DOI:
10.1039/C5CE00871A
(Paper)
CrystEngComm, 2015,
17, 6355-6369
Aspects of crystal engineering arising from packing behavior of functional mono para-substituted trityl compounds†
Received
4th May 2015
, Accepted 15th July 2015
First published on 16th July 2015
Abstract
Four trityl compounds differing both in the functional group (OH, OMe, NH2) at the specific trityl carbon and a para substituent, being bromine or phenyl at one of the phenyl groups, have been prepared and structurally studied by means of single crystal X-ray diffraction and making use of Hirshfeld surface analysis. Compared to the structures of corresponding prototypes and analogues, specific interaction modes and packing motifs including cluster aggregates as well as non-cluster type structures depending on the substitution and involving polymorphism were found, opening potential trend prediction with reference to crystal engineering being useful in this compound class.
Introduction
Aside from their important property to be a parent substance of the well known class of triphenylmethane dyes,1 trityl compounds are also useful agents for functional group protection in the synthesis of alcohols and amines.2,3 In other respects, the basic compound triphenylmethanol has been shown to selectively enclathrate methanol and DMSO in its crystal lattice4 while more bulkily substituted triphenylmethanol derivatives and analogous triarylmethanols demonstrate corresponding inclusion formation with other guest solvents.5 Conversely, triphenylmethane derivatives as the guest were found to be inclusion-complexed with crown compounds6,7 and, as a further subject, the packing modes of trityl alcohols have been studied in dependency of specific para substitution involving different halogen atoms.8,9 In all these latter structures, O–H⋯π10 and halogen⋯π11,12 interactions have proven to dominate the crystal lattices while conventional O–H⋯O hydrogen bonding13 is prevented from steric reason. However, in most of the previous cases, the trityl alcohols are symmetrically para substituted compound examples and corresponding tritylamines are rarely described in the literature.14–16
Here we report the X-ray crystal structures of four specific trityl compounds (1–4, Fig. 1). Although being in close structural relation, they systematically differ both in the main functional group (hydroxyl, methoxy, amino) and the para substituent (bromo, phenyl) being present at one of the phenyl units. Based on a comparative discussion of the structural results, including previous findings and making use of Hirshfeld surface analysis,17 conclusions can be drawn that can be helpful for future aspects of crystal engineering involving this interesting field of compounds.18–20 Moreover, previous findings in the context have shown that different melting behavior of corresponding compounds are rather frequent indicating the potential existence of polymorphous structures,21–24 which is another inviting point of consideration for carrying out this study.
 |
| | Fig. 1 Chemical formula structures of the compounds studied. | |
Results and discussion
Synthesis and X-ray structural study
The compounds 1–4, studied in this paper, were synthesized using respectively approaching described methods (1–4)21,25 or following standard preparative procedures (2, 3)26,27 and are given in the experimental section. The crystal structures of the compounds were performed using an X-ray diffraction technique. Crystallographic data, experimental parameters and selected details of the refinement calculations are summarized in Table 1. Because of the high content of aromatic units, the geometric features of the molecules can be expressed by a set of dihedral angles between the arene rings. These parameters together with relevant torsion angles are summarized in Table 2, while information regarding possible non-covalent interactions in the crystal structures are listed in Table 3. Perspective views of the molecular structures including atom labelling and ring specification as well as illustrations of the packing structures are presented in Fig. 2–8.
Table 1 Crystallographic and structure refinement data of the compounds studied
| Compound |
1
|
2B
|
2C
|
3
|
4
|
|
P = (Fo2 + 2Fc2)/3
|
|
|
| Empirical formula |
C19H15OBr |
C25H20O |
C25H20O |
C20H17OBr |
C19H16NBr |
|
|
| Formula weight |
339.22 |
336.41 |
336.41 |
353.25 |
338.24 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Orthorhombic |
Monoclinic |
| Space group |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21 |
Pna21 |
P21/c |
|
a/Å |
10.7736(2) |
15.6058(3) |
8.9849(6) |
12.1520(3) |
10.7837(2) |
|
b/Å |
15.2684(3) |
18.7804(4) |
5.7029(3) |
13.7838(3) |
15.2773(4) |
|
c/Å |
18.6621(3) |
21.1189(4) |
17.1999(11) |
9.5790(3) |
18.8871(4) |
|
α/° |
90.0 |
68.874(1) |
90.0 |
90.0 |
90.0 |
|
β/° |
104.275(1) |
74.947(1) |
99.978(4) |
90.0 |
104.273(1) |
|
γ/° |
90.0 |
74.080(1) |
90.0 |
90.0 |
90.0 |
|
V/Å3 |
2975.05(9) |
5462.16(19) |
867.99(9) |
1604.49(7) |
3015.52(12) |
|
Z
|
8 |
12 |
2 |
4 |
8 |
|
F(000) |
1376 |
2136 |
356 |
720 |
1376 |
|
D
c/Mg m−3 |
1.515 |
1.227 |
1.287 |
1.462 |
1.490 |
|
μ/mm−1 |
2.759 |
0.073 |
0.077 |
2.561 |
2.719 |
| Data collection |
|
|
|
|
|
| Temperature/K |
90(2) |
100(2) |
100(2) |
173(2) |
100(2) |
| No. of collected reflections |
30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 095 095 |
107343 |
12855 |
8781 |
59570 |
| Within the θ-limit/° |
1.8–29.2 |
1.2–28.1 |
2.3–30.1 |
2.6–28.4 |
1.7–28.6 |
| Index ranges ± h, ±k, ±l |
−14/14, −20/20, −25/25 |
−20/20, −24/24, −27/27 |
−12/12, −8/8, −24/24 |
−8/16, −14/18, −12/7 |
−14/14, −20/20, −25/25 |
| No. of unique reflections |
7905 |
26583 |
4710 |
3219 |
7712 |
|
R
int
|
0.0244 |
0.0314 |
0.0222 |
0.0185 |
0.0223 |
| Refinement calculations: full-matrix least-squares on all F2 values |
|
|
|
|
|
| Weighting expression wa |
[σ2(Fo2) + (0.0435P)2 + 1.4259P]−1 |
[σ2(Fo2) + (0.0595P)2 + 3.4661P]−1 |
[σ2(Fo2) + (0.0561P)2 + 0.1424P]−1 |
[σ2(Fo2) + (0.0282P)2 + 0.3652P]−1 |
[σ2(Fo2) + (0.0428P)2 + 2.5837P]−1 |
| No. of refined parameters |
387 |
1411 |
240 |
201 |
395 |
| No. of F values used [I > 2σ(I)] |
6551 |
17994 |
4409 |
2913 |
6771 |
| Final R-Indices |
|
|
|
|
|
|
R(=∑|ΔF|/∑|Fo|) |
0.0292 |
0.0521 |
0.0358 |
0.0231 |
0.0272 |
| wR on F2 |
0.0801 |
0.1522 |
0.0952 |
0.0594 |
0.0782 |
|
S (=Goodness of fit on F2) |
1.035 |
1.005 |
1.037 |
0.940 |
1.006 |
| Final Δρmax/Δρmin (e Å−3) |
0.60/−0.92 |
0.65/−0.43 |
0.34/−0.19 |
0.45/−0.44 |
0.92/−0.64 |
| KPI (%) |
69.8 |
67.7 |
71.4 |
68.6 |
69.7 |
| Total potential solvent accessible |
|
|
|
|
|
| Void volume/Å3 |
— |
81.2 |
— |
— |
— |
| Void volume per unit cell/% |
— |
1.5 |
— |
— |
— |
Table 2 Relevant conformational parameters of the compounds studied
| Compound |
1
|
2B
|
2C
|
3
|
4
|
| Molecule 1 |
Molecule 2 |
Molecule 1 |
Molecule 2 |
Molecule 3 |
Molecule 4 |
Molecule 5 |
Molecule 6 |
|
|
Molecule 1 |
Molecule 2 |
|
mpla means least-squares plane through the aromatic ring. 1, 3, 4: Ring A: C(1)⋯C(6); ring B: C(7)⋯C(12); ring C: C(13)⋯C(18). 2: Ring A: C(1)⋯C(6); ring B: C(7)⋯C(12); ring C: C(14)⋯C(19); ring D: C(20)⋯C(25).
|
| Dihedral angles (°)a |
|
|
|
|
|
|
|
|
|
|
|
|
| mpla(A)–mpla(B) |
73.6(1) |
72.9(1) |
84.7(1) |
86.9(1) |
80.1(1) |
88.3(1) |
83.0(1) |
85.9(1) |
66.5(1) |
81.0(1) |
48.7(1) |
87.3(1) |
| mpla(A)–mpla(C) |
71.2(1) |
77.7(1) |
86.2(1) |
74.2(1) |
71.2(1) |
70.8(1) |
80.9(1) |
74.2(1) |
84.6(1) |
61.4(1) |
65.0(1) |
74.2(1) |
| mpla(B)–mpla(C) |
87.2(1) |
82.3(1) |
87.5(1) |
84.6(1) |
89.4(1) |
83.3(1) |
89.7(1) |
64.0(1) |
75.1(1) |
70.1(1) |
53.8(1) |
53.8(1) |
| mpla(C)–mpla(D) |
|
|
30.0(1) |
44.6(1) |
37.2(1) |
22.90(1) |
35.5(1) |
28.0(1) |
4.0(1) |
|
|
|
|
|
| Torsion angles |
|
|
|
|
|
|
|
|
|
|
|
|
| C(1)–C(19)–O(1)–H(1) |
−82.3(1) |
78.8(1) |
|
|
|
|
|
|
|
|
|
|
| C(14)–C(13)–O(1)–H(1) |
|
|
−70.2(1) |
−80.5(1) |
−75.5(1) |
−72.0(1) |
−82.3(1) |
−67.0(1) |
−82.2(1) |
|
|
|
| C(1)–C(19)–O(1)–C(20) |
|
|
|
|
|
|
|
|
|
−75.7(1) |
|
|
| C(1)–C(19)–N(1)–H(1A) |
|
|
|
|
|
|
|
|
|
|
166.3(1) |
−53.0(1) |
| C(1)–C(19)–N(1)–H(1B) |
|
|
|
|
|
|
|
|
|
|
53.0(1) |
−165.3(1) |
Table 3 Geometric parameters for hydrogen bond type contacts of the compounds studied
| Atoms involved |
Symmetry |
Distance/Å |
Angle/° |
| D–H⋯A |
|
D–H |
D⋯A |
H⋯A |
D–H⋯A |
| C–Br⋯Br |
|
C–Br |
C⋯Br |
Br⋯Br |
C–Br⋯Br |
|
Means centre of the aromatic ring. 1: Ring CA: C(13A)⋯C(18A). 2: Ring B: C(7)⋯C(12); ring C: C(14)⋯C(19); ring BA: C(7A)⋯C(12A); ring CC: C(14C)⋯C(19C); ring BD: C(7D)⋯C(12D); ring AE: C(1E)⋯C(6E). 3, 4: Ring A: C(1)⋯C(6); ring B: C(7)⋯C(12); ring AA: C(1A)⋯C(6A); ring CA: C(13A)⋯C(18A).
To achieve reasonable hydrogen bond geometries, individual atoms instead of ring centroids were choosen as acceptors.
|
|
1
|
|
|
|
|
|
| O(1)–H(1)⋯C(14A)b |
1 − x, 1 − y, 2 − z |
0.83(1) |
3.428(2) |
2.670(2) |
153.2(2) |
| O(1A)–H(1A)⋯C(12)b |
−x, 1 − y, 2 − z |
0.83(1) |
3.230(2) |
2.492(2) |
148.5(2) |
| C(8)–H(8)⋯O(1) |
x, y, z |
0.95 |
2.689(2) |
2.34 |
101 |
| C(18A)–H(18A)⋯O(1A) |
x, y, z |
0.95 |
2.660(2) |
2.30 |
102 |
| C(5)–H(5)⋯O(1A) |
x, y, z |
0.95 |
3.669(2) |
2.75 |
164 |
| C(3A)–H(3A)⋯O(1) |
x, 1 + y, z |
0.95 |
3.674(2) |
2.78 |
157 |
| C(14)–H(14)⋯centroid(CA)a |
1 − x, 1 − y, 2 − z |
0.95 |
3.544(3) |
2.86 |
130 |
| C(15)–H(15)⋯C(6)b |
1 − x, 1 − y, 2 − z |
0.95 |
3.639(2) |
2.73 |
160 |
| C(6A)–H(6A)⋯C(16)b |
1 − x, 1 − y, 2 − z |
0.95 |
3.590(2) |
2.86 |
135 |
| C(9A)–H(9A)⋯C(18)b |
−x, 0.5 + y, 1.5 − z |
0.95 |
3.600(2) |
2.88 |
133 |
| C(11A)–H(11A)⋯C(3A)b |
−x, 2 − y, 2 − z |
0.95 |
3.634(2) |
2.78 |
150 |
| C(4)–Br(1)⋯Br(1A) |
1 − x, −0.5 + y, 2.5 − z |
1.898(2) |
4.245(2) |
3.869(2) |
87.9(2) |
| C(4A)–Br(1A)⋯Br(1) |
1 − x, 0.5 + y, 2.5 − z |
1.902(2) |
5.613(2) |
3.869(2) |
151.4(2) |
|
|
|
2B
|
|
|
|
|
|
| O(1)–H(1)⋯O(1B) |
x, y, −1 + z |
0.84 |
2.929(2) |
2.17 |
151 |
| O(1A)–H(1A)⋯O(1) |
x, y, z |
0.84 |
2.737(2) |
2.02 |
143 |
| O(1B)–H(1B)⋯O(1A) |
x, y, 1 + z |
0.84 |
2.790(2) |
2.09 |
141 |
| O(1C)–H(1C)⋯O(1D) |
x, y, z |
0.84 |
2.743(2) |
1.99 |
150 |
| O(1D)–H(1D)⋯O(1E) |
x, y, z |
0.84 |
2.735(2) |
2.00 |
147 |
| O(1E)–H(1E)⋯O(1C) |
x, y, z |
0.84 |
2.741(2) |
1.96 |
155 |
| C(3)–H(3)⋯centroid(BA)a |
1 − x, −y, −z |
0.95 |
3.639(3) |
2.70 |
172 |
| C(8)–H(8)⋯C(22E)b |
1 − x, −y, 1 − z |
0.95 |
3.678(3) |
2.86 |
145 |
| C(23)–H(23)⋯C(5B)b |
1 − x, 1 − y, 1 − z |
0.95 |
3.553(3) |
2.85 |
132 |
| C(5A)–H(5A)⋯C(5B)b |
1 − x, −y, 1 − z |
0.95 |
3.592(3) |
2.82 |
139 |
| C(10A)–H(10A)⋯C(22C)b |
2 − x, −y, −z |
0.95 |
3.595(3) |
2.84 |
137 |
| C(22A)–H(22A)⋯centroid(CC)a |
x, y, z |
0.95 |
3.663(3) |
2.76 |
160 |
| C(3B)–H(3B)⋯C(4)b |
x, y, 1 + z |
0.95 |
3.565(3) |
2.72 |
149 |
| C(4B)–H(4B)⋯C(24A)b |
−1 + x, y, 1 + z |
0.95 |
3.683(3) |
2.89 |
142 |
| C(9B)–H(9B)⋯centroid(B)a |
1 − x, −y, 1 − z |
0.95 |
3.663(3) |
2.76 |
160 |
| C(15B)–H(15B)⋯O(1A) |
x, y, 1 + z |
0.95 |
3.532(3) |
2.62 |
161 |
| C(12C)–H(12C)⋯O(1C) |
x, y, z |
0.95 |
2.724(2) |
2.39 |
101 |
| C(5C)–H(5C)⋯centroid(AE)a |
2 − x, 1 − y, 1 − z |
0.95 |
3.456(3) |
2.72 |
134 |
| C(8C)–H(8C)⋯C(22B)b |
2 − x, 1 − y, 1 − z |
0.95 |
3.633(3) |
2.85 |
141 |
| C(24C)–H(24C)⋯C(18)b |
x, y, z |
0.95 |
3.570(3) |
2.81 |
138 |
| C(2D)–H(2D)⋯C(9D)b |
1 − x, 1 − y, 1 − z |
0.95 |
3.682(3) |
2.86 |
146 |
| C(5D)–H(5D)⋯C(10C)b |
2 − x, 1 − y, 1 − z |
0.95 |
3.535(3) |
2.71 |
146 |
| C(8D)–H(8D)⋯C(18D)b |
1 − x, 1 − y, 1 − z |
0.95 |
3.563(3) |
2.75 |
145 |
| C(19D)–H(19D)⋯C(3D)b |
1 − x, 1 − y, 1 − z |
0.95 |
3.673(3) |
2.90 |
139 |
| C(21D)–H(21D)⋯C(25E)b |
x, y, z |
0.95 |
3.632(3) |
2.83 |
143 |
| C(2E)–H(2E)⋯O(1E) |
x, y, z |
0.95 |
2.712(2) |
2.37 |
101 |
| C(2E)–H(2E)⋯C(6D)b |
x, y, z |
0.95 |
3.671(3) |
2.78 |
157 |
| C(4E)–H(4E)⋯C(16B)b |
x, y, z |
0.95 |
3.669(3) |
2.84 |
146 |
| C(9E)–H(9E)⋯centroid(BD)a |
2 − x, 1 − y, 1 − z |
0.95 |
3.482(3) |
2.75 |
134 |
| C(24E)–H(24E)⋯C(22C)b |
x, y, z |
0.95 |
3.660(3) |
2.82 |
148 |
|
|
|
2C
|
|
|
|
|
|
| O(1)–H(1)⋯C(11)b |
x, −1 + y, z |
0.85(2) |
3.398(2) |
2.62(10) |
153(2) |
| C(6)–H(6)⋯O(1) |
x, 1 + y, z |
0.95 |
3.371(2) |
2.53 |
148 |
| C(15)–H(15)⋯O(1) |
x, y, z |
0.84 |
2.805(2) |
2.47 |
101 |
| C(4)–H(4)⋯centroid(C)a |
−1 + x, y, z |
0.95 |
3.704(2) |
2.92 |
140 |
| C(9)–H(9)⋯C(1)b |
−x, 0.5 + y, 1 − z |
0.95 |
3.619(2) |
2.77 |
149 |
| C(11)–H(11)⋯C(10)b |
1 − x, 0.5 + y, 1 − z |
0.95 |
3.725(2) |
2.89 |
147 |
|
|
|
3
|
|
|
|
|
|
| C(8)–H(8)⋯C(15)b |
1 − x, 2 − y, −z |
0.95 |
3.705(3) |
2.85 |
150 |
| C(16)–H(16)⋯centroid(B)a |
0.5 − x, 0.5 + y, −0.5 + z |
0.95 |
3.594(3) |
2.75 |
149 |
|
|
|
4
|
|
|
|
|
|
| N(1)–H(1A)⋯centroid(AA)a |
−x, 1 − y, 2 − z |
0.89(1) |
3.518(3) |
2.682(2) |
157(2) |
| N(1)–H(2A)⋯C(14A)b |
−x, 1 − y, 2 − z |
0.89(1) |
3.363(3) |
2.582(2) |
148(2) |
| N(1A)–H(1AA)⋯C(8)b |
1 − x, 1 − y, 2 − z |
0.88(1) |
3.579(3) |
2.799(2) |
148(2) |
| N(1A)–H(1AB)⋯centroid(A)a |
1 − x, 1 − y, 2 − z |
0.89(1) |
3.649(3) |
2.826(2) |
156 |
| C(6)–H(6)⋯N(1) |
x, y, z |
0.95 |
2.785(3) |
2.45 |
101 |
| C(12)–H(12)⋯N(1) |
x, y, z |
0.95 |
2.741(3) |
2.35 |
104 |
| C(2A)–H(2AA)⋯N(1A) |
x, y, z |
0.95 |
2.799(3) |
2.47 |
100 |
| C(18A)–H(18A)⋯N(1A) |
x, y, z |
0.95 |
2.773(3) |
2.40 |
103 |
| C(14)–H(14)⋯C(15A)b |
−x, −0.5 + y, 1.5 − z |
0.95 |
3.650(3) |
2.86 |
141 |
| C(17)–H(17)⋯C(3)b |
−x, 1 − y, 2 − z |
0.95 |
3.637(3) |
2.79 |
149 |
| C(18)–H(18)⋯centroid(CA)a |
−x, 1 − y, 2 − z |
0.95 |
3.698(3) |
2.95 |
136 |
| C(9A)–H(9A)⋯C(6A)b |
1 − x, 2 − y, 2 − z |
0.95 |
3.618(3) |
2.70 |
163 |
| C(4)–Br(1)⋯Br(1A) |
1 − x, −0.5 + y, 2.5 − z |
1.901(2) |
5.622(3) |
3.856(2) |
153.5(2) |
| C(4A)–Br(1A)⋯Br(1) |
1 − x, 0.5 + y, 2.5 − z |
1.900(2) |
4.251(3) |
3.856(2) |
88.4(2) |
 |
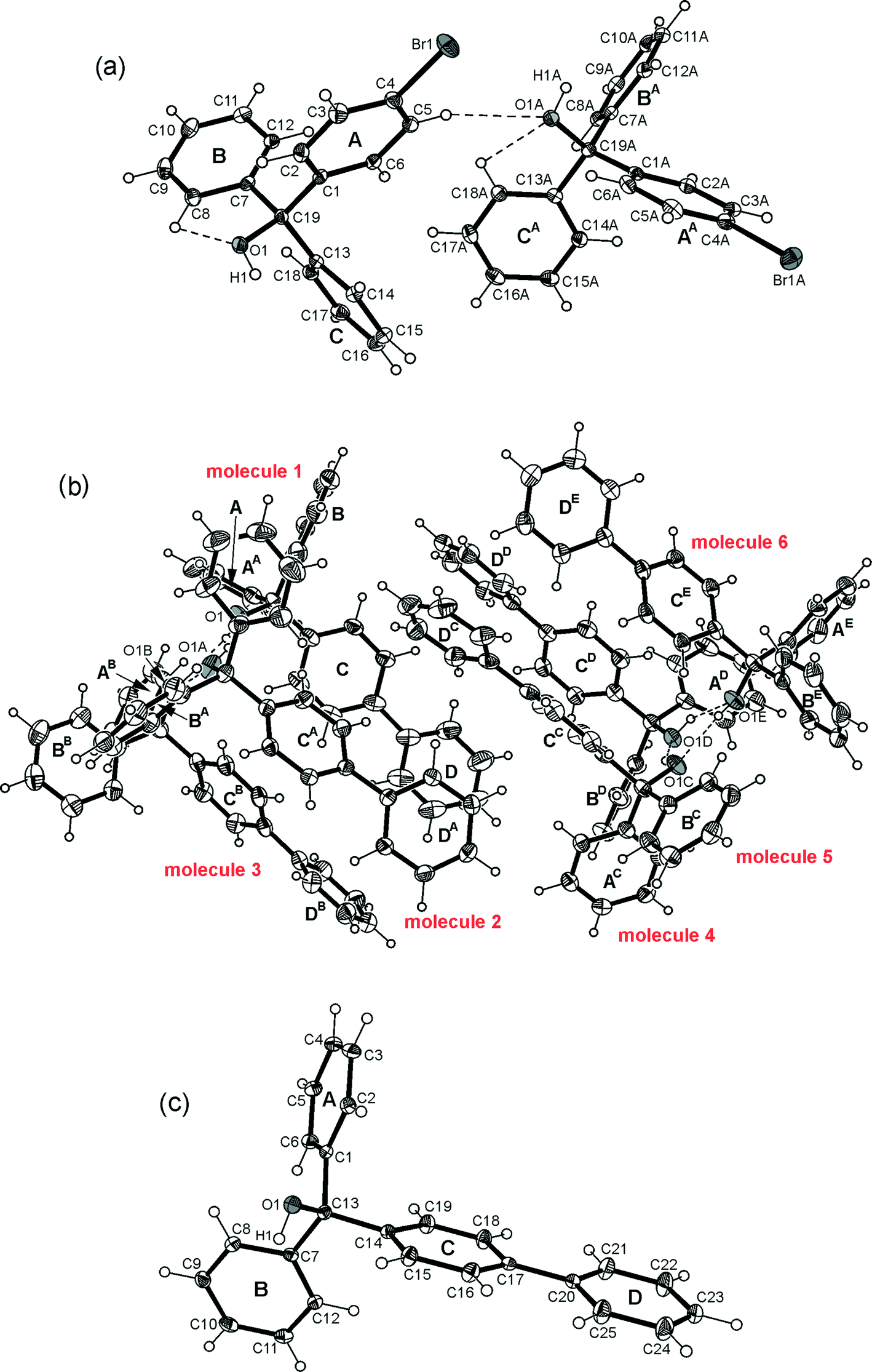
| | Fig. 2 Illustration of the molecular structures of 1 (a) and of the polymorphs 2B, 2C (b, c) including numbering of relevant atoms and ring specification. Thermal ellipsoids are drawn at 50% probability level. Grey ellipsoids represent hetero atoms. Broken lines represent C–H⋯O and O–H⋯O hydrogen bonds. | |
 |
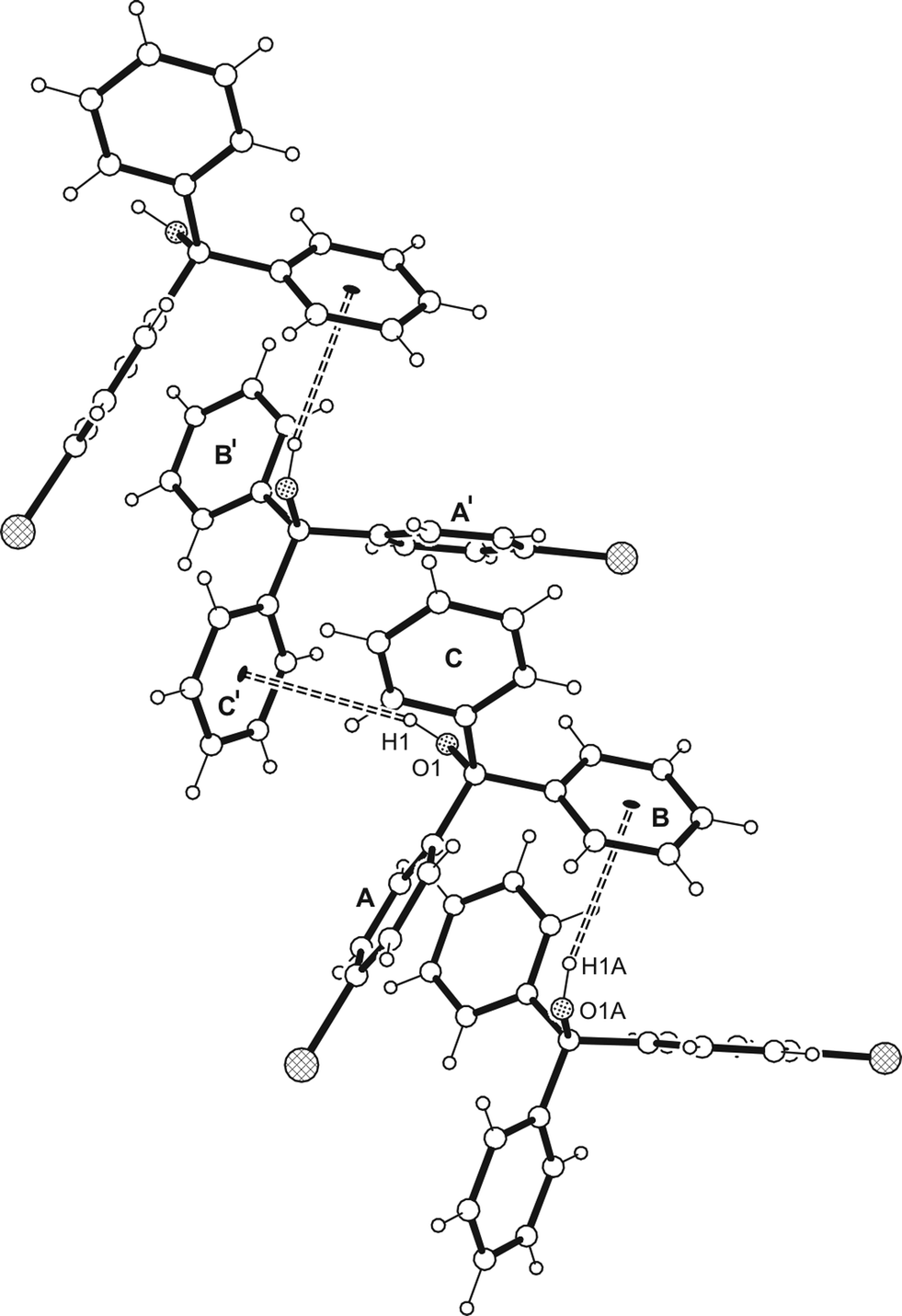
| | Fig. 3 Structure of the supramolecular strand of 1 with labelling of relevant atoms. The oxygens are displayed as dotted, the bromine atoms as cross-hatched circles. Broken double lines represent O–H⋯π interactions. | |
 |
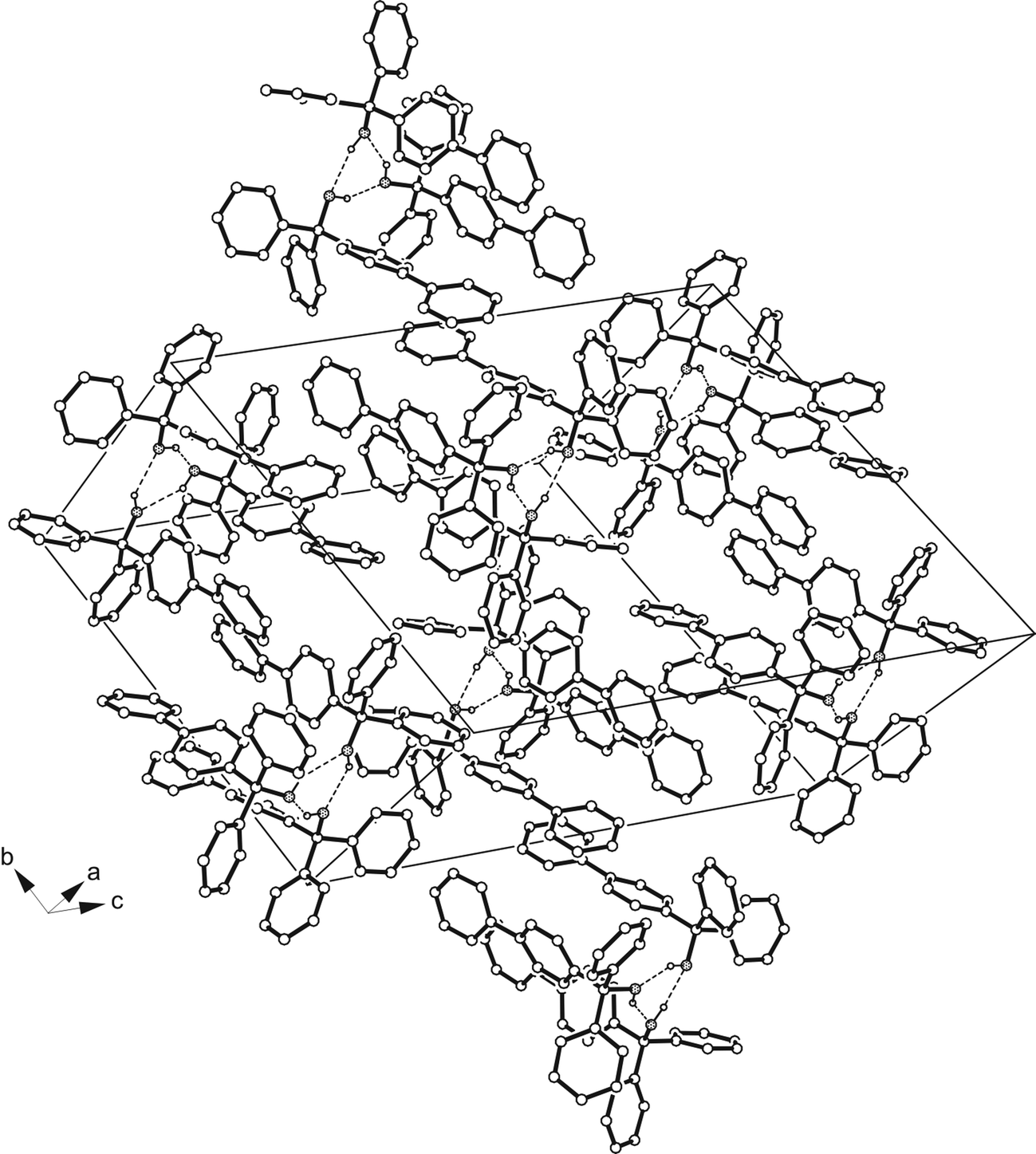
| | Fig. 4 Packing diagram of 2B. The hydrogen atoms of the aromatic units are omitted for clarity. The oxygens are displayed as dotted circles. Broken lines represent hydrogen bond type interactions. | |
 |
| | Fig. 5 Packing diagram of 2C. The oxygen atoms are displayed as dotted circles. Broken lines represent C–H⋯O hydrogen bonds, broken double lines O/C–H⋯π type interactions. | |
 |
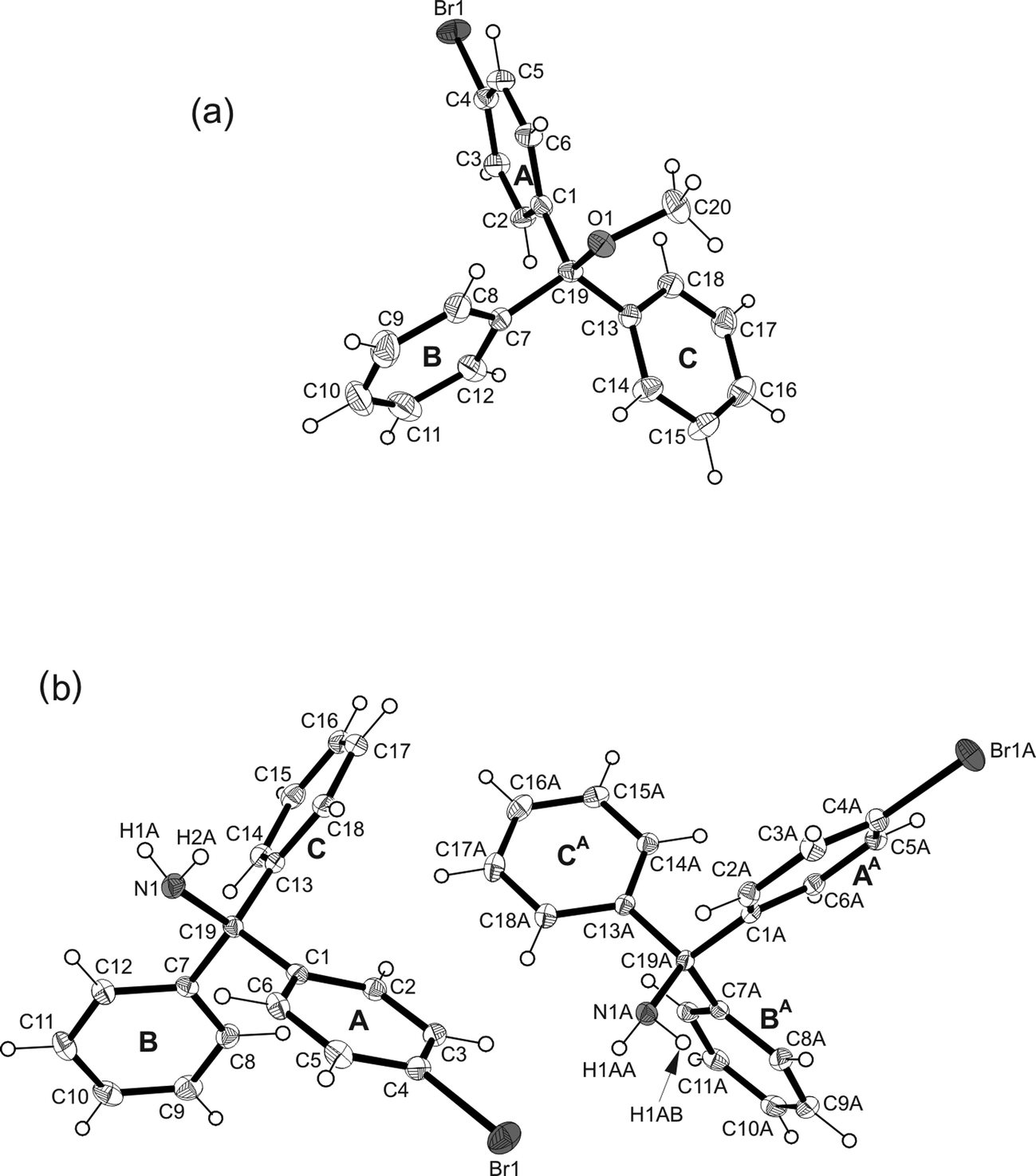
| | Fig. 6 Illustration of the molecular structures of 3 (a) and 4 (b). Thermal ellipsoids are drawn at 50% probability level. The oxygens/nitrogens are displayed as light grey, the bromine atoms as dark grey ellipsoids. | |
 |
| | Fig. 7 Packing illustration of 3 viewed down the crystallographic c-axis. The oxygen atoms are displayed as dotted, the bromine atoms as cross-hatched circles. Dotted double lines represent C–H⋯π interactions. | |
 |
| | Fig. 8 Structure of the supramolecular strand of 4 with labelling of relevant atoms. The nitrogens are displayed as hatched, the bromine atoms as cross-hatched circles. Broken double lines represent N–H⋯π interactions. | |
Crystallization of the bromo substituted triarylmethanol 1 from a solvent mixture of n-hexane and diethyl ether (9:1) yields colorless plates of the monoclinic space group P21/c with two crystallographically independent molecules in the asymmetric unit. The perspective view of the molecular structure is presented in Fig. 2a. The unsubstituted arene rings of the molecules are arranged nearly orthogonal to one another showing interplanar angles of 87.2(1) and 82.3(1)°, respectively. The conformation of the molecule is stabilized by a relatively short intramolecular C–H⋯O interaction10 [d(H⋯C) 2.34, 2.30 Å]. Contrary to expectations, the crystal structure lacks conventional hydrogen bonding.13 Instead, the hydroxy hydrogen of the molecules is used for the formation of a O–H⋯π contact10 to the bromophenyl ring of an adjacent molecule [O–H⋯C(arene) 2.64 Å, 156.0°; 2.46 Å, 153.6°] resulting in the creation of supramolecular strands, the structure of which is shown in Fig. 3. Interstrand association is accomplished by a close network of weak C–H⋯π contacts28,29 [d(H⋯C(aryl)) 2.73–2.88 Å] and C–Br⋯Br–C interactions of the type II geometry30,31 (θ1 = 151.4°, θ2 = 87.9°).
Crystallization of the biphenyl containing alcohol 2 from CHCl3 yields colorless crystals of the space group P with the asymmetric part of the unit cell comprising six independent molecules (Z = 12, Z′ = 6). Crystal structures featuring large values of Z′, particularly in connection with polymorphism, have received increasing interest in the last years being discussed in a topical review article.32 In the present structure, the molecules are assembled to form two trimers (Fig. 2b), each of them held together by a cyclic array of O–H⋯O hydrogen bonds [d(O⋯O) 2.735(3)–2.928(3) Å] and weak C–H⋯O [d(H⋯O) 2.37–2.38 Å] as well as C–H⋯π contacts [d(H⋯C) 2.72–2.84 Å]. The conformation of a trimer is such that its molecules are oriented in one direction with a nearly parallel alignment of their biphenyl axes. As is evident from geometric parameters (Table 2), the conformations of the molecules significantly deviate, which in particular can be seen from tilt angles between aromatic rings of the biphenyl units ranging from 22.9(1) to 44.6(1)° (Sup 1). It should be noted at this point that a known crystal structure of 2 (2A) reported by Ferguson et al.33 exists in the non-centrosymmetric orthorhombic space group P212121 (Z = 4, Z′ = 1), so that the present crystal structure represents a second polymorph (2B) of this compound. The difference in space group symmetries of the polymorphs suggests fundamental differences regarding the packing structures and modes of molecular association. In the present crystal structure, the molecules of adjacent trimers adopt an antiparallel arrangement (Fig. 4) thus allowing close packing and formation of multiple C–H⋯π-interactions. Nevertheless, the crystal of 2B contains solvent accessible lattice voids with a volume of 81.2 Å3 per unit cell (~1.5% of the cell volume). By way of contrast, neither conventional intramolecular nor intermolecular hydrogen bonds are observed in the previously reported structure of 2A.33 The crystal of this polymorph is composed of isolated molecules with only weak C–H⋯π contacts [d(H⋯C) 2.80–2.92 Å] acting between them to form molecular strings.
In the course of our crystal growing experiments we noticed that crystals of 2 obtained from diethyl ether existing in the monoclinic space group P21 with one molecule in the asymmetric unit represent another new polymorph 2C (Fig. 2c). The twist angle between the aromatic rings of the biphenyl element is 4.0(1)°. Remarkably, the packing of molecules as well as the pattern of intermolecular non-covalent bonding of this polymorph are also fundamentally different from that of the aforementioned structures of 2A and 2B. As illustrated in Fig. 5, the crystal is constructed of supramolecular strands running along the b-axis. Within a given strand, the molecules are connected by C–H⋯O hydrogen bonding [d(H⋯C) 2.53 Å]. The hydroxy hydrogen of the molecule participates in O–H⋯π hydrogen bond formation [d(H⋯π) 2.62(1) Å, ∠O–H⋯π 153(2)°] between the molecules of adjacent strands. Interstand association is completed by weak C–H⋯π contacts [d(H⋯π) 2.77–2.92 Å, ∠C–H⋯π 140–149°].
Crystallization of the methyl ether derivative of 1, that is 3, from methanol yields colorless rods of the orthorhombic space group Pna21 with one molecule in the asymmetric part of the unit cell (Fig. 6a). The pair of phenyl rings are oriented at 70.1(1)° to one another, while these rings are inclined at angles of 81.0(1) and 61.4(1)° with respect to the plane of the bromophenyl ring. As the molecule 3 lacks a strong hydrogen donor, the crystal structure is characterized by poor molecular association. A view of the packing structure along the crystallographic c-axis (Fig. 7) reveals that neither the ether oxygen nor the bromine atoms participate in molecular cross-linking, Hence, interactions between the molecules are restricted to weak C–H⋯π contacts only.
The compound 4, being the amine analogue of 1, crystallizes from n-hexane as colorless plates of the monoclinic space group P21/c with two independent molecules in the asymmetric unit (Fig. 6b). The molecules markedly deviate in their conformations as can be seen from the dihedral angles of 53.8(1) and 70.7(1)° formed between the unsubstituted aromatic rings. The bond lengths C(aryl)–C(sp3) and C–N are 1.537(2)–1.542(2) and 1.479(2)/1.484(2) Å and agree well with those found in the crystal structures of the polymorphic forms of plain triphenylmethylamine.22–24 The coordination behavior of the amine 4 resembles that of the alcohol 1, i.e. the strong hydrogen donors are involved in weak N–H⋯π contacts [d(H⋯π) 2.62–2.83 Å] giving rise to the formation of supramolecular strands (Fig. 8). Owing to the similarity of cell parameters between 1 and 4 the packing of molecules in the respective crystal structures is essentially of the same kind. More strictly speaking, in 4 one of the unsubstituted arene rings and the bromophenyl ring of each molecule act as acceptors for hydrogen bonding. Interstrand association is realized by C–Br⋯Br–C contacts of type-II geometry [θ1 = 153.5°; C(7)–Br(1)⋯Br(2) θ2 = 91.0°] and arene–arene interactions [C–H⋯π 2.70–2.86 Å, 141–163°].
Hirshfeld surface analysis
Hirshfeld surfaces17 represent an effective way to gain insight in the packing behavior of molecules in the crystalline state.34 In order to visualize quantitatively the different types of intermolecular interactions (hydrogen bonding, halogen-based interactions, C–H⋯π contacts, etc.) two-dimensional fingerprint plots,35 derived from Hirshfeld surfaces, were prepared by using the CrystalExplorer program (version 3.1).36 In these plots the distance de (distance of the Hirshfeld surface from the nearest nucleus outside the surface) is plotted against di (the corresponding distance to the nearest nucleus inside the surface).
For the structure 1, there are two crystallographically independent molecules in the asymmetric part of the unit cell. Thus, we have produced two fingerprint plots for the molecules (labelled as 1 and 2 in Fig. 9) which are very similar. Due to the high content of hydrogen atoms in the crystal structure, the 2D-fingerprint plots are dominated by H⋯H interactions comprising 58.4 and 56.6% of the surface, respectively (peak 1). Peak 2 represents C⋯H interactions (including C–H⋯π and O–H⋯π contacts) which comprise 38.0 and 40.7% of the surface. Br⋯H interactions (de + di ≈ 3.0 Å, peak 3) make up 17.2 and 14.1% of the Hirshfeld surface. The bright streak along the diagonal (de = di ≈ 1.9 Å) indicate the presence of weak Br⋯Br contacts. The sum (de + di) = 2.30 Å for H⋯H interactions as well as the diffuse pattern of points at the upper right region of the plots (de ≈ 2.6, di ≈ 2.75 Å) indicate that the packing is moderately efficient. The lattice energy calculated for 1 is −133.9 kJ mol−1.
 |
| | Fig. 9 2D-fingerprint plots for the molecules 1 and 2 in the asymmetric unit of compound 1. Spikes labelled as 1–3 correspond to H⋯H, C⋯H and Br⋯H interactions, respectively. | |
Polymorph 2A, derived from the CSD (Refcode YUHGOX), and polymorph 2C crystallize with one molecule in the asymmetric unit of the cell. Their 2D-fingerprint plots (Fig. 10) exhibit similarities which are reflected by nearly identical values for the different kinds of interactions, in which H⋯H contacts make up 58.4/56.6%, C⋯H interactions 38.0/40.7% and O⋯H interactions 3.1/3.1%. Nevertheless, the plots display differences. The fingerprint plot of 2A reveals a diffuse blue tail of points at the upper right (di = 2.4–2.8 Å), which indicates a non-optimal packing of molecules. The plot of polymorph 2C is characterized by two sharp features (de + di = 2.5 Å) which, apart from CH⋯π bonding, represent O–H⋯π contacts, the latter being absent in polymorph 2A. The structural similarities of the polymorphs are reflected by nearly identical lattice energies of −165.5 (2A) and −169.9 kJ mol−1 (2C).
 |
| | Fig. 10 2D-fingerprint plots for the molecules 2 in the polymorphs 2A and 2C. Spikes labelled as 1 and 2 correspond to H⋯H and C⋯H interactions, respectively. | |
The polymorph 2B refers to the structure with Z′ = 6. We therefore computed Hirshfeld surfaces choosing each individual molecule in turn as a target and obtained six distinct fingerprint plots (Fig. 11). Each fingerprint plot displays three characteristic patterns labelled 1–3 for molecule 3. The plots are similar in that they all indicate a large proportion for H⋯H interactions varying from 57.0 to 63.6%, followed by C⋯H interactions (32.2–39.1%) and O–H⋯O hydrogen bonds (2.8–3.4%), the latter appearing as a pair of spikes, the upper left (de > di) corresponding with the hydrogen bond donor, the lower one (de < di) with the hydrogen bond acceptor. The values for de + di are in the range between 1.95 and 2.15 Å. Peak 1 of the plots represents H⋯H interactions with some of them showing anomalous short distances near 2.0 Å which suggests a tight packing of molecules. Compared with 2A and 2C, the increased content of H⋯H interactions which is accompanied by a decrease of C⋯H interactions may be attributed to completely different packing modes which in 2B includes compact trimers of O–H⋯O bonded molecules. Obviously, the unique alignment of the biphenyl parts within the trimers enhances the degree of C–H⋯π interactions. They are visible in the 2D-fingerprint plots as “wings” (peak 2). It should be noted here, that O–H⋯O hydrogen bonds have a structure directing effect, although they represent only a small proportion of the Hirshfeld surfaces. Furthermore, we see in some of the fingerprint plots (molecules 1–3) a rather diffuse collection of points at larger values for de and di, indicating that parts of the Hirshfeld surface of the molecules are without close contacts indicating the presence of voids in the crystal lattice. Unfortunately, we could not compute the lattice energy for polymorph 2B, because the number of atoms exceeded the limit allowed by the program OPIX (Gavezotti).37
 |
| | Fig. 11 2D-fingerprint plots for the six molecules 2 in the asymmetric unit of 2B. | |
The structure 3 bears resemblance to the structure of 1 in that the only strong features are H⋯H (52.8%) and C⋯H contacts (24.9%). The fingerprint plot of the molecule (Fig. 12) is characterized by two “wings” (de + di ≈ 2.7 Å) which can be ascribed to C–H⋯π interactions. The lattice energy is computed to −130.1 kJ mol−1.
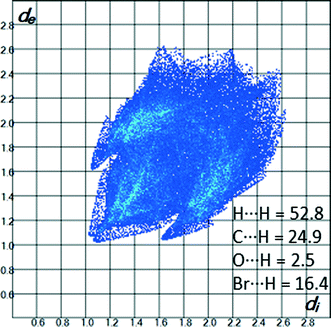 |
| | Fig. 12 2D-fingerprint plot for the molecule in the structure of 3. | |
The 2D-fingerprint plots for the two independent molecules of the amine 4 (Z′ = 2) (Fig. 13) show proportions of 51.4 and 46.6% for H⋯H interactions. C⋯H represent 31.5 and 33.3% of the Hirshfeld surface, respectively. The C–H⋯π and N–H⋯π interactions in the crystal structure appear as distinct “wings” at the upper left and lower right region of the plot. The Br⋯H interactions also have a relatively significant contribution to the total Hirshfeld surfaces comprising 14.3 and 17.0%, respectively. Similar to the crystal structure of 1, Br⋯Br contacts are clearly visible as a bright streak (de = di ≈ 1.9 Å). The lattice energy of the structure amounts to −137.0 kJ mol−1.
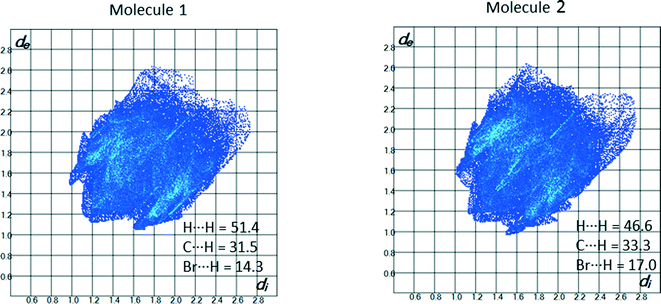 |
| | Fig. 13 2D-fingerprint plots for the molecules 1 and 2 in the asymmetric unit of compound 4. | |
Comparative reflection and conclusions
Based on the above discussion and including previous structural results of appropriate compounds of comparison, remarks can be made and conclusions drawn as follows. Considering the construction mode observed in the crystal structure of the prototype tritylalcohol (Ph3OH), molecular tetramers with the hydroxyl groups forming a pyramidal motif of O–H⋯O hydrogen bonds are found.38 Though behaving in a special manner, conventional hydrogen bonding is taking a main effect in this crystal. By way of contrast, the crystal structure of 1 shows O–H⋯π bonded molecular strands interlinked via weak Br⋯Br interactions. Thus, compared to the prototype just one single bromo substituent as in 1 induces loss of conventional hydrogen bonding between OH groups which in the main may be attributed to effects of molecular packing possibly supported by the opening of a specific Br⋯Br interaction. A similar way of acting is reported for the tribromo and triiodo derivatives of tritylalcohol in their crystal structures that are characterized by O–H⋯π bonded molecular chains with the halogens being here involved in Hal⋯π instead of Hal⋯Hal contacts.39 Even more puzzling facts have been found for the biphenyl derivative 2 of which we succeeded to crystallize two polymorphous structures (2B from CHCl3, 2C from n-hexane/diethyl ether, 9:1) differing from that of the known polymorph (2A) obtained from nitromethane.33 While in 2A the crystal structure is only stabilized by weak C–H⋯π interactions, surprisingly enough, in the polymorphous crystal of 2B the molecules are assembled to O–H⋯O bonded trimers which are further stabilized by C–H⋯O and C–H⋯π contacts. Hence, 2B behaves similarly to the unsubstituted prototype compound in O–H⋯O bonded cluster formation though giving rise to trimer instead of tetramer clustered building units typical of the prototype. This may be due to the spatial demand of the additional phenyl substituent in 2B, but enabling as a secondary effect favourable weak intermolecular contacts in the packing. As opposed to 2B, in the polymorphous structure 2C the molecules are assembled to C–H⋯O bonded strands which are connected by O–H⋯π hydrogen bonding interactions and weak C–H⋯π contacts to a three-dimensional supramolecular network. Hence grown from different solvents, 2 can be obtained as crystals 2A–2C showing distinctly different modes of intermolecular interactions. Considering the thermal behavior of the polymorphs, a detailed investigation has been performed by differential scanning calorimetry (DSC). The onset temperatures for the melting of 135.3 °C (2A), 105.0 °C (2B) and 134.6 °C (2C) as well as enthalpy values of 86.1 (2A), 77.5 (2B) and 82.4 J g−1 (2C) have been determined. Actually, the temperature difference between the polymorphs 2A and 2C is very small, which can be explained by structural similarities.
It stands to reason that the methyl ether analogous compound 3 can not form corresponding OH involved interactions. However, Hal⋯Hal contacts typical of 1 are also absent in 3 while only weak C–H⋯π interactions, comparable to 2A determine the crystal structure. Two polymorphic crystal structures are also known for the parent compound of 4, i.e. the unsubstituted triphenylmethylamine. One is composed of N–H⋯N bonded molecular dimers with the amino hydrogens disordered over two positions23,24 whereas in the other polymorph, molecules are interlinked by weak N–H⋯π contacts to 2D supramolecular aggregates.15 On the other side, a molecular organization analogous to that of 1 is found in the crystal structure of the corresponding amine 4 with both amino hydrogens being involved in chain formation via N–H⋯π interaction. So, following the relation of O–H⋯O to O–H⋯π interactions in the tritylalcohols, interchangeability of N–H⋯N for N–H⋯π contacts, according to substitution and polymorphism obviously is also a characteristic feature in the crystal formation of the tritylamines.
In summary, the impression is given that compounds involving both trityl alcohols and amines rather sensitively respond to substitution or change of substituent as well as the solvent used for crystallization in the formation of an appropriate crystal structure. Therefore, to exert control on a desired crystal structure including examples of these compound classes is not without its problems. Nevertheless, deduced from the given discussion certain trends in forming cluster aggregates via strong hydrogen bonding or non-cluster type structures being founded on weak hydrogen bond type and related interactions can be read from the comments of this paper that could be supportive for purposes of crystal engineering in the field.
Experimental
General
Melting points (uncorrected) were determined using a microscope heating stage PHMK Rapido (VEB Wägetechnik). IR spectra were measured on a FT-IR 510 Nicolet as KBr pellets. 1H and 13C NMR spectra were recorded in chloroform solution at room temperature on a Bruker Avance DPX 400 at 400 MHz, respectively. Elemental analyses were performed on a Hereus CHN rapid analyzer. Mass spectra were obtained using a Hewlett-Packard GC-MS 5890. Column chromatography was performed on a silica gel (particle size 0.063–0.1 mm, Merck). For analytical thin layer chromatography silica gel 60 F254 (Merck) was used.
Materials
Organic solvents were purified by standard procedures. Starting compounds bromobenzene and 4-bromobenzophenone were purchased from Acros Organics. 4-Biphenylyl phenyl ketone was synthesized as described.3
Preparation of the trityl compounds 1–4
(4-Bromophenyl)diphenylmethanol (1).
To a solution of phenyl magnesium bromide (8.8 g, 48.6 mmol) in dry diethyl ether (50 ml), prepared from bromobenzene (7.6 g, 48.6 mmol), a solution of 4-bromobenzophenone (10.83 g, 44.2 mmol) in dry diethyl ether (100 ml) was added dropwise under an atmosphere of argon. The mixture was refluxed for 1 h and then stirred at room temperature for 2 h. After that, the mixture was cooled in ice and quenched with a saturated aqueous solution of NH4Cl. The organic layer was separated and the aqueous layer extracted with diethyl ether. The combined organic phases were washed with water, dried (Na2SO4) and evaporated. The crude product (yellow-orange oil) was crystallized from diethyl ether-n-hexane (1:9, ν/ν) to yield 7.5 g (50%) colorless crystals. M.p. 75 °C (lit.21 74 °C, lit.22 99–100 °C). Found: C, 67.48; H, 4.53; C19H15OBr requires C, 67.27; H, 4.46%; νmax (KBr cm−1) 3566 (OH), 3079, 3056, 3022 (CAr–H), 1597, 1489, 1448 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, Ar), 1394, 1154, 1033, 1008, 821 (disubst. Ar), 764, 698 (monosubst. Ar); δH (400 MHz; CDCl3; Me4Si) 2.77 (s, 1H, OH), 7.16 (d, 3JHH = 8.0 Hz, 2H, Ar–H), 7.22–7.30 (m, 10H, Ar–H), 7.47 (d, 3JHH = 8.8 Hz, 2H, Ar–H); δC (100 MHz; CDCl3; Me4Si) 81.7 (C–OH), 121.4 (C–Br), 127.5–129.7, 131.0, 145.9, 146.4 (Ar–C); m/z: 338 [M–H]+, 261, 183, 155, 105 (100%), 77, 51.
C, Ar), 1394, 1154, 1033, 1008, 821 (disubst. Ar), 764, 698 (monosubst. Ar); δH (400 MHz; CDCl3; Me4Si) 2.77 (s, 1H, OH), 7.16 (d, 3JHH = 8.0 Hz, 2H, Ar–H), 7.22–7.30 (m, 10H, Ar–H), 7.47 (d, 3JHH = 8.8 Hz, 2H, Ar–H); δC (100 MHz; CDCl3; Me4Si) 81.7 (C–OH), 121.4 (C–Br), 127.5–129.7, 131.0, 145.9, 146.4 (Ar–C); m/z: 338 [M–H]+, 261, 183, 155, 105 (100%), 77, 51.
4-Biphenylyldiphenylmethanol (2).
Solutions of phenyl magnesium bromide (8.8 g, 48.6 mmol) and of 4-biphenylyl phenyl ketone (12.50 g, 44.2 mmol) in dry diethyl ether were reacted and worked up following the above procedure. Crystallization of the crude product from chloroform yielded 73% colorless crystals. M.p. 104–105 °C. νmax (KBr cm−1) 3441 (OH), 3027 (CAr–H), 1598, 1486, 1445 (CC, Ar), 1401, 1157, 1005, 898 (disubst. Ar), 762, 695 (monosubst. Ar); δC (500.1 MHz; CDCl3; Me4Si) 81.9 (C–OH), 126.6, 127.1, 127.3, 127.9, 128.0, 128.3, 128.8, 140.0, 140.6, 145.9, 146.8 (Ar–C); δH (400 MHz; CDCl3; Me4Si) 2.83 (s, 1H, OH), 7.25–7.37 (m, 13H, Ar–H), 7.43 (t, 2 H, Ar–H), 7.54 (d, 3JHH = 8.8 Hz, 2H, Ar–H), 7.59 (d, 3JHH = 8.8 Hz, 2H, Ar–H); m/z: 336 [M–H]+, 259, 181, 152, 105, 77, 51.
[(4-Bromophenyl)diphenylmethyl] methyl ether (3).
To a mixture of sodium hydride (1.2 g, 50 mmol, suspension in paraffin oil) in dry THF was added 1 (1.6 g, 5 mmol) under an atmosphere of argon. After having stirred for 30 min, methyl iodide (4.25 ml, 66 mmol) was added dropwise and the mixture stirred for 16 h at 40 °C. The mixture was quenched with water, the organic layer separated and the aqueous layer extracted with diethyl ether. The combined organic phases were washed with water, dried (Na2SO4) and evaporated. The crude product (colorless oil) was crystallized from DMF to yield 1.7 g (96%) colorless crystals. M.p. 69 °C. Found: C, 68.19; H, 5.00; C20H17OBr requires C, 68.00; H, 4.85%; νmax (KBr cm−1) 3085, 3024, (CAr–H), 2950, 2851 (CH3), 1583, 1491, 1451 (CC, Ar), 1075, 1014, 823 (1,4-disubst. Ar), 773, 706 (monosubst. Ar); δH (400 MHz; CDCl3; Me4Si) 3.04 (s, 3H, OCH3), 7.23–7.34 (m, 12H, Ar–H), 7.40 (d, 3JHH = 8.8 Hz, 2H, Ar–H); δC (100 MHz; CDCl3; Me4Si) 52.1 (OCH3), 86.7 (C–OCH3), 121.0 (C–Br), 127.2–130.2, 130.9, 143.2, 143.6 (Ar–C); m/z: 352 [M–H]+, 352, 321, 273, 239, 197, 165, 105 (100%), 77.
(4-Bromophenyl)diphenylmethanamine (4).
To a solution of 1 (3.6 g, 10 mmol) in dry dichloromethane (50 ml) was added freshly destilled thionyl chloride (4 ml, 55 mmol). The mixture was stirred for 8 h at room temperature. The excess thionyl chloride and the solvent were removed under vacuum. The residue was dissolved in dichloromethane (50 ml) and to the solution was added conc. aqueous ammonia (100 ml) at 0 °C. The mixture was stirred for 48 h at room temperature and then extracted with dichloromethane. The combined organic phases were washed with water, dried (Na2SO4) and evaporated. Purification of the crude product by column chromatography on silica gel using dichloromethane as eluent yielded 2.8 g (83%) colorless crystalline solid. M.p. 79 °C. Found: C, 67.37; H, 4.73; N, 4.10; C20H17NBr requires C, 67.47; H, 4.77; N, 4.14%; νmax (KBr cm−1) 3357, 3294 (NH2), 3088, 3050, 3018 (CAr–H), 1603 (NH2), 1581, 1492, 1445 (CC, Ar), 1074, 1030, 1008, 821 (1,4-disubst. Ar), 767, 704 (monosubst. Ar); δH (400 MHz; CDCl3; Me4Si) 2.30 (s, br, 2H, NH2), 7.15–7.29 (m, 12H, Ar–H), 7.40 (d, 3JHH = 8.8 Hz, 2H, Ar–H); δC (100 MHz; CDCl3; Me4Si) 66.0 (C–NH2), 120.7 (C–Br), 126.8–130.0, 130.9, 147.6, 148.1 (Ar–C); m/z: 337 [M–H]+, 260, 182 (100%), 104, 77, 51.
X-ray structure determination
The intensity data of 1–4 were collected on a Kappa APEX II diffractometer (Bruker AXS) with MoKα radiation (λ = 0.71073 Å). Reflections were corrected for background, Lorentz and polarisation effects. Preliminary structure models were derived by application of direct methods40 and were refined by full-matrix least-squares calculation based on F2 for all reflections.41 With the exception of the hydroxy hydrogens in 1 and 2C as well as the amino hydrogens in 4, all other hydrogen atoms were included in the models in calculated positions and were refined as constrained to bonding atoms. Crystallographic data for the structures in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication numbers CCDC 1062479 (1), 1062483 (2B), 1062482 (2C), 1062481 (3) and 1062480 (4).
Acknowledgements
We thank Prof. L. R. Nassimbeni and Dr. E. Batisai (University of Cape Town) for performing the Hirshfeld surface analyses and their help with discussion of the results.
References
-
V. G. Witterholt, in Kirk-Othmer Encyclopedia of Chemical Technology, 2nd edn, Wiley, New York, 1969, vol. 20, p. 672 Search PubMed.
-
T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, 3rd edn, New York, 1999 Search PubMed.
- G. V. M. Sharma, T. Rajendra Prasad, Rakesh and B. Srinivas, Synth. Commun., 2004, 34, 941 CrossRef CAS PubMed.
- E. Weber, K. Skobridis and I. Goldberg, J. Chem. Soc., Chem. Commun., 1989, 1195 RSC.
- E. Weber, K. Skobridis, A. Wierig and I. Goldberg, J. Incl. Phenom. Mol. Recogn., 1997, 28, 163 CrossRef CAS.
- M. S. Fonari, Y. A. Simonov, W.-J. Wang, S.-W. Tang and E. V. Ganin, CrystEngComm, 2009, 11, 94 RSC.
- M. S. Fonari, E. V. Ganin and W.-J. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o431 Search PubMed.
- D. Schollmeyer, O. V. Shishkin, T. Rühl and M. O. Vysotsky, CrystEngComm, 2008, 10, 715 RSC.
- N. Hayashi, T. Mori and K. Matsumoto, Chem. Commun., 1998, 1905 RSC.
-
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- M. D. Prasanna and T. N. Guru Row, Cryst. Eng., 2000, 3, 135 CrossRef CAS.
-
P. Metrangolo, G. Resnati, T. Pilati and S. Biella, in Halogen Bonding, Structure and Bonding, ed. P. Metrangolo and G. Resnati, Berlin-Heidelberg, 2008, vol. 126, p. 105 Search PubMed.
-
G. A. Jeffrey, An Introduction to Hydrogen Bonding, Wiley, Chichester, 1997 Search PubMed.
- M. Canle, L. W. Clegg, I. Demirtas, M. R. J. Elsegood and H. Maskill, J. Chem. Soc., Perkin Trans. 2, 2000, 85 RSC.
- C. Glidewell and G. Ferguson, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 924 CrossRef.
- F. H. Allen and O. Kennard, J. Mol. Graphics, 1993, 8, 31 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378 RSC.
-
Organic Crystal Engineering, ed. R. T. Tiekink, J. J. Vittal and M. Zaworotko, Wiley, Chichester, 2010 Search PubMed.
- C. B. Aakeröy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22 RSC.
-
Making Crystals by Design: Methods, Techniques and Applications, ed. D. Braga and F. Grepioni, Wiley-VCH, Weinheim, 2007 Search PubMed.
- M. Gomberg and L. H. Cone, Ber. Dtsch. Chem. Ges., 1906, 39, 3274 CrossRef CAS PubMed.
- C. Manning, M. R. McClory and J. J. McCullough, J. Org. Chem., 1981, 46, 919 CrossRef CAS.
-
W. Clegg and M. R. G. Elsewood, Private Communication, 2005 Search PubMed.
- V. N. Khrustalev, I. V. Borisova, N. N. Zemlyansky and M. Yu. Antipin, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o31 CAS.
- V. Theodorou, K. Skobridis and A. Karkatsoulis, Tetrahedron, 2007, 63, 4284 CrossRef CAS PubMed.
-
K. Nützel, H. Gilman and G. F. Wright, in Methoden Org. Chem. (Houben-Weyl), Thieme, Stuttgart, 1973, vol. 13/2a, pp. 49–527 Search PubMed.
-
H. Feuer and J. Hooz, in Chemistry of the Ether Linkage, ed. S. Patai, Wiley, New York, 1967, pp. 445–498 Search PubMed.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC.
- V. R. Pedireddi, D. S. Reddy, B. S. Goud, D. C. Craig, A. D. Rae and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353 RSC.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952 CrossRef CAS PubMed.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895 CrossRef CAS PubMed.
- G. Ferguson, C. D. Carroll, C. Glidewell, C. M. Zakaria and A. J. Lough, Acta Crystallogr., Sect. B: Struct. Sci., 1995, 51, 367 Search PubMed.
- E. Batisai, L. R. Nassimbeni and E. Weber, CrystEngComm, 2015, 17, 4205 RSC.
- J. J. McKinnon, M. A. Spackman and A. S. Mitchell, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 627 CrossRef PubMed.
-
S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2010 Search PubMed.
-
A. Gavezotti, OPIX: A computer program package for the calculation of intermolecular interactions and crystal energies, University of Milan, 2003 Search PubMed.
- G. Ferguson, J. F. Gallagher, C. Glidewell, J. N. Low and S. N. Scrimgeour, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 1272 CrossRef.
- D. Schollmeyer, O. V. Shishkin, T. Rühl and M. O. Vysotsky, CrystEngComm, 2008, 10, 715 RSC.
-
G. M. Sheldrick, SHELXS-97: Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 2008 Search PubMed.
-
G. M. Sheldrick, SHELXS-97: Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 2013 Search PubMed.
Footnote |
| † CCDC 1062479–1062483. Overlay scheme of molecular conformation (Sup 1) and crystallographic data. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00871a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence