménez-Saiz, C. J. Gómez-García, P. Deplano, M. L. Mercuri, A. Serpe, L. Pilia, C. Faulmann and E. Canadell, Inorg. Chem., 2006, 46, 4446–4457 CrossRef PubMed.
- F. Riobé, F. Piron, C. Réthoré, A. M. Madala, C. J. Gómez-García, J. Lacour, J. D. Wallis and N. Avarvari, New J. Chem., 2011, 35, 2279–2286 RSC.
- L. Martin, P. Day, H. Akutsu, J.-I. Yamada, S.-I. Nakatsuji, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, P. McMillan and S. Firth, CrystEngComm, 2007, 865–867 RSC.
- N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061–4076 RSC; E. Coronado and J. R. Galón-Mascaros, J. Mater. Chem., 2005, 15, 66–74 RSC.
- S. Yang, A. C. Brooks, L. Martin, P. Day, H. Li, P. Horton, L. Male and J. D. Wallis, CrystEngComm, 2009, 993–996 RSC.
- M. R. Tomcsi and F. J. Stoddart, J. Org. Chem., 2007, 72, 9335–9338 CrossRef CAS PubMed; B. W. Laursen, S. Nygaard, J. O. Jeppesen and F. J. Stoddart, Org. Lett., 2004, 6, 4167–4170 CrossRef PubMed.
- R. C. Teitelbaum, T. J. Marks and C. K. Johnson, J. Am. Chem. Soc., 1980, 102, 2986–2989 CrossRef CAS; K. Honda, M. Goto, M. Kurahashi, H. Anzai, M. Tokumoto, H. Bando and T. Ishiguro, Bull. Chem. Soc. Jpn., 1987, 60, 782–784 CrossRef; D. V. Konarev, S. S. Khasanov, A. Otsuka, G. Saito and R. N. Lyubovskaya, Synth. Met., 2005, 151, 231–238 CrossRef PubMed.
- M. A. Guziak, J. D. Wallis and L. Martin, paper in preparation.
- S. C. Nyberg and C. H. Faerman, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 274–279 CrossRef.
- S. Matsuzaki, T. Moriyama and K. Toyoda, Solid State Commun., 1980, 34, 857–859 CrossRef CAS; M. Miizutani, K. Tanaka and K. Kawabata, Synth. Met., 1991, 44, 321–325 CrossRef; H. H. Wang, J. R. Ferraro, J. M. Williams, U. Geiser and J. A. Schlueter, J. Chem. Soc., Chem. Commun., 1994, 1893–1894 RSC; I. Olejniczak, A. Frąckowiak, J. Matysiak, A. Madalan, F. Pop and N. Avarvari, Cent. Eur. J. Phys., 2013, 12, 215–220 Search PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- R. J. Cernik, W. Clegg, C. R. A. Catlow, G. Bushnell-Wye, J. V. Flaherty, G. N. Greaves, I. Burrows, D. J. Taylor, S. J. Teat and M. Hamichi, J. Synchrotron Radiat., 1997, 4, 279–286 CrossRef CAS PubMed; R. J. Cernik, W. Clegg, C. R. A. Catlow, G. Bushnell-Wye, J. V. Flaherty, G. N. Greaves, I. Burrows, D. J. Taylor, S. J. Teat and M. Hamichi, J. Synchrotron Radiat., 2000, 7, 40 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † CCDC 1413630–1413635. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01430a |
| This journal is © The Royal Society of Chemistry 2015 |