
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt(II/III), nickel(II) and copper(II) coordination clusters employing a monoanionic Schiff base ligand: synthetic, topological and computational mechanistic aspects†
Edward
Loukopoulos
a,
Benjamin
Berkoff
a,
Kieran
Griffiths
a,
Victoria
Keeble
a,
Vassiliki N.
Dokorou
a,
Athanassios C.
Tsipis
*b,
Albert
Escuer
c and
George E.
Kostakis
 *a
*a
aDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, UK. E-mail: G.Kostakis@sussex.ac.uk
bLaboratory of Inorganic and General Chemistry, Department of Chemistry, University of Ioannina, 451 10 Ioannina, Greece. E-mail: attsipis@uoi.gr
cDepartamento de Quimica Inorganica, Universitat de Barcelona, Diagonal 645, 08028 Barcelona, Spain
First published on 3rd August 2015
Abstract
Nine mono-, di- and tetranuclear coordination clusters (M = CoII/III, NiII, CuII) using a monoanionic Schiff base ligand were synthesized and characterized by X-ray crystallography. A series of transformations occur in the ligand in certain compounds for which theoretical studies are presented. Synthetic aspects, topological issues and magnetic studies are discussed.
Introduction
Despite the long history and activity of polynuclear metal complexes, also known as coordination clusters (CCs),1 their coordination chemistry continues to be a field that attracts great interest today. Among the different categories of ligands used in this field, perhaps one of the most significant is Schiff bases, which have received a large amount of attention especially in the past decade. This is due to their wide range of useful properties such as strong biological activity,2–4 ease of access and versatile coordination abilities. As a result, CCs using ligands of this nature are of special importance and many examples of their applications in materials science,5–7 catalysis,8–12 biological processes,13–15 molecular magnetic materials,16–23 photochemistry,24,25 and nanostructure studies26,27 have been explored.In particular, Schiff bases formed from o-vanillin as starting material have been of significant interest and their coordination abilities along with various metal centres have been investigated thoroughly, leading to very interesting results.28–41 For example, the usage of a Schiff base formed between L-glutamic acid and o-vanillin along with Ni(NO3)2·6H2O results in an intriguing 15-nickel metallomacrocyclic complex.31 Ferromagnetic and ferroelectric properties were observed in two enantiomerically pure nanoscale manganese CCs supported by chiral Schiff base ligands,29 while the pentanuclear compound Mn3IIICaIINaI appeared to be a sufficient catalyst for water oxidation.42
However, according to a CSD search43 all previously reported o-vanillin-based Schiff base ligands used for the synthesis of polynuclear CCs were derived mainly through a condensation of o-vanillin along with either amino alcohol such as 2-aminoethanol44 or amino acid31,45 to produce di- or triprotic organic species. Based on this observation, we recently decided to study the coordination abilities of a Schiff base ligand derived from the condensation of o-vanillin and 4-aminoantipyrine and named (E)-4-(2-hydroxy-3-methoxybenzylideneamino)-2,3-dimethyl-1-phenyl-1,2-dihydropyrazol-5-one (HL1, Scheme 1).46 Despite providing similar coordination modes to other diprotic ligands, i.e. those derived from o-vanillin and 2-aminoethanol or 2-aminophenol, HL1 is monoanionic and thus is anticipated to lead to unprecedented topologies. Indeed, the employment of HL1 in CoII/DyIII chemistry resulted in a series of polynuclear CoIIxDyIIIy CCs displaying previously unseen topologies and interesting magnetic properties.46,47 The formation of the aforementioned high nuclearity CC, using solely HL1, is in contrast to what has been achieved with other diprotic ligands that provide similar coordination environment, which resulted in tri- or tetranuclear CCs.37,48–50
 | ||
| Scheme 1 The protonated form of (E)-4-(2-hydroxy-3-methoxybenzylideneamino)-2,3-dimethyl-1-phenyl-1,2-dihydropyrazol-5-one (HL1) ligand used in this study. The different transformations of the HL1 ligand observed in compounds 5–7 (H2L2, H3L3, H2L4). Transformed organic molecules are presented in their protonated form. | ||
In this study, we report the initial employment of HL1 in CoII,III, NiII and CuII chemistry and thus present the synthesis, characterization and crystal structure of nine CCs formulated as [CoIIL12]·5MeCN (1·5MeCN), [NiIIL12]·5MeCN (2·5MeCN), [CuIIL12]·MeCN (3·MeCN), [CoII4L14(MeO)2]·2(ClO4) (4), [CoII4L24(H2O)4] (5), [CoII4L34(H2O)4] (6), [CoIIIL1L4]·2MeCN (7·2MeCN), [NiII2L13(MeOH)]·(ClO4)·2MeOH (8·2MeOH), and [CuII4L14(MeO)2]·2(ClO4)·6MeOH (9·6MeOH) as well as the magnetic properties for representative compounds 4 and 8. Synthetic and topological issues are discussed. In addition, we report interesting cases of ligand transformation found in 4–7 supported by theoretical density functional theory studies, which give valuable insight into the direct mechanism taking place during these transformations.
Experimental
Materials and methods
Synthetic procedures
Synthesis of [CoIIL12]·5MeCN (1·5MeCN). 0.55 mmol (0.190 g) of HL1 and 0.55 mmol (77.1 μL) of Et3N were dissolved in 20 ml of MeCN. The resulting yellow solution was brought to reflux and stirred for 10 minutes. To this, 0.23 mmol (0.050 g) of CoBr2 were added and the resulting red solution was refluxed for another 20 minutes, filtered and kept in a vial and subjected to slow evaporation. Red crystals formed within 1 day. Yield: 35% (based on Co). Selected IR peaks (cm−1): 2929 (w), 2820 (w), 1590 (s), 1536 (m), 1492 (m), 1462 (m), 1434 (s), 1389 (s), 1300 (m), 1237 (m), 1209 (s), 1185(s), 1108 (m), 1080 (m), 1045 (m), 972 (m), 921 (w), 878 (w), 853 (w), 786 (w), 740 (s), 702 (s), 678 (m). Analogous reaction with Co(ClO4)2·6H2O instead of CoBr2 at room temperature afforded a powder crystalline material which was found to be isostructural to 1 by determining the unit cell of the crystals and recording the IR spectra.
Synthesis of [NiIIL12]·5MeCN (2·5MeCN). 0.25 mmol (0.084 g) of HL1 and 0.25 mmol (34.9 μL) of Et3N were dissolved in 20 ml of MeCN while stirring to produce a yellow solution. To this, 0.1 mmol (0.037 g) of Ni(ClO4)2·6H2O were added. The resulting yellow solution was stirred for a further 15 minutes, filtered, and then kept in a vial and stored at room temperature. Large green needles were obtained after 2 days. Yield: 55% (based on Ni). Selected IR peaks (cm−1): 2927 (w), 2822 (w), 2247 (w), 1593 (s), 1541 (m), 1496 (m), 1450 (m), 1438 (s), 1393 (m), 1298 (m), 1242 (m), 1208 (s), 1185(s), 1109 (m), 1076 (m), 1047 (m), 974 (s), 922 (w), 878 (w), 847 (w), 820 (w), 781 (w), 741 (s), 702 (s), 686 (m).
Synthesis of [CuIIL12]·MeCN (3·MeCN). 0.25 mmol (0.084 g) of HL1 and 0.5 mmol (69.0 μL) of Et3N were dissolved in 20 ml of MeCN while stirring to produce a yellow solution. To this, 0.1 mmol (0.020 g) of Cu(OAc)2·H2O were added. The resulting dark brown solution was stirred for a further 15 minutes, filtered, and then kept in a vial and stored at room temperature. Dark brown block crystals were obtained after 1 day. Yield: 45% (based on Cu). Selected IR peaks (cm−1): 2932 (w), 2830 (w), 1662 (s), 1583 (s), 1538 (m), 1492 (m), 1456 (s), 1434 (m), 1389 (m), 1298 (m), 1242 (m), 1208 (s), 1155 (w), 1109 (m), 1073 (s), 1043 (s), 976 (s), 928 (w), 878 (w), 850 (w), 820 (w), 789 (m), 742 (s), 707 (s), 688 (m). Elemental analysis for C40H39CuN7O6: calcd. C 61.81, H 5.06, N 12.61; found C 61.67, H 5.27, N 12.51.
Synthesis of [CoII4L14(MeO)2]·2(ClO4) (4). 0.25 mmol (0.084 g) of HL1 and 0.25 mmol (34.9 μL) of Et3N were dissolved in 20 ml MeOH while stirring to produce a yellow solution. To this, 0.1 mmol (0.037 g) of Co(ClO4)2·6H2O were added. The resulting red solution was stirred for a further 15 minutes, filtered, and then kept in a vial and stored at room temperature. Red prismatic crystals were obtained after 5 days. Yield: 25% (based on Co). Selected IR peaks (cm−1): 2935 (w), 2817 (w), 1603 (s), 1583 (s), 1558 (m), 1544 (m), 1489 (w), 1456 (s), 1433 (s), 1391 (m), 1241 (m), 1213 (s), 1194 (m), 1081 (s), 1036 (s), 969 (s), 913 (w), 869 (w), 855 (w), 788 (m), 741 (s), 701 (s), 677 (m).
Synthesis of [CoII4L24(H2O)4] (5). 0.30 mmol (0.100 g) of HL1 and 0.30 mmol (41.8 μL) of Et3N were dissolved in 20 ml MeCN while stirring to produce a yellow solution. To this, 0.24 mmol (0.060 g) of Co(OAc)2·4H2O were added. The resulting red solution was stirred for a further 20 minutes, filtered, and then kept in a vial and stored at room temperature. Red block crystals were obtained after 3 days. Yield: 20% (based on Co). Selected IR peaks (cm−1): 2929 (w), 2834 (w), 1623 (s), 1592 (s), 1540 (m), 1497 (m), 1440 (s), 1404 (m), 1311 (m), 1235 (m), 1209 (s), 1072 (s), 1024 (s), 971 (s), 877 (w), 857 (w), 787 (w), 738 (s), 722 (s), 684 (m).
Synthesis of [CoII4L34(H2O)4] (6). Method 1: (6) was prepared in the same ratio and solvent as (3) by using Co(OAc)2·4H2O and then carefully layering the solution over Et2O at a respective ratio of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Red block crystals were obtained after 1 day. Yield: 35% (based on Co). Selected IR peaks (cm−1): 2935 (w), 2828 (w), 1621 (s), 1592 (s), 1544 (m), 1494 (m), 1445 (s), 1403 (m), 1307 (m), 1239 (m), 1210 (s), 1074 (s), 1026 (s), 967 (s), 875 (w), 856 (w), 811 (w), 737 (s), 719 (s), 690 (m). Method 2: 0.56 mmol (0.190 g) of L and 1.11 mmol (154.3 μL) of Et3N were dissolved in 20 ml MeCN. The resulting yellow solution was brought to reflux and stirred for 10 minutes. To this, 0.46 mmol (0.157 g) of Co(BF4)2·6H2O were added and the resulting red solution was refluxed for another 20 minutes, filtered and kept in a vial and subjected to slow evaporation. Red crystals formed within 1 day. Yield: 20% (based on Co). Method 3: 0.56 mmol (0.190 g) of L and 2.21 mmol (308.6 μL) of Et3N were dissolved in 20 ml MeCN. The resulting yellow solution was brought to reflux and stirred for 10 minutes. To this, 0.45 mmol (0.166 g) of Co(ClO4)2·6H2O were added and the resulting red solution was refluxed for another 20 minutes, filtered and kept in a vial and subjected to slow evaporation. Red crystals formed within 1 day. Yield: 20% (based on Co). Elemental analysis for C76H76Co4N12O24: calcd. C 51.37, H 4.31, N 9.46; found C 47.67, H 4.15, N 8.74. This result corresponds to the presence of eight water molecules. C76H76Co4N12O24 (H2O)8: C 47.49, H 4.82, N 8.75. The crystalline material collected using Methods 2 and 3 was found to be isostructural to that collected using Method 1 by determining the unit cell of the crystals and recording the IR spectra.
:2. Red block crystals were obtained after 1 day. Yield: 35% (based on Co). Selected IR peaks (cm−1): 2935 (w), 2828 (w), 1621 (s), 1592 (s), 1544 (m), 1494 (m), 1445 (s), 1403 (m), 1307 (m), 1239 (m), 1210 (s), 1074 (s), 1026 (s), 967 (s), 875 (w), 856 (w), 811 (w), 737 (s), 719 (s), 690 (m). Method 2: 0.56 mmol (0.190 g) of L and 1.11 mmol (154.3 μL) of Et3N were dissolved in 20 ml MeCN. The resulting yellow solution was brought to reflux and stirred for 10 minutes. To this, 0.46 mmol (0.157 g) of Co(BF4)2·6H2O were added and the resulting red solution was refluxed for another 20 minutes, filtered and kept in a vial and subjected to slow evaporation. Red crystals formed within 1 day. Yield: 20% (based on Co). Method 3: 0.56 mmol (0.190 g) of L and 2.21 mmol (308.6 μL) of Et3N were dissolved in 20 ml MeCN. The resulting yellow solution was brought to reflux and stirred for 10 minutes. To this, 0.45 mmol (0.166 g) of Co(ClO4)2·6H2O were added and the resulting red solution was refluxed for another 20 minutes, filtered and kept in a vial and subjected to slow evaporation. Red crystals formed within 1 day. Yield: 20% (based on Co). Elemental analysis for C76H76Co4N12O24: calcd. C 51.37, H 4.31, N 9.46; found C 47.67, H 4.15, N 8.74. This result corresponds to the presence of eight water molecules. C76H76Co4N12O24 (H2O)8: C 47.49, H 4.82, N 8.75. The crystalline material collected using Methods 2 and 3 was found to be isostructural to that collected using Method 1 by determining the unit cell of the crystals and recording the IR spectra.
Synthesis of [CoIIIL1L4]·2MeCN (7·2MeCN). Compound 7 was prepared in the same ratio, metal salt and solvent as (5) by refluxing the solution for 20 minutes. The resulting red solution was then filtered and kept in a vial stored at room temperature. Red block crystals were obtained after 3 days. Yield: 30% (based on Co). Selected IR peaks (cm−1): 2935 (w), 1639 (s), 1621 (s), 1591 (s), 1547 (m), 1492 (m), 1442 (s), 1401 (m), 1302 (m), 1238 (m), 1213 (s), 1077 (s), 1028 (s), 968 (s), 875 (w), 856 (w), 811 (w), 737 (s), 719 (s), 690 (m).
Synthesis of [NiII2L13(MeOH)]·(ClO4)·2MeOH (8·2MeOH). Compound 8 was prepared in the same ratio and solvent as (4) by using Ni(ClO4)2·6H2O and then keeping the filtered solution in a vial stored at room temperature. Large dark yellow crystals were obtained after 2 days. Yield: 35% (based on Ni). Selected IR peaks (cm−1): 2935 (w), 2816 (w), 1603 (s), 1585 (s), 1559 (m), 1543 (m), 1492 (w), 1453 (s), 1436 (s), 1391 (m), 1241 (m), 1213 (s), 1193 (m), 1081 (s), 970 (s), 913 (w), 856 (w), 789 (m), 741 (s), 701 (s), 677 (m). Elemental analysis for C60H66ClN9Ni2O16: calcd. C 54.51, H 5.03, N 9.53; found C 54.33, H 4.88, N 9.41.
Synthesis of [CuII4L14(MeO)2]·2(ClO4)·6MeOH (9·6MeOH). 0.1 mmol (0.034 g) of HL1 and 0.1 mmol (13.9 μL) of Et3N were dissolved in 20 ml of MeOH while stirring to produce a yellow solution. To this, 0.2 mmol (0.074 g) of Cu(ClO4)2·6H2O were added. The resulting dark green solution was stirred for a further 20 minutes, filtered, and then kept in a vial stored at room temperature. Brown block crystals were obtained after 2 days. Yield: 20% (based on Cu). Selected IR peaks (cm−1): 2939 (w), 2817 (w), 1603 (s), 1576 (s), 1558 (m), 1548 (m), 1492 (w), 1451 (s), 1433 (s), 1391 (m), 1241 (m), 1213 (s), 1195 (m), 1081 (s), 974 (s), 916 (w), 869 (w), 855 (w), 791 (m), 742 (s), 701 (s), 677 (m). Elemental analysis for C84H102Cl2Cu4N12O28: calcd. C 42.15, H 4.01, N 8.19; found C 41.50, H 4.04, N 7.62.
Crystallography
Data for 1, 2, 3, 5, 6, 9 and 10 were collected (ω-scans) at the University of Sussex using an Agilent Xcalibur Eos Gemini Ultra diffractometer with a CCD plate detector under a flow of nitrogen gas at 173(2) K using Mo Kα radiation (λ = 0.71073 Å). CrysAlis CCD and RED software were used for data collection and processing, respectively. Reflection intensities were corrected for absorption by the multi-scan method. Data for 4, 7 and 8 were collected at the National Crystallography Service, University of Southampton.59 All structures were determined using Olex2,60 solved using either Superflip61 or SHELXT62,63 and refined with SHELXL-2014.64 All non-H atoms were refined with anisotropic thermal parameters, and H atoms were introduced at calculated positions and allowed to ride on their carrier atoms. In compounds 8 and 9, atoms of minor components (several with partial occupancy) were sensibly refined isotropically. Crystal data and structure refinement parameters for all compounds are given in Tables S1 and S2.† Geometric/crystallographic calculations were performed using PLATON,65 Olex2,60 and WINGX62 packages; graphics were prepared with Crystal Maker and MERCURY.66Results and discussion
Crystal structure description
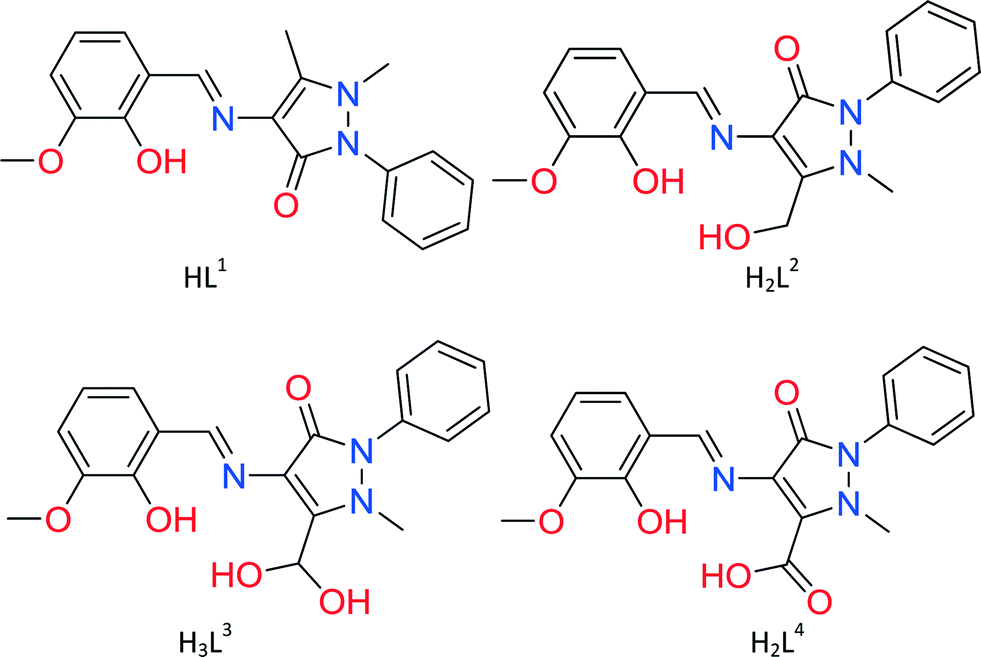
Compounds 1–3 are monomers synthesized using a 4:5:5, 2:5:10 or 2:5:5 metal–ligand–base ratio and acetonitrile as solvent (Fig. 1). All compounds crystallize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and contain one molecule in the asymmetric unit. Compounds 1 and 2 are isostructural and thus only the former will be further described. Compound 1 is a CoII monomer in which the metal centre has a distorted octahedral geometry, coordinating to two ligand molecules and a total of 6 atoms. Each ligand coordinates to CoIIvia the phenoxide oxygen atoms (O2 and O5), the imine group nitrogen atoms (N1 and N4) and the carbonyl group oxygen atoms (O3 and O6) (Scheme S1,† mode A). The mean M–Ophenoxide distances are 2.0076(17) and 2.0141(16) Å, while the M–Nimine distances were measured at 2.0873(19) and 2.0855(18) Å. M–Ocarbonyl distances are slightly longer at 2.2458(16) and 2.2286(16) Å, respectively, indicative of a CoII. Similar bond distances for 2 can be found in the ESI† (Table S3). Five acetonitrile molecules are also present in the crystal lattice. Each molecule is isolated within the crystal structure as there are no hydrogen bonds or other supramolecular interactions formed.
space group and contain one molecule in the asymmetric unit. Compounds 1 and 2 are isostructural and thus only the former will be further described. Compound 1 is a CoII monomer in which the metal centre has a distorted octahedral geometry, coordinating to two ligand molecules and a total of 6 atoms. Each ligand coordinates to CoIIvia the phenoxide oxygen atoms (O2 and O5), the imine group nitrogen atoms (N1 and N4) and the carbonyl group oxygen atoms (O3 and O6) (Scheme S1,† mode A). The mean M–Ophenoxide distances are 2.0076(17) and 2.0141(16) Å, while the M–Nimine distances were measured at 2.0873(19) and 2.0855(18) Å. M–Ocarbonyl distances are slightly longer at 2.2458(16) and 2.2286(16) Å, respectively, indicative of a CoII. Similar bond distances for 2 can be found in the ESI† (Table S3). Five acetonitrile molecules are also present in the crystal lattice. Each molecule is isolated within the crystal structure as there are no hydrogen bonds or other supramolecular interactions formed.
 | ||
| Fig. 1 (Left) The structure of compounds 1 and 2. M = CoII (1), NiII (2). (Right) The structure of compound 3. H atoms and solvent molecules are omitted for clarity. Colour code: M, grey; Cu, light blue; O, red; C, black; N, blue. | ||
Compound 3 is a Cu monomer in which the metal centre coordinates to two ligand molecules and a total of 4 atoms, having a distorted square planar geometry. Each ligand coordinates to CuIIvia the phenoxide oxygen atoms (O2 and O5) and the imine group nitrogen atoms (N1 and N4) (Scheme S1,† mode B). The mean M–Ophenoxide distances are 1.8991(13) Å and 1.9006(14) Å, while the M–Nimine distances were measured at 1.9671(14) and 1.9532(17) Å, all slightly lower than the respective distances in compounds 1 and 2. One acetonitrile molecule is also present in the crystal lattice. As in 1 and 2, no hydrogen bonding or other supramolecular interaction can be found within the crystal structure.
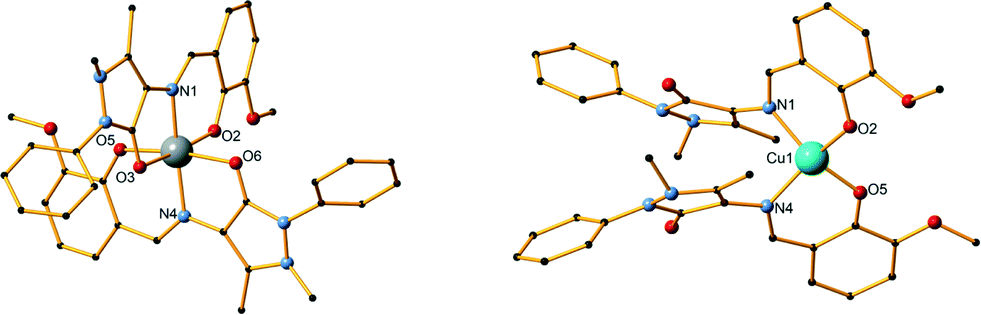
Compound 4 is synthesized using a 2:5:5 metal–ligand–base ratio and methanol as solvent and crystallizes in the monoclinic P21/c space group. The asymmetric unit contains four metal centres, four ligand molecules, two bridging methoxides and two perchlorate counter ions (Fig. 2, top).
 | ||
| Fig. 2 The structure of compounds 4 (top) and 5 (middle). Symmetry operations: (i) 1 − x, 3/2 − y, z; (ii) 5/4 − y, 1/4 + x, 1/4 − z; (iii) y − 1/4, 5/4 − x, 1/4 − z. (Bottom) The structure of compound 6. Symmetry operations: (i) 1 − x, 3/2 − y, z; (ii) y − 1/4, 5/4 − x, 5/4 − z; (iii) 5/4 − y, 1/4 + x, 5/4 − z. H atoms and lattice molecules are omitted for clarity. Colour code: Co, purple; O, red; C, black; N, blue. | ||
The main core of the cluster is a deformed cubane-like Co4O4 consisting of four CoII centres, two bridging μ3-O methoxido atoms, and two bridging μ3-O phenoxido atoms. Each of the CoII centres coordinates to six atoms and displays a distorted octahedral geometry. The four organic ligands exhibit two different coordination modes, two ligands per mode. In the first mode, the phenoxide oxygen atom and the imine nitrogen atom coordinate to one CoII centre, while the carbonyl oxygen atom coordinates to a second CoII centre (Scheme S1,† mode C). In the second mode, one CoII centre is coordinated to the phenoxide oxygen atom, the imine nitrogen atom and the carbonyl oxygen atom, while the phenoxide atom is further bridging two CoII centres and the methyl ether group oxygen atom also coordinates to the third CoII centre (Scheme S1,† mode D). The CoII⋯CoII distances range from 3.018(7) to 3.350(7) Å. Selected bond lengths and angles for 4 are given in Table S10.† No hydrogen bonds or other supramolecular interactions can be found between the molecules within the crystal structure.
Compound 5 is synthesized using a 4:5:5 metal–ligand–base ratio and acetonitrile as solvent (Fig. 2, middle). As in 4, a CoII4O4 cubane-like core is formed; however, a transformation of the ligand has taken place. The CH3 group of the C atom of the pyrazolone ring is oxidized to a CH2OH, as confirmed by X-ray crystallography (Scheme 1, H2L2). Compound 5 has crystallographically imposed ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) symmetry and crystallizes in the tetragonal I41/a space group and the asymmetric unit consists of one CoII centre, one L2 ligand and one water molecule. There are no solvent molecules or counter ions present in the structure. The cubane-like CoII4O4 consists of the four CoII and four bridging μ3-O methoxido atoms of the CH2OH transformed group. Only one type of coordination mode is present in the structure, as the ligand coordinates to the metal centre via the imine nitrogen atom (N1), the phenoxide oxygen atom (O2) and the CH2OH transformed group oxygen atom (O4) (Scheme S1,† mode G). A water molecule also coordinates to each of the cobalt centres, thus fulfilling its distorted octahedral geometry. The mean M–Nimine distance was measured at 2.110(2) Å, which is the longest M–Nimine bond observed in this study. Co⋯Co distances range from 3.2064(10) to 3.2157(6) Å. Additional bond distances and angles are given in Table S10.† The crystal structure of 5 is stabilized by a strong O–H⋯O intermolecular hydrogen bond which involves a water oxygen atom (O5) as a donor and the phenoxide oxygen atom (O2) as an acceptor. The parameters of this hydrogen bond can be found in Table S4.† Compound 6 has crystallographically imposed symmetry and is isoskeletal to 4 and 5. It was synthesized using a 2:5:10 metal–ligand–base ratio and acetonitrile as solvent (Fig. 2, right). Reactions with different conditions under a 4:5:10 and a 4:5:20 ratio afforded the same product but in a lower yield. As in 4 and 5, a CoII4O4 cubane-like core is formed; however, a different transformation of the ligand is observed. The methyl group connected to the pyrazolone ring is, in the present case, transformed to a CH(OH)2 group (Scheme 1, H3L3). The two C–O bonds were crystallographically refined, without any restrictions, to 1.346(7) and 1.398(4) Å for O4 and O5, respectively, typical of a single C–O bond value. The compound crystallizes in the tetragonal I41/a space group and the asymmetric unit consists of one CoII centre, one L3 ligand and one water molecule. There are no solvent molecules or counter ions present in the structure. The cubane-like CoII4O4 consists of the four CoII centres and four bridging μ3-O atoms; the oxygen atom belongs to the transformed CH(OH)2 group. Only one type of coordination mode is present in the structure, as the ligand coordinates to the metal centre via the imine nitrogen atom (N1), the phenoxide oxygen atom (O2) and an oxygen atom (O4) (Scheme S1,† mode H).
symmetry and crystallizes in the tetragonal I41/a space group and the asymmetric unit consists of one CoII centre, one L2 ligand and one water molecule. There are no solvent molecules or counter ions present in the structure. The cubane-like CoII4O4 consists of the four CoII and four bridging μ3-O methoxido atoms of the CH2OH transformed group. Only one type of coordination mode is present in the structure, as the ligand coordinates to the metal centre via the imine nitrogen atom (N1), the phenoxide oxygen atom (O2) and the CH2OH transformed group oxygen atom (O4) (Scheme S1,† mode G). A water molecule also coordinates to each of the cobalt centres, thus fulfilling its distorted octahedral geometry. The mean M–Nimine distance was measured at 2.110(2) Å, which is the longest M–Nimine bond observed in this study. Co⋯Co distances range from 3.2064(10) to 3.2157(6) Å. Additional bond distances and angles are given in Table S10.† The crystal structure of 5 is stabilized by a strong O–H⋯O intermolecular hydrogen bond which involves a water oxygen atom (O5) as a donor and the phenoxide oxygen atom (O2) as an acceptor. The parameters of this hydrogen bond can be found in Table S4.† Compound 6 has crystallographically imposed symmetry and is isoskeletal to 4 and 5. It was synthesized using a 2:5:10 metal–ligand–base ratio and acetonitrile as solvent (Fig. 2, right). Reactions with different conditions under a 4:5:10 and a 4:5:20 ratio afforded the same product but in a lower yield. As in 4 and 5, a CoII4O4 cubane-like core is formed; however, a different transformation of the ligand is observed. The methyl group connected to the pyrazolone ring is, in the present case, transformed to a CH(OH)2 group (Scheme 1, H3L3). The two C–O bonds were crystallographically refined, without any restrictions, to 1.346(7) and 1.398(4) Å for O4 and O5, respectively, typical of a single C–O bond value. The compound crystallizes in the tetragonal I41/a space group and the asymmetric unit consists of one CoII centre, one L3 ligand and one water molecule. There are no solvent molecules or counter ions present in the structure. The cubane-like CoII4O4 consists of the four CoII centres and four bridging μ3-O atoms; the oxygen atom belongs to the transformed CH(OH)2 group. Only one type of coordination mode is present in the structure, as the ligand coordinates to the metal centre via the imine nitrogen atom (N1), the phenoxide oxygen atom (O2) and an oxygen atom (O4) (Scheme S1,† mode H).
A water molecule also coordinates to each of the cobalt centres to fulfil the distorted octahedral geometry. The mean M–Nimine distance was measured at 2.107(3) Å. Co⋯Co distances range from 3.2207(11) to 3.2277(9) Å. Additional bond distances and angles are given in Table S10.† The crystal structure of 6 is stabilized by a strong O–H⋯O intermolecular hydrogen bond, which involves a water oxygen atom (O6) as a donor and a carbonyl oxygen atom (O3) as an acceptor. The structure is further stabilized by a strong O–H⋯O intramolecular hydrogen bond within two ligands, in which an oxygen atom of the transformed group (O5) participates as a donor. The parameters of these hydrogen bonds are listed in Table S5.†
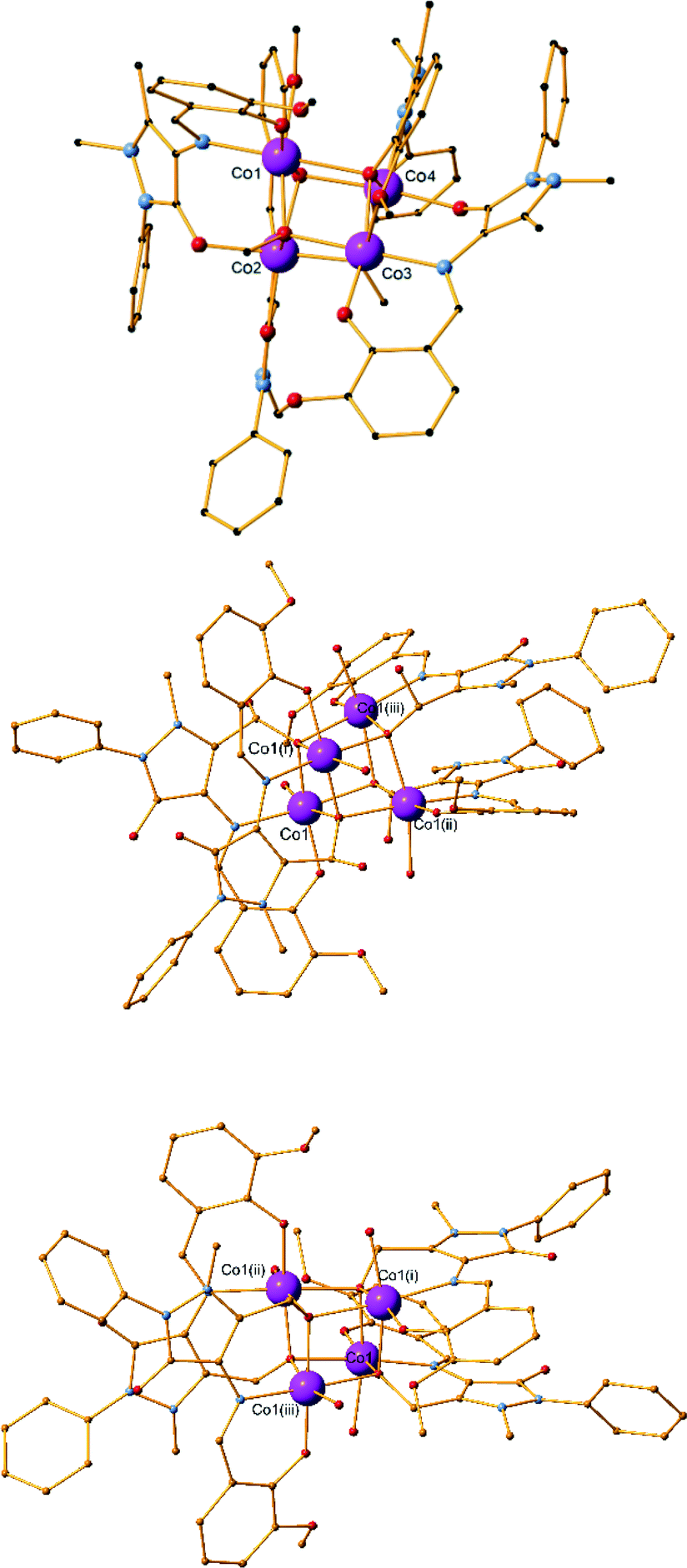
Compound 7 was synthesized using a 4:5:5 metal–ligand–base ratio and acetonitrile as solvent (Fig. 3). It crystallizes in the triclinic P space group and contains two molecules in the asymmetric unit. Unlike the three previous structures, bond valence sum (BVS) calculations67 are indicative of oxidation state III for both Co centres (3.22 for Co1 and 3.24 for Co2). The ligand undergoes another oxidation (H2L4, Scheme 1) and the methyl group connected to the pyrazolone ring is transformed to a carboxylate, as assigned by the 1.238(5)Å and 1.275(4)Å bond distances for C31–O7 and C31–O6, respectively. Each molecule contains a CoIII centre, one L1 ligand and one L4 ligand as well as two acetonitrile solvent molecules. The CoIII centre has a distorted octahedral geometry and is coordinated to one L1 molecule via the phenoxide oxygen atom, the imine group nitrogen atom and the carbonyl group oxygen atom (Scheme S1,† mode A) as well as one L4 molecule via the imine nitrogen atom, the phenoxide oxygen atom and the carboxyl group oxygen atom (Scheme S1,† mode I). The mean M–Nimine distances are relatively similar for both ligands (1.929(3) Å for L4 coordination, 1.933(3) and 1.937(3) Å for L1 coordination), while the M–Ophenoxide distances are smaller in the case of the L1 ligand (1.869(3) Å for L1 coordination, 1.873(3) and 1.885(3) Å for L4 coordination). The mean M–Ocarbonyl distances are 1.970(3) and 1.985(3) Å, significantly larger than the M–Ocarboxyl distances which were measured at 1.904(3) and 1.912(3) Å. Two acetonitrile molecules are also present in the crystal lattice. As in the previous monomeric compounds (1–3), there are no hydrogen bonds or other supramolecular interactions formed between the molecules within the crystal structure.
 | ||
| Fig. 3 The structure of compound 7. H atoms and solvent molecules are omitted for clarity. Color code: Co, pink; O, red; C black; N, blue. | ||
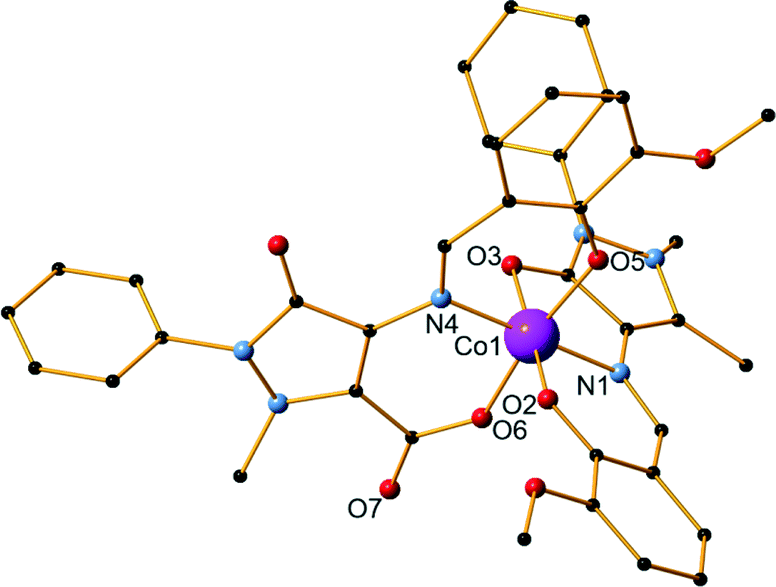
Compound 8 is a Ni dimer, synthesized using a 2:5:5 metal–ligand–base ratio and methanol as solvent (Fig. 4). It crystallizes in the monoclinic I2/c space group and contains one molecule in the asymmetric unit. Each nickel centre is coordinated to six atoms and displays a distorted octahedral geometry. There are three ligand molecules in the structure and each exhibits a different coordination mode (Scheme 1,† modes A, E, and F). Ni1 is coordinated to two ligands, while Ni2 is coordinated to all three. In detail, Ni1 is coordinated to the carbonyl oxygen atom of one ligand (O6) and the imine nitrogen atoms (N4, N7) as well as the phenoxide oxygen atoms (O5, O8) of two ligand molecules. A terminal methanol solvent molecule also coordinates to the metal centre through the oxygen atom O10. Ni2 is coordinated to phenoxide oxygen atoms from all three ligands (O2, O5, O8), the imine nitrogen atom and the phenoxide oxygen atom of one ligand (N1 and O3, respectively), as well as the methyl ether group oxygen atom of a second ligand molecule (O4). Out of the respective Ni–O distances, Ni2–O4 was the longest and was measured at 2.295(3) Å, while Ni1–O5 was the shortest at 1.984(3) Å. Comparing the respective Ni–N distances, the mean Ni1–N4 distance was the longest at 2.085(3) Å, while Ni2–N1 was the shortest at 2.020(3) Å. Selected bond lengths and angles are given in the ESI† (Table S6). Two perchlorate counter ions are also present in the structure along with two lattice solvent molecules. The crystal structure of 8 is stabilized by strong O–H⋯O intermolecular hydrogen bonds, which are formed between either two lattice methanol molecules or one lattice methanol and a perchlorate anion. Further stabilization occurs with the formation of a strong O–H⋯O intramolecular hydrogen bond within the ligands. The parameters of these hydrogen bonds are listed in Table S7.† Compound 9 is synthesized using a 4:2:2 metal–ligand–base ratio and methanol as solvent and is isoskeletal68 to compound 4 previously described in detail. Bond angles and distances can be found in the ESI† (Tables S8 and S9).
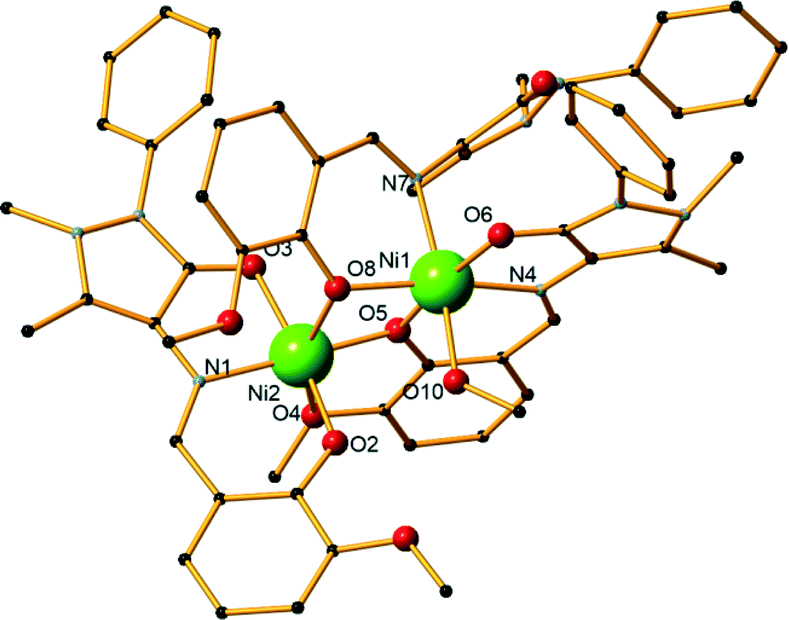 | ||
| Fig. 4 The structure of compound 8. H atoms and lattice molecules are omitted for clarity. Color code: Ni, green; O, red; C, black; N, blue. | ||
Topological features
HL1 offers similar coordination environment to other diprotic ligands; however, in order to identify its unique and different coordination abilities we performed an extended literature CSD search43 seeking the coordination abilities of any ligand that resembles those in Scheme 2, bottom. Then, we further categorized these findings employing our topological approach to describe CCs,69 which resulted in a library of all CCs with nuclearity over 3 (ESI,† Excel). The library consists of 100 CCs possessing 29 different motifs. Only 28 entries contain solely 3d (Mn, Fe, Co, Ni and Cu) metal centres. Utilizing HL1 along with CoII and CuII, in methanolic solution, results in cubane Co4II (4) and Cu4II (9) structures (3M4–1, Scheme 2),69 in the presence of MeO bridges, and along with Ni(ClO4)2·6H2O in a NiII dimer (8) (1M2–1, Scheme 2). In contrast, the reaction of diprotic ligands with Ni results in a NiII4 cubane (3M4–1),30 built solely by the organic ligand, while Cu forms either a defective dicubane70 (2,3M4–1, Scheme 2) or a cubane (3M4–1) motif.44,71 No data were found for Co chemistry, and thus for a structural comparison, we performed the reaction of the diprotic ligand [(E)-2-(2-hydroxy-3-methoxybenzylidene-amino)phenol, H2L5] with Co(ClO4)2·6H2O. The latter reaction resulted in a compound formulated as [CoIII2CoII2(MeO)4(L5)2Cl2] (10) (Fig. S4, Table S11†) possessing a defective dicubane or 2,3M4–1 topology, indicating sensitivity to oxidation. The transformation of the perchlorate anion to a chlorine anion is not unusual and has been seen before.72,73 More crystallographic data are necessary to perform a complete structural comparison; however, the present findings indicate that HL1, despite offering similar coordination sites to other similar diprotic ligands, coordinates completely different from them and thus its coordination chemistry can lead to new, interesting and unprecedented topologies.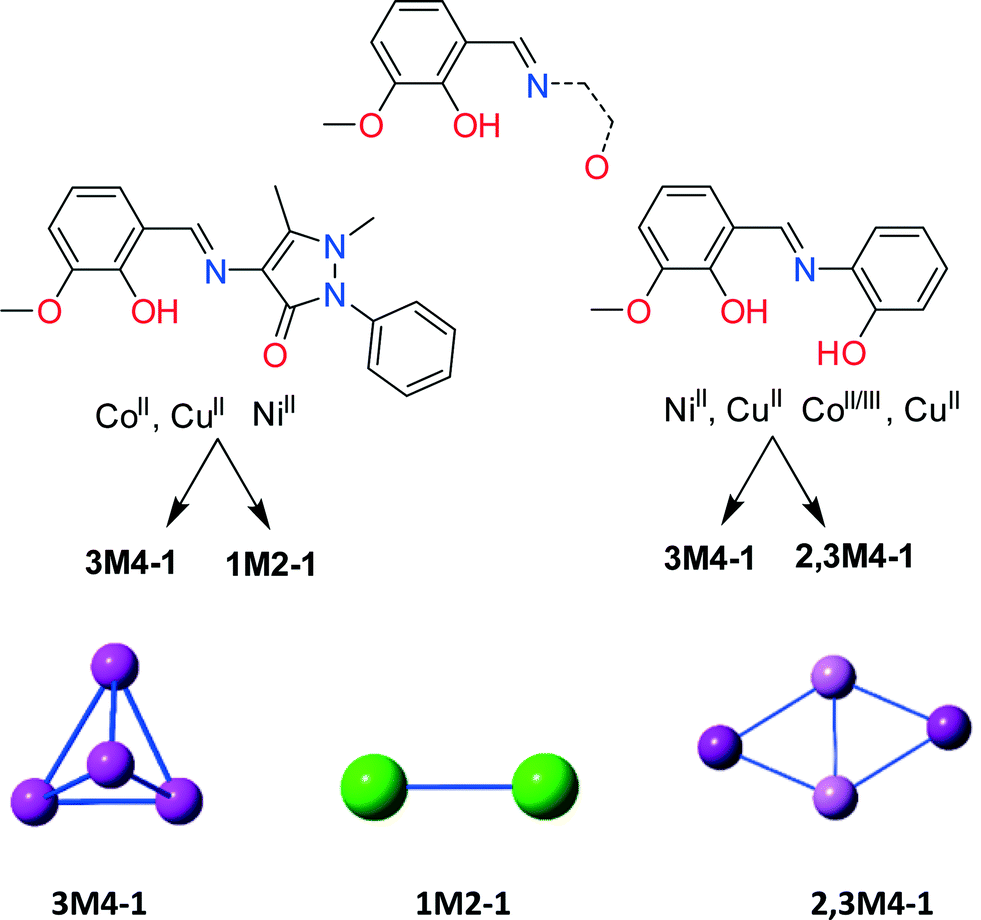 | ||
| Scheme 2 (Top) A drawing of the organic molecules with similar coordination environment to HL1 used for the CSD search. (Bottom) A cartoon representation of the Co-, Ni-, and Cu-based CCs obtained using different ligands, indicated in the centre, and their motif. | ||
Synthetic issues
A detailed analysis of the synthetic aspects for the afforded compounds 1–9 provides very interesting points. There are a few parameters that influence the resulting structures. Among these parameters are a) the solvent which was used, b) the temperature in which the syntheses took place and c) the metal source used. From the resulting compounds it becomes evident that these attributes not only affect the metal nuclearity and the coordination modes of the ligand to the metal centres but also facilitate the ligand transformations which were observed in compounds 4–7.With regard to the solvent which was used, the afforded compounds can be divided into two categories: a) those which were obtained using a polar solvent such as methanol (compounds 4, 8, 9) and b) those which were obtained using a non-polar solvent, i.e. acetonitrile (compounds 1–3, 5–7). An immediate observation is the change in the metal nuclearity which also seems to be connected to the ligand transformations; the reaction in protic solvent affords compounds 4, 8 and 9 which are either dimers or tetramers (Co4, Ni2, Cu4); however, upon using acetonitrile, the respective monomeric Co (1), Ni (2), and Cu (3) compounds are obtained. In addition, the room temperature reaction of Co(ClO4)2·6H2O with HL1 in MeCN results in a powder crystalline material in low yield which was found to be the mononuclear compound 1; however, a similar reaction in MeOH results in the formation of the tetranuclear CoII4.
It is well known that Co-based catalysts have been used in the oxidation of alkanes for the synthesis of terephthalic acid or adipic acid involving molecular O2.74 In Table 1, a synthetic overview of the synthesis of compounds 1, 4, 5, 6 and 7 is given, where transformation of the organic ligand is observed. With regard to the temperature in which the syntheses of the aforementioned compounds took place two major conclusions can be drawn. a) The room temperature reaction of HL1 with Co(ClO4)2·6H2O in MeCN afforded compound 1 (entry 2); however, when a similar reaction is performed under reflux, a ligand transformation occurs (H3L3, Scheme 1), affording compound 6 (entry 7). b) A comparison of the synthetic protocol of compounds 5 and 7 indicates that these two molecules were derived using the same metal salt Co(OAc)2·4H2O, solvent (MeCN) and metal–ligand–base ratio (4:5:5); however, room temperature conditions favor the formation of a cobalt tetramer with the transformed L2 ligand found in 5 (entry 4), while reflux conditions lead towards the synthesis of a cobalt monomer with a mix of L1 and L4 ligands, found in 7 (entries 4 and 8, respectively).
| Entries | Metal salt | T | Solvent | Ratio (M:L:B) |
Compound | Ligand |
|---|---|---|---|---|---|---|
| 1 | CoBr2 | Reflux | MeCN | 4:5:5 |
1 | L1 |
| 2 | Co(ClO4)2·6H2O | r.t. | MeCN | 4:5:5 |
1 | L1 |
| 3 | Co(ClO4)2·6H2O | r.t. | MeOH | 2:5:5 |
4 | L1 |
| 4 | Co(OAc)2·4H2O | r.t. | MeCN | 4:5:5 |
5 | L2 |
| 5 | Co(OAc)2·4H2O | r.t. | MeCN, Et2O | 2:5:10 |
6 | L3 |
| 6 | Co(BF4)2·6H2O | Reflux | MeCN | 4:5:10 |
6 | L3 |
| 7 | Co(ClO4)2·6H2O | Reflux | MeCN | 4:5:20 |
6 | L3 |
| 8 | Co(OAc)2·4H2O | Reflux | MeCN | 4:5:5 |
7 | L1, L4 |
From this table, another two important notes can be pointed out, both related with the synthesis of compound 6. First, repeating the room temperature reaction that affords 5 and layering the resultant solution with Et2O affords compound 6 (entry 5). Second, the latter compound can be obtained from the reaction of HL1 with Co(BF4)2·6H2O in MeCN under reflux conditions (entry 6). With regard to the organic ligand's transformations found in 5, 6 and 7 (entries 4–8), these are dependent on i) the temperature (entries 4 and 8), ii) the Co source used (entries 6–8) and iii) the presence of Et2O (entry 5).
Computational mechanistic studies
A comprehensive and consistent picture of the transformations that the coordinated ligands to Co complexes under study undergo (cf.Scheme 1) has been derived by means of DFT calculations employing the PBE0/Def2-TZVP(Co) ∪ 6-31G(d,p)(E) (E = main group element) computational protocol. All crucial reaction steps have been scrutinized by examining the participation of Co–O2 intermediates in terms of located structures, energies and activation barriers. First we calculated the equilibrium geometries and the electronic structure of the “free” ligands Ln (n = 1–4) in MeCN solutions. Fig. 5 shows the ground-state geometries of the “free” ligands Ln, the natural atomic charges on selected atoms and the frontier molecular orbitals relevant to coordination of the ligands in MeCN solutions calculated by the PBE0/6-31G(d,p)/PCM computational protocol.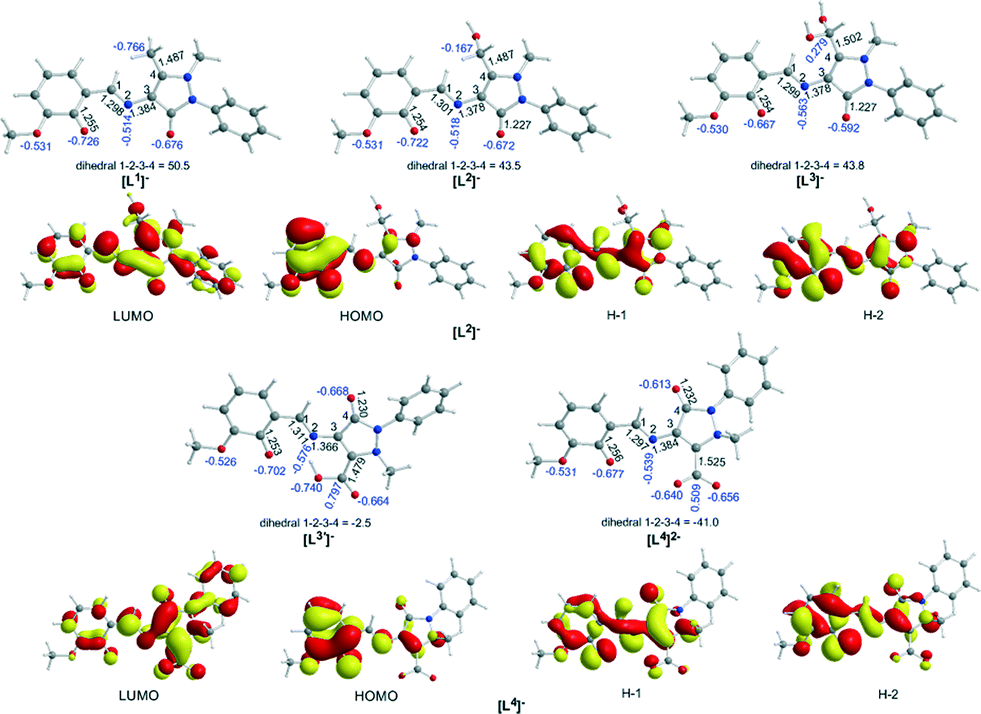 | ||
| Fig. 5 Equilibrium geometries of the “free” ligands Ln, the natural atomic charges on selected atoms (in blue) and the frontier molecular orbitals relevant to coordination of the ligands in MeCN solutions calculated by the PBE0/6-31G(d,p)/PCM computational protocol. | ||
Perusal of the highest occupied molecular orbitals (HOMOs) of the “free” Ln ligands reveals that the electron density is primarily localized on the phenoxide moieties including also the localization on the 2p AOs of the O and N donor atoms of the ligands. These atoms acquire negative natural atomic charges, namely −0.667–0.722|e| on the phenoxide O atom, −0.592–0.677|e| on the carbonyl group O atom, −0.526–0.531|e| on the methoxide O atom and −0.514–0.576|e| on the imine group nitrogen atom. Both the nature of the HOMOs and the negative natural atomic charges show that all these atoms could participate in the coordination of the Ln ligands to the Co central atom, thus explaining all coordination modes observed experimentally (Scheme S1†).
To probe the “distant” intramolecular C–H bond activation by the coordinated superoxide ligand of the C–H bonds of the –CH3, –CH2OH and –CH(OH)2 substituents on the C atom of the pyrazolone ring of the ligands Ln that promotes the L1 → L2 → L3 → L3′ → L4 ligand transformations, the reaction trajectory was explored through DFT calculations of the potential energy surfaces (PESs) and monitoring the geometric and energy reaction profiles (Fig. 6–8). A possible reaction mechanism is shown in Scheme 3.
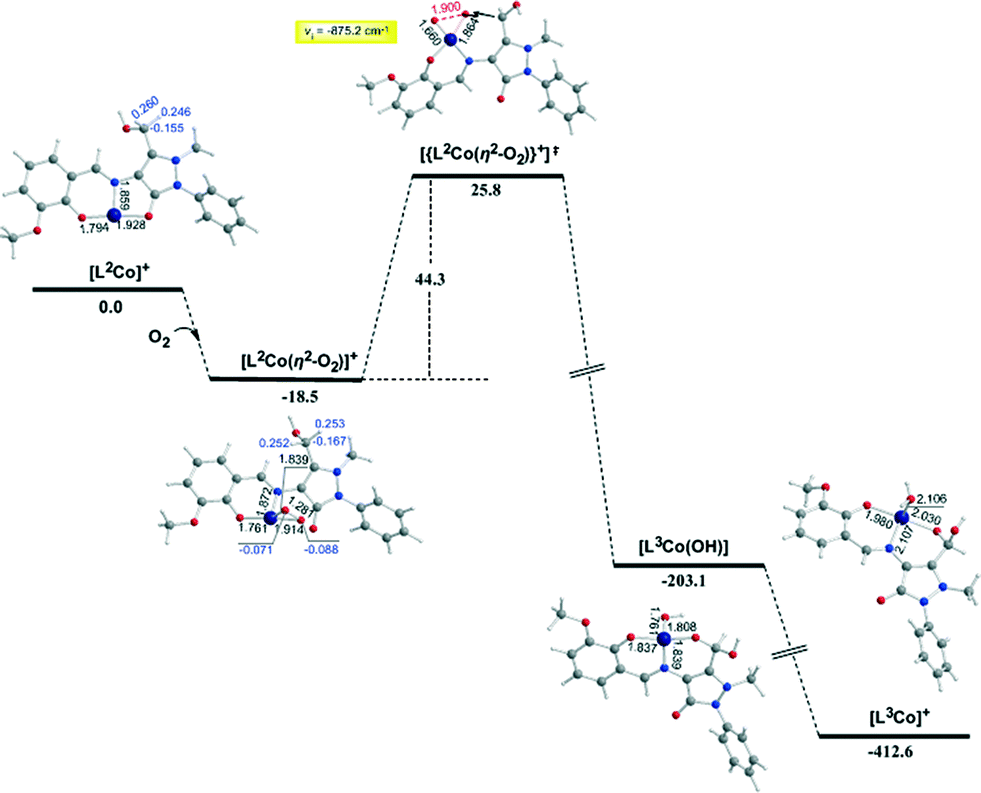
 | ||
| Fig. 6 Geometric and energy profiles of the reaction trajectory for the intramolecular transformation of coordinated ligand L1 to L2 calculated by the PBE0/Def2-TZVP(Co) ∪ 6-31G(d,p)(E)/PCM (E = main group element) computational protocol. | ||
 | ||
| Fig. 7 Geometric and energy profiles of the reaction trajectory for the intramolecular transformation of coordinated ligand L2 to L3 calculated by the PBE0/Def2-TZVP(Co) ∪ 6-31G(d,p)(E)/PCM (E = main group element) computational protocol. | ||
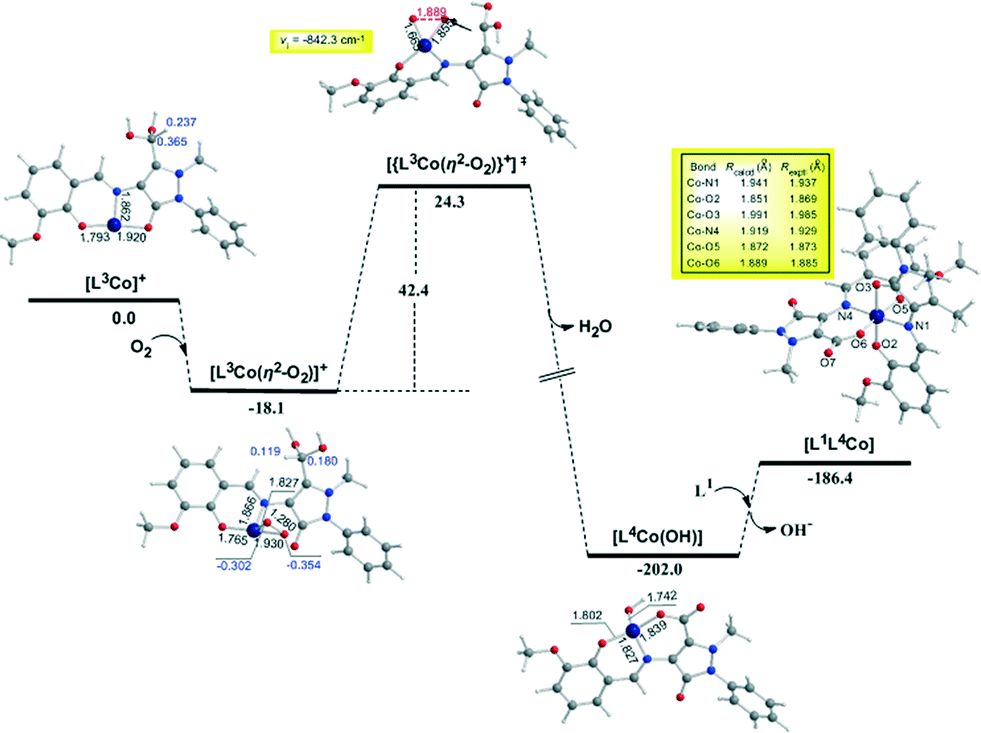 | ||
| Fig. 8 Geometric and energy profiles of the reaction trajectory for the intramolecular transformation of coordinated ligand L3 to L4 calculated by the PBE0/Def2-TZVP(Co) ∪ 6-31G(d,p)(E)/PCM (E = main group element) computational protocol. | ||
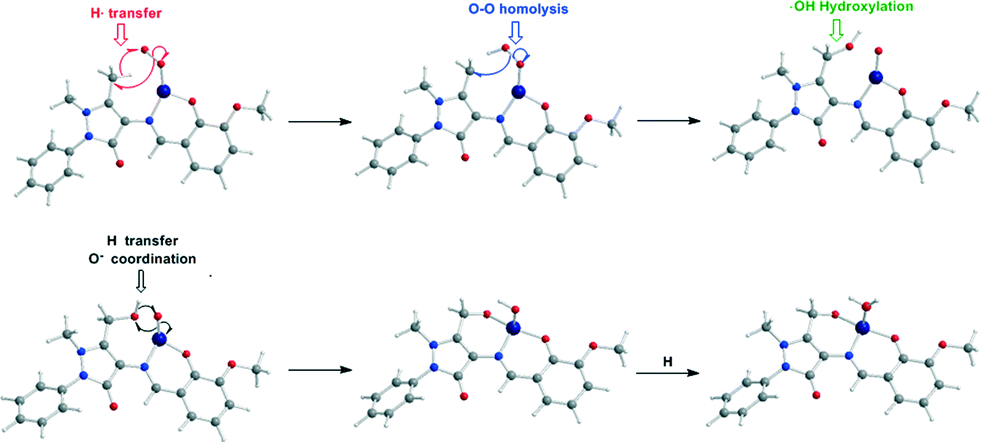 | ||
| Scheme 3 Proposed mechanism for the intramolecular ligand transformations induced by the [LnCoII]+ model complexes. | ||
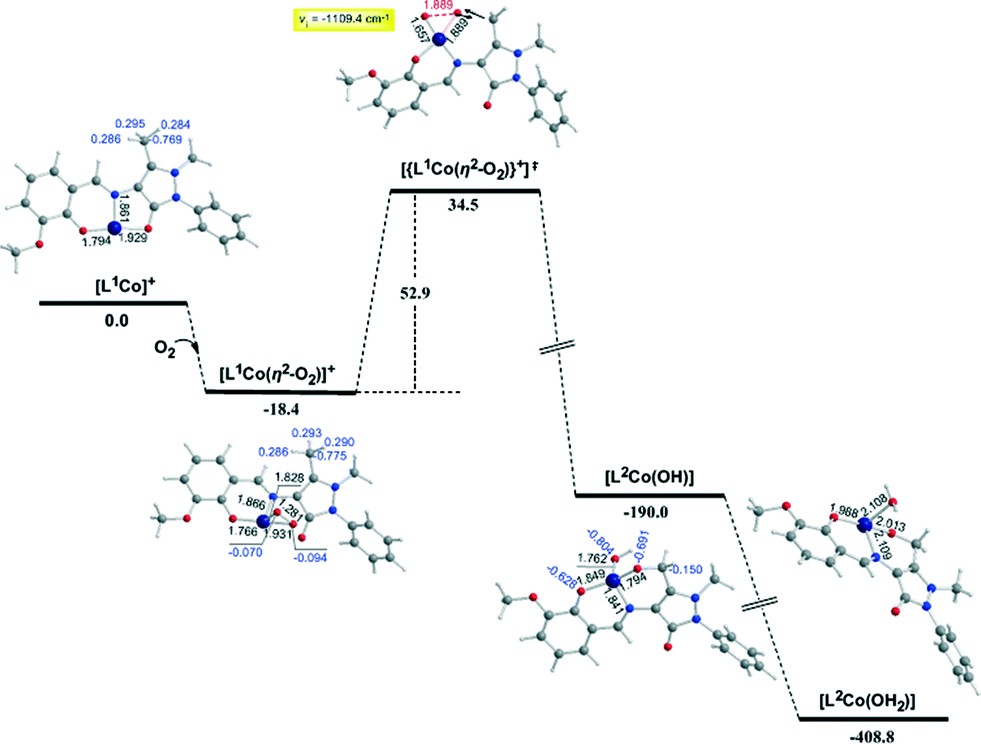
In the first step, the [LnCoII]+ complexes interact with dioxygen forming the superoxo [LnCo(O–O˙−)]+ adducts. The formation of the superoxo [LnCo(O–O˙−)]+ adducts correspond to exothermic processes, and the estimated exothermicities are around −18.1–18.5 kcal mol−1 at the PBE0/Def2-TZVP(Co) ∪ 6-31G(d,p)(E)/PCM level of theory. In the superoxo [LnCo(O–O˙−)]+ complexes the spin density, which was localized at the metal centre in the precursor [LnCoII]+ complexes, is now localized at the superoxide ligand (Scheme 4).
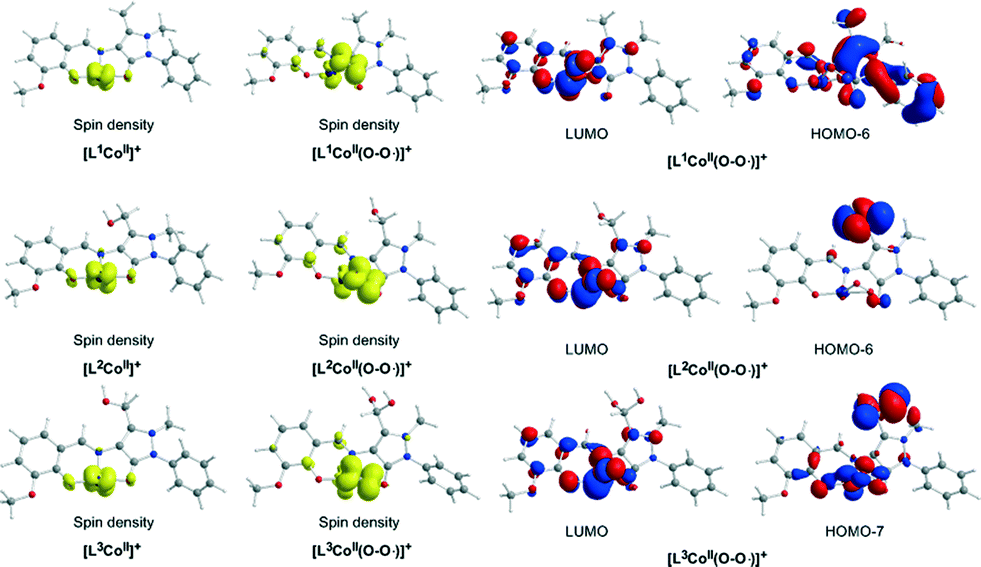 | ||
| Scheme 4 3D plots of the spin density distribution (isospin surfaces = 0.002) in the [LnCoII]+ and [LnCo(O–O˙−)]+ (n = 1–3) model complexes along with the 3D plots of the molecular orbitals of the [LnCo(O–O˙−)]+ complexes which support the hydrogen transfer. | ||
An inspection of Fig. 6–8 reveals that the [LnCo]+ (n = 1–3) complexes of CoII in their doublet ground states bind O2 in an asymmetric side-on η2-O2 coordination mode with an O–O bond length of 1.28 Å, intermediate between the values of 1.21 Å for O2 and 1.34 Å for O22−. This bonding mode involving a one-electron transfer from the CoII metal centre to the dioxygen ligand gives rise to the formation of superoxo [LnCo(O–O˙−)]+ complexes. In the superoxo [LnCo(O–O˙−)]+ complexes the Co–O bond lengths for the shorter Co–O bonds are 1.866, 1.839, 1.827 and 1.822 Å for the L1Co(O–O˙−)]+, L2Co(O–O˙−)]+, L3Co(O–O˙−)]+ and L3′Co(O–O˙−)]+complexes, respectively, and for the longer Co–O bonds are 1.931, 1.914, 1.930 and 1.937 Å, respectively.
It is worth noting that in 1:1 metal–O2 complexes both the end-on (η1-) and the side-on (η2-) bonding modes have been identified so far and the corresponding adducts were defined as superoxo or peroxo complexes, respectively, based primarily on the X-ray structural data (O–O bond distance) and vibrational spectra (O–O stretching frequency, νO–O).75–79
In particular, when the O–O bond length is ≈1.4–1.5 Å and the νO–O ≈ 800–930 cm−1 the compounds are designated as peroxides, whereas when O–O ≈ 1.2–1.3 Å and νO–O ≈ 1050–1200 cm−1 the compounds are characterized as superoxides. In the model [LnCo(O–O˙−)]+ complexes the O–O bond length of 1.28 Å and the unscaled νO–O stretching vibrational frequencies around 1299–1317 cm−1 illustrate their superoxo character. Although for the superoxide ligand the more common coordination is the end-on coordination (η1-O2) it is also well-coordinated in the side-on fashion (η2-O2),80,81 as in the case of the model [LnCo(O–O˙−)]+ complexes. The calculated Co–Nimine distances in the [LnCo]+ and [LnCo(O2)]+ model complexes are found in the range 1.859–1.867 Å and 1.849–1.872 Å, respectively, while the Co–Ophenoxide distances are found in the range 1.918–1.929 Å and 1.917–1.937 Å, respectively.
The coordinated superoxo radical abstracts a hydrogen atom through a homolytic C–H bond cleavage (H· transfer) supported by both electrostatic and orbital interactions. An intramolecular electrophilic attack of the C atom of the –CH3 and –CH2OH groups by the negatively charged coordinated superoxo radicals is precluded due to the negative natural atomic charges bearing the C atoms of these groups (−0.775 and −0.167|e|, respectively). The orbital interactions supporting the hydrogen transfer in the [LnCo(O–O˙−)]+ complexes correspond predominantly to HOMO-6 ↔ LUMO and HOMO-7 ↔ LUMO interactions (Scheme 4). After the hydrogen abstraction of the C–H bond, a homolytic O–O bond cleavage (O–O homolysis) in the CoII–OOH species takes place, affording CoIII = O (↔CoII–O) and HO˙ radical, which attacks the C atoms of the CH3, CH2OH and CH(OH)2 groups in a nearly concerted fashion (˙OH hydroxylation), yielding the hydroxylated ligands L2, L3 and L3′ predominantly (Fig. 6–8). Furthermore, the active CoIII = O (↔CoII–O) species generated after the homolytic O–O bond cleavage could perform cooperatively a second C–H bond cleavage step (H· transfer) yielding the [LnCo(OH)] (n = 2–4) intermediates, which subsequently are transformed to the [L2Co(OH2)], [L3Co(OH2)] and [L1L4Co] products. These transformations are predicted to be highly exothermic; the estimated exothermicities are −190.0, −203.1 and −202.0 kcal mol−1 for the L1 → L2, L2 → L3 and L3 → L4 ligand transformations, respectively. The superoxo [LnCo(O–O˙−)]+ complexes are transformed to [LnCo(OH)] (n = 2–4) intermediates through the [{LnCo(O2)}+]≠ transition states surmounting an activation barrier of 52.9, 44.3 and 42.4 kcal mol−1 for the L1 → L2, L2 → L3 and L3 → L4 ligand transformations, respectively. The lowering of the activation barrier along the L1 → L2, L2 → L3 and L3 → L4 ligand transformations could be attributed to the increase in the electrophilic character of the C atom of the –CH3, –CH2OH and –CH(OH)2 groups, which facilitates the ˙OH hydroxylation process. An analogous reaction trajectory was proposed by Wada and co-workers82 for the stoichiometric oxidation of substrates by the non-heme mononuclear hydroperoxo–FeIII complex and more recently by Li et al.83 in the proposed mechanism of dioxygen-activating non-heme enzymes.
Magnetic studies
The room temperature χT value for compound 4 under an applied field of 3000 G is 8.64 cm3 K mol−1 which is higher than that of the spin only value of 7.48 cm3 K mol−1 for four CoII (CoII; S = 3/2 and g = 2.0). The χT value gradually decreases with decreasing temperature, reaching a plateau at around 10–20 K and then sharply falls down to 5.17 cm3 K mol−1 at 1.8 K. (Fig. 9). The non-continuous χT decay evidences some coupling inside the cube in addition to the usual Dion. To have a rough approach to the CoII⋯CoII interactions, fitting of the experimental data was performed with the PHI program,84 assuming a distorted cube of four S = 3/2 spins on the basis of the Hamiltonian H = −J1(S1·S4 + S2·S3) − J2(S1·S2 + S1·S3) − J3(S2·S3 + S2·S4), which corresponds to a cube with similar interactions in opposite faces. Taking into account a Dion term, an excellent fit was obtained for the parameters J1 = +4.4 cm−1, J2 = +1.3 cm−1, J3 = −1.4 cm−1, g = 2.12 and Dion = 18.8 cm−1. The positive J1 interaction can be assigned to the pair of faces with smaller Co–O–Co bond angles, close to 94–95°. Magnetization experiments performed at 2 K show a constant increase of magnetization up to a quasi-saturated value equivalent to 5.91 electrons under the maximum external field of 5 T, (Fig. 9, inset). Each CoII can be treated as an effective S = 1/2 spin at low temperature due to the ZFS, and thus the magnetization must behave as a strongly anisotropic effective S = 2 spin level. The fit of the magnetization, assuming the above response, gives an excellent agreement for DS = 2 = −1.7 cm−1 and g = 3.10.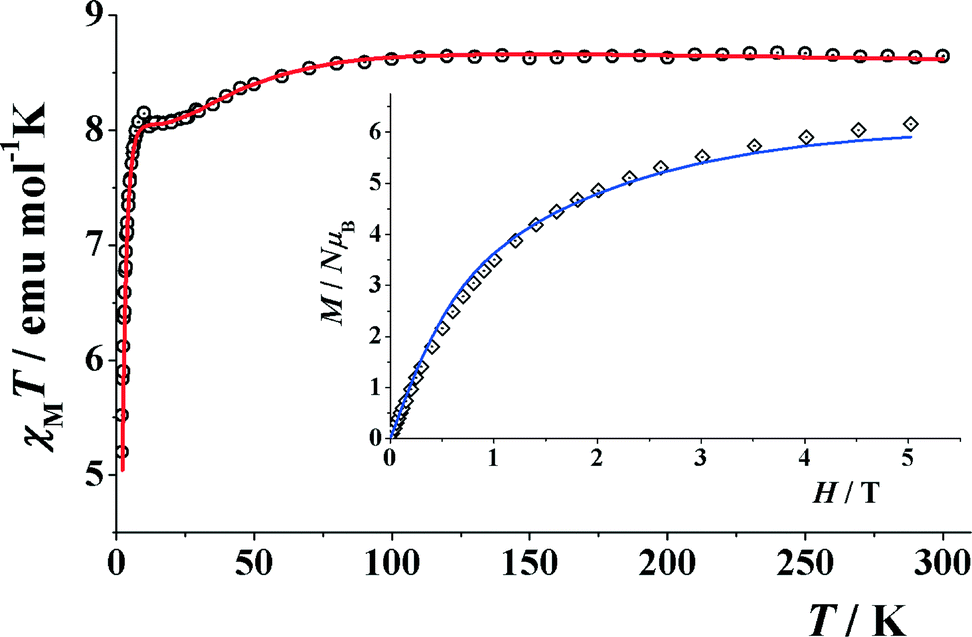 | ||
| Fig. 9 Product of χMvs. T for compound 4. (Inset) Magnetization plot in the range 0–5 T. Solid lines show the best obtained fits. | ||
The room temperature χMT value for compound 8 is 2.57 cm3 K mol−1, which is higher than that of the spin only value of 2.33 cm3 K mol−1 for two NiII (Fig. 10). Upon cooling, this value starts to increase to a maximum value of 2.62 cm3 K mol−1 at 20 K and then decreases to 1.68 cm3K mol−1 at 2 K. Magnetization measurements show a continuous increase of magnetization up to a maximum value equivalent to 3.2 electrons. This behavior suggests weak ferromagnetic coupling with an S = 2 ground state and a significant D contribution. Simultaneous fit of the experimental data was performed, applying the Hamiltonian H = −J(S1·S2) and including a Dion parameter. Least-squares fitting of the experimental data gave the common values of J = +1.2 cm−1, g = 2.255, D = 3.79 cm−1 for both measurements. The fit confirms an S = 2 ground state for 8.
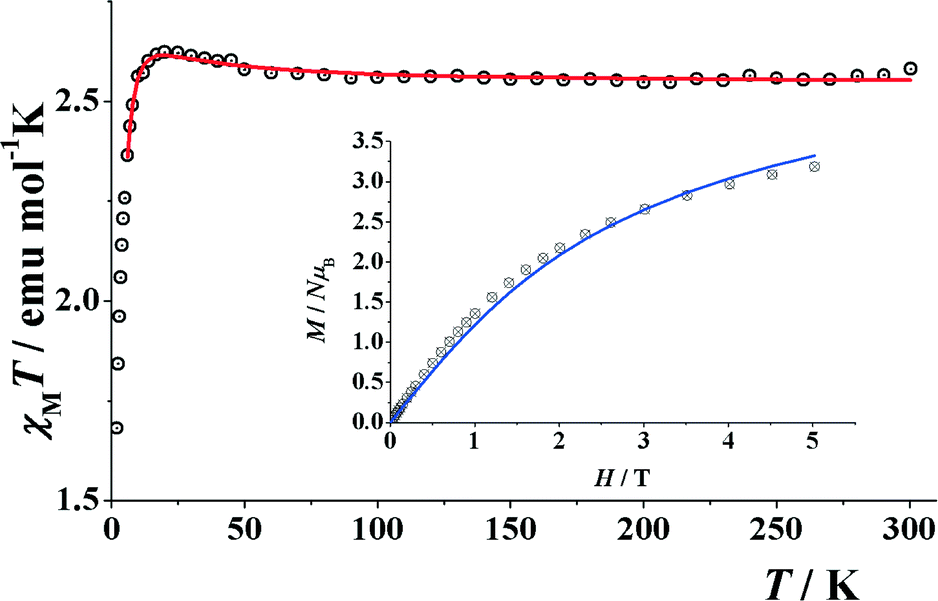 | ||
| Fig. 10 Product of χMvs. T for compound 8. (Inset) Magnetization plot in the range 0–5 T. Solid lines show the best obtained simultaneous fit. | ||
Conclusions
In this work we have successfully, for the first time, employed the monoanionic Schiff base ligand (HL1, Scheme 1) derived from the condensation of o-vanillin and 4-aminoantipyrine in cobalt, nickel and copper chemistry to access nine new CCs. A topological evaluation of all polynuclear CCs obtained is reported. In addition, a structural comparison of the present polynuclear CCs along with the compounds derived from structurally related ligands is attempted (Scheme 2). We conclude that a) HL1 coordinates completely different from them and thus its coordination chemistry can lead to new, interesting and unprecedented topologies and b) more synthetic studies are required to reach a point wherein the design of such species can be undertaken.85 In addition, we report interesting cases of ligand transformations (Scheme 1), which take place in certain experiments with cobalt sources (4–7). These transformations occur in one of the methyl groups of the pyrazolone ring of the HL ligand under specific conditions such as the use of a particular metal salt, ratio and solvent, altering drastically the coordination mode of the ligand and resulting in different structures. DFT calculations of the potential energy surfaces (PESs) and monitoring the geometric and energetic reaction profiles of the intramolecular L1 → L2 → L3 → L3′ → L4 ligand transformations revealed a possible reaction mechanism that involves the formation of superoxo [LnCo(O–O˙−)]+ adducts, which are transformed to [LnCo(OH)] (n = 2–4) intermediates through the [{LnCo(O2)}+]≠ transition states, surmounting an activation barrier of 52.9, 44.3 and 42.4 kcal mol−1 for the L1 → L2, L2 → L3 and L3 → L4 ligand transformations, respectively. These transformations involving concomitant H transfer, O–O homolysis and ˙OH hydroxylation processes afford the [LnCo(OH)] (n = 2–4) intermediates that subsequently are transformed to the final products. The present findings indicate that the coordination chemistry of HL1 can lead to unprecedented CCs bearing interesting properties, and thus our future studies will be focused on further exploring it.Acknowledgements
We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for compounds 4, 7 and 8. Research development funding from the University of Sussex is gratefully acknowledged (V. N. D.). V. K. acknowledges University of Sussex for a Junior Research Award summer fellowship. Support from CICYT Project CTQ2012-30662 is acknowledged (A. E.)Notes and references
- L. Cronin and J. Fielden, in Coordination Clusters in Encyclopedia of Supramolecular Chemistry, Taylor & Francis, London, 2007, pp. 1–10 Search PubMed.
- E. Ritter, P. Przybylski, B. Brzezinski and F. Bartl, Curr. Org. Chem., 2009, 13, 241–249 CrossRef CAS.
- M. S. Karthikeyan, D. J. Prasad, B. Poojary, K. Subrahmanya Bhat, B. S. Holla and N. S. Kumari, Bioorg. Med. Chem., 2006, 14, 7482–7489 CrossRef CAS PubMed.
- M. J. O'Donnell, Acc. Chem. Res., 2004, 37, 506–517 CrossRef PubMed.
- R. J. P. Corriu, E. Lancelle-Beltran, A. Mehdi, C. Reyé, S. Brandès and R. Guilard, J. Mater. Chem., 2002, 12, 1355–1362 RSC.
- R. J. P. Corriu, E. Lancelle-Beltran, A. Mehdi, C. Reyé, S. Brandès and R. Guilard, Chem. Mater., 2003, 15, 3152–3160 CrossRef CAS.
- F. Faridbod, M. R. Ganjali, R. Dinarvand, P. Norouzi and S. Riahi, Sensors, 2008, 8, 1645–1703 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- P. G. Cozzi, Chem. Soc. Rev., 2004, 33, 410–421 RSC.
- D. Ramakrishna, B. R. Bhat and R. Karvembu, Catal. Commun., 2010, 11, 498–501 CrossRef CAS.
- Z. Ma, L. Wei, E. C. B. A. Alegria, L. M. D. R. S. Martins, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Dalton Trans., 2014, 43, 4048–4058 RSC.
- D. S. Nesterov, E. N. Chygorin, V. N. Kokozay, V. V. Bon, R. Boča, Y. N. Kozlov, L. S. Shul'pina, J. Jezierska, A. Ozarowski, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2012, 51, 9110–9122 CrossRef CAS PubMed.
- C. M. Da Silva, D. L. Da Silva, L. V. Modolo, R. B. Alves, M. A. De Resende, C. V. B. Martins and Â. De Fátima, J. Adv. Res., 2011, 2, 1–8 CrossRef.
- P. Vicini, A. Geronikaki, M. Incerti, B. Busonera, G. Poni, C. A. Cabras and P. La Colla, Bioorg. Med. Chem., 2003, 11, 4785–4789 CrossRef CAS PubMed.
- W. Chen, Y. Li, Y. Cui, X. Zhang, H. L. Zhu and Q. Zeng, Eur. J. Med. Chem., 2010, 45, 4473–4478 CrossRef CAS PubMed.
- H. Miyasaka, R. Clérac, W. Wernsdorfer, L. Lecren, C. Bonhomme, K.-I. Sugiura and M. Yamashita, Angew. Chem., Int. Ed., 2004, 43, 2801–2805 CrossRef CAS PubMed.
- S. Mandal, G. Rosair, J. Ribas and D. Bandyopadhyay, Inorg. Chim. Acta, 2009, 362, 2200–2204 CrossRef CAS.
- Z. Lü, M. Yuan, F. Pan, S. Gao, D. Zhang and D. Zhu, Inorg. Chem., 2006, 45, 3538–3548 CrossRef PubMed.
- N. C. Anastasiadis, D. A. Kalofolias, A. Philippidis, S. Tzani, C. Raptopoulou, V. Psycharis, C. J. Milios, A. Escuer and S. P. Perlepes, Dalton Trans., 2015, 44, 10200–10209 RSC.
- A. A. Athanasopoulou, M. Pilkington, C. P. Raptopoulou, A. Escuer and T. C. Stamatatos, Chem. Commun., 2014, 50, 14942–14945 RSC.
- D. I. Alexandropoulos, T. N. Nguyen, L. Cunha-Silva, T. F. Zafiropoulos, A. Escuer, G. Christou and T. C. Stamatatos, Inorg. Chem., 2013, 52, 1179–1181 CrossRef CAS PubMed.
- A. B. Canaj, D. I. Tzimopoulos, A. Philippidis, G. E. Kostakis and C. J. Milios, Inorg. Chem., 2012, 51, 10461–10470 CrossRef CAS PubMed.
- A. B. Canaj, D. I. Tzimopoulos, A. Philippidis, G. E. Kostakis and C. J. Milios, Inorg. Chem. Commun., 2012, 51, 7451–7453 CrossRef CAS PubMed.
- K.-H. Chang, C.-C. Huang, Y.-H. Liu, Y.-H. Hu, P.-T. Chou and Y.-C. Lin, Dalton Trans., 2004, 1731–1738 RSC.
- H.-C. Lin, C.-C. Huang, C.-H. Shi, Y.-H. Liao, C.-C. Chen, Y.-C. Lin and Y.-H. Liu, Dalton Trans., 2007, 781–791 RSC.
- L. A. Saghatforoush, R. Mehdizadeh and F. Chalabian, Transition Met. Chem., 2010, 35, 903–910 CrossRef CAS.
- B. Tamami and S. Ghasemi, Appl. Catal., A, 2011, 393, 242–250 CrossRef CAS.
- E. C. Constable, C. E. Housecroft, J. A. Zampese and G. Zhang, Polyhedron, 2012, 44, 150–155 CrossRef CAS.
- C.-M. Liu, R.-G. Xiong, D.-Q. Zhang and D.-B. Zhu, J. Am. Chem. Soc., 2010, 132, 4044–4045 CrossRef CAS PubMed.
- S. Saha, S. Pal, C. J. Gómez-García, J. M. Clemente-Juan, K. Harms and H. P. Nayek, Polyhedron, 2014, 74, 1–5 CrossRef CAS.
- Y. Thio, S. W. Toh, F. Xue and J. J. Vittal, Dalton Trans., 2014, 43, 5998–6001 RSC.
- H. Oshio, N. Hoshino, T. Ito, M. Nakano, F. Renz and P. Gütlich, Angew. Chem., Int. Ed., 2003, 42, 223–225 CrossRef CAS PubMed.
- N. Hoshino, A. M. Ako, A. K. Powell and H. Oshio, Inorg. Chem., 2009, 48, 3396–3407 CrossRef CAS PubMed.
- M. Sarwar, A. M. Madalan, C. Tiseanu, G. Novitchi, C. Maxim, G. Marinescu, D. Luneau and M. Andruh, New J. Chem., 2013, 37, 2280–2292 RSC.
- Y.-N. Guo, X.-H. Chen, S. Xue and J. Tang, Inorg. Chem., 2012, 51, 4035–4042 CrossRef CAS PubMed.
- H. Ke, S. Zhang, W. Zhu, G. Xie and S. Chen, J. Coord. Chem., 2015, 68, 808–822 CrossRef CAS.
- H. Ke, W. Zhu, S. Zhang, G. Xie and S. Chen, Polyhedron, 2015, 87, 109–116 CrossRef CAS.
- Y.-Z. Zheng, Y. Lan, C. E. Anson and A. K. Powell, Inorg. Chem., 2008, 47, 10813–10815 CrossRef CAS PubMed.
- B. Gole, K. C. Mondal and P. S. Mukherjee, Inorg. Chim. Acta, 2014, 415, 151–164 CrossRef CAS.
- K. C. Mondal, G. E. Kostakis, Y. Lan and A. K. Powell, Polyhedron, 2013, 66, 268–273 CrossRef CAS.
- M. Hasanzadeh, M. Salehi, M. Kubicki, S. M. Shahcheragh, G. Dutkiewicz, M. Pyziak and A. Khaleghian, Transition Met. Chem., 2014, 39, 623–632 CrossRef CAS.
- S. Nayak, H. P. Nayek, S. Dehnen, A. K. Powell and J. Reedijk, Dalton Trans., 2011, 40, 2699–2702 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- P. Bhowmik, N. Aliaga-Alcalde, V. Gómez, M. Corbella and S. Chattopadhyay, Polyhedron, 2013, 49, 269–276 CrossRef CAS.
- R. Ganguly, B. Sreenivasulu and J. J. Vittal, Coord. Chem. Rev., 2008, 252, 1027–1050 CrossRef CAS.
- E. Loukopoulos, B. Berkoff, A. Abdul-Sada, G. J. Tizzard, S. J. Coles, A. Escuer and G. E. Kostakis, Eur. J. Inorg. Chem., 2015, 2646–2649 CrossRef CAS.
- B. Berkoff, K. Griffiths, A. Abdul-Sada, G. J. Tizzard, S. Coles, A. Escuer and G. E. Kostakis, Dalton Trans., 2015, 44, 12788–12795 RSC.
- K. C. Mondal, A. Sundt, Y. Lan, G. E. Kostakis, O. Waldmann, L. Ungur, L. F. Chibotaru, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2012, 51, 7550–7554 CrossRef CAS PubMed.
- I. Nemec, M. Machata, R. Herchel, R. Boča and Z. Trávníček, Dalton Trans., 2012, 41, 14603–14610 RSC.
- K. C. Mondal, G. E. Kostakis, Y. Lan, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2011, 50, 11604–11611 CrossRef CAS PubMed.
- M. Frisch, G. Trucks and H. Schlegel, Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, Chem. Phys. Lett., 1997, 274, 242–250 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- C. Adamo, G. E. Scuseria and V. Barone, J. Chem. Phys., 1999, 111, 2889 CrossRef CAS.
- C. Adamo and V. Barone, Theor. Chem. Accounts Theory, Comput. Model. (Theoretica Chim. Acta), 2000, 105, 169–172 CrossRef CAS.
- V. Vetere, C. Adamo and P. Maldivi, Chem. Phys. Lett., 2000, 325, 99–105 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- O. V. Dolomanov, A. J. Blake, N. R. Champness and M. Schröder, J. Appl. Crystallogr., 2003, 36, 1283–1284 CrossRef CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- H. H. Thorp, Inorg. Chem., 1992, 31, 1585–1588 CrossRef CAS.
- J. L. Atwood and J. W. Steed, in Supramolecular Chemistry, 2nd Edition, 2009, p. 402 Search PubMed.
- G. E. Kostakis, V. A. Blatov and D. M. Proserpio, Dalton Trans., 2012, 41, 4634–4640 RSC.
- H. Xiang, Y. Lan, L. Jiang, W.-X. Zhang, C. E. Anson, T.-B. Lu and A. K. Powell, Inorg. Chem. Commun., 2012, 16, 51–54 CrossRef CAS.
- Z. Puterová-Tokárová, V. Mrázová, J. Kožíšek, J. Valentová, B. Vranovičová and R. Boča, Polyhedron, 2014, 70, 52–58 CrossRef.
- B. Bhattacharya, R. Dey, D. K. Maity and D. Ghoshal, CrystEngComm, 2013, 15, 9457–9464 RSC.
- K. Griffiths, C. W. D. Gallop, A. Abdul-Sada, A. Vargas, O. Navarro and G. E. Kostakis, Chem. – Eur. J., 2015, 21, 6358–6361 CrossRef CAS PubMed.
- P. J. Perez, Alkane C–H Activation by Single-Site Metal Catalysis, Springer Netherlands, Dordrecht, 2012, vol. 38 Search PubMed.
- M. H. Gubelmann and A. F. Williams, Struct. Bonding, 1983, 55, 2–65 Search PubMed.
- H. A. O. Hill and D. G. Tew, Comprehensive Coordination Chemistry, Pergamon Press, Oxford, 1987 Search PubMed.
- C. J. Cramer, W. B. Tolman, K. H. Theopold and A. L. Rheingold, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3635–3640 CrossRef CAS PubMed.
- J. W. Egan, B. S. Haggerty, A. L. Rheingold, S. C. Sendlinger and K. H. Theopold, J. Am. Chem. Soc., 1990, 112, 2445–2446 CrossRef CAS.
- A. Hess, M. R. Hörz, L. M. Liable-Sands, D. C. Lindner, A. L. Rheingold and K. H. Theopold, Angew. Chem., Int. Ed., 1999, 38, 166–168 CrossRef CAS.
- J. S. Valentine, Chem. Rev., 1973, 73, 235–245 CrossRef CAS.
- L. Vaska, Acc. Chem. Res., 1976, 9, 175–183 CrossRef CAS.
- A. Wada, S. Ogo, S. Nagatomo, T. Kitagawa, Y. Watanabe, K. Jitsukawa and H. Masuda, Inorg. Chem., 2002, 41, 616–618 CrossRef CAS PubMed.
- F. Li, K. K. Meier, M. A. Cranswick, M. Chakrabarti, K. M. Van Heuvelen, E. Münck and L. Que, J. Am. Chem. Soc., 2011, 133, 7256–7259 CrossRef CAS PubMed.
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed.
- E. V. Alexandrov, A. P. Shevchenko, A. A. Asiri and V. A. Blatov, CrystEngComm, 2015, 17, 2913–2924 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format for structures 1–10. Crystallographic parameters can be found in Tables S1 and S2. Scheme S1. Selected bond distances and angles for 1–9 can be found in Tables S3–S10. Cartesian coordinates and energies of the Ln (n = 1–4) ligands, the reactant, intermediate, transition state and product cobalt complexes located on the potential energy surfaces of the transformations the coordinated ligands undergo (Table S12). A library of the 100 polynuclear CCs with nuclearity over 3 is provided as an Excel file. CCDC 1400124–1400133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce01294e |
| This journal is © The Royal Society of Chemistry 2015 |