
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFeatures of styryl dye crystal packings and their influence on [2 + 2] photocycloaddition reaction with single crystal retention†
Lyudmila G.
Kuz'mina
*a,
Artem I.
Vedernikov
b,
Judith A. K.
Howard
c,
Michael V.
Alfimov
b and
Sergey P.
Gromov
*b
aN. S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninsky prosp. 31, Moscow 119991, Russian Federation. E-mail: kuzmina@igic.ras.ru; Fax: +7 (495) 954 1279
bPhotochemistry Center, Russian Academy of Sciences, Novatorov str. 7A-1, Moscow 119421, Russian Federation. E-mail: spgromov@mail.ru; Fax: +7 (495) 936 1255
cDepartment of Chemistry, University of Durham, Science Laboratories, South Road, Durham DH1 3LE, England, UK. E-mail: j.a.k.howard@durham.ac.uk
First published on 18th May 2015
Abstract
A new styryl dye of the 2-benzothiazole series which contains three methoxy groups and an iodide anion was synthesized. Three triclinic crystal forms of this dye were investigated by single crystal X-ray diffraction. All the modifications were shown to contain centrosymmetrically related stacks of cations with the syn-“head-to-tail” mutual arrangement. Stacks of cations are arranged as dimeric pairs with short interatomic distances between the ethylene group carbon atoms, d1 = 3.54–3.75 Å and significantly longer analogous distances between the adjacent pairs, d2 = 4.61–4.68 Å. The main difference of the packing consists in various orientations of N-ethyl substituents with respect to the centre of the dimer. The solid phase [2 + 2] photocycloaddition (PCA) reaction resulting in rctt isomer of the cyclobutane derivative, only proceeds in pairs where N-substituents are oriented outward from the centre of the dimeric pair. PCA is accomplished as a “single crystal-to-single crystal” transformation.
Introduction
The topochemical [2 + 2] photocycloaddition (PCA) reaction of ethylene-containing organic compounds developed in 1960s1,2 has stimulated considerable interest.3–46 According to this reaction, two ethylene compounds under irradiation give a cyclobutane product via simultaneous formation of two σ-bonds (Scheme 1). | ||
| Scheme 1 PCA reaction of ethylene compounds.1 | ||
An investigation of the PCA reaction is important for synthetic and medicinal chemistry, for understanding some chemical processes in nature and also those underlying the behavior of light sensitive resistors. The PCA reaction is also challenging for creation of information optical recording systems.8,16
Many reactions of cycloaddition are known in organic chemistry, for instance – [2 + 3] cyloaddition,47 [3 + 4] cycloaddition,48 [4 + 2] cycloaddition.49 However, these reactions are thermal and they proceed in solution. Whereas, PCA represents a temperature independent process. It is photochemically reversible, often regio- and stereoselective and it proceeds in both solution and in the solid phase. Commonly the PCA reaction is initiated by visible light or near UV region light (340–500 nm), and the back reaction (retro-PCA), by a shorter wave UV radiation (~220–270 nm). In order to achieve a solid phase PCA, pre-organization of the ethylene fragments is required in which these fragments are situated effectively parallel to one above another at a distance between the ethylene carbon atoms (d1) not exceeding 4.2 Å (Schmidt's criterion1). The PCA reactions that take place in single crystals without their decomposition are of special interest and by now many such cases are known.3–7,9,17–20,22,28,29,38,43,45,46
In studies of PCA reactions it is important to a priori achieve the formation of a “pre-organized” dimeric pair which shows the stacking interaction of the two molecules. Molecular design is one of several methods to achieve this. Many examples of pre-organized dimeric pairs are described in the literature via the coordination of the initial ethylene systems with metal cations11,30,32,34,38,40,43,50 or the formation of hydrogen bonds between the components,7,9,10,12,14,15,17,20,21,23–25,29,30,40,41,46,50 as well as entering two structural units in cavitand cavity – cyclodextrins, calixarenes, cucurbit[8]uril, nanocages.17,35,36,39,40
Another approach to create the “pre-organized” dimeric pair is only possible for the PCA reaction in the solid state. This is crystal engineering.51 This approach is based on the assumption that crystal packing may involve “pre-organized” pairs as its elements, mainly due to stacking interaction of the component conjugated systems.3–7,13,16,18,19,22,26–28,31,33,37,42,44,45 In this case the problem consists in directed design to create the required crystal packing. With this approach, the X-ray structural analysis turns out to be the most important and informative method.
An analysis of large number of the packing arrangements of flat conjugated molecule crystal available from the Cambridge Structural Database (CSD)52 indicates six canonic types (Fig. 1).
 | ||
| Fig. 1 Canonical types of crystal packings of flat conjugated molecules; the lines represent side projections of the molecules; the red brackets indicate the dimeric pairs. | ||
Stacking pattern 1 and 2, as well as parallel-dimeric packings 3 and 4 contain “pre-organized” dimeric pairs as their elements. Packings 5 and 6 do not contain these pairs, hence, the PCA reaction in this arrangement is impossible.
Our previous investigations3–7,17,18,28,45,46 have shown that brightly colored protonated styrylheterocycles and quaternary salts of styrylheterocycles (styryl dyes) Het+–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Ar X− (X− = I−, ClO4−, BF4−, TsO−) are very suitable species for studying PCA reaction in the solid state, including the “single crystal-to-single crystal” (SCSC) processes (i.e., the processes that proceed without single crystal destruction).
CH–Ar X− (X− = I−, ClO4−, BF4−, TsO−) are very suitable species for studying PCA reaction in the solid state, including the “single crystal-to-single crystal” (SCSC) processes (i.e., the processes that proceed without single crystal destruction).
First their advantage is the potential to create the stacking packing motifs 1 or 2, and the rarely parallel-dimeric motifs 3 or 4. Second advantage is a large colour contrast between the initial compounds and PCA reaction products (cyclobutane derivatives) which makes visual control of the process possible.
Earlier, we have studied in detail the peculiarities and conditions for PCA reactions in crystals and in polycrystalline films of styryl dyes of the 4-pyridine series,5,6,45,46 a selection of the 4-quinoline series,4 various solvate forms of two dyes of the 4-pyridine (ClO4− anion)7 and 2-benzothiazole (TsO− anion)28 series, as well as in the protonated 4-styrylpyridines (ClO4− anions).18 For two of mentioned dyes we first proved a feasibility of both direct PCA and retro-PCA reactions in the same crystal without its decomposition using X-ray structure determination.28,45
In this current study, we continued to investigate solid phase PCA using the example of the styryl dye of 2-benzothiazole series (1), with the I− anion and methoxy groups in position 5 of benzothiazole residue and positions 3′ and 4′ of benzene ring.
Results and discussion
Styryl dye 1 was synthesized by a condensation of 3-ethyl-5-methoxy-2-methylbenzothiazolium iodide with 3,4-dimethoxybenzaldehyde by heating in acetic anhydride (Scheme 2). The dye was characterized by 1H and 13C NMR spectroscopy data; it has the E configuration of the ethylene bond, judging by the spin–spin coupling constant 3Jtrans-HCCH = 15.7 Hz. According to elemental analysis data, dye 1 was obtained as a mixed salt with 15% impurity of I3− anions. Apparently, in the course of heating in Ac2O, the iodide anions partly oxidized to give I2. However, single crystals of this dye were found to contain I− anions only (see below).
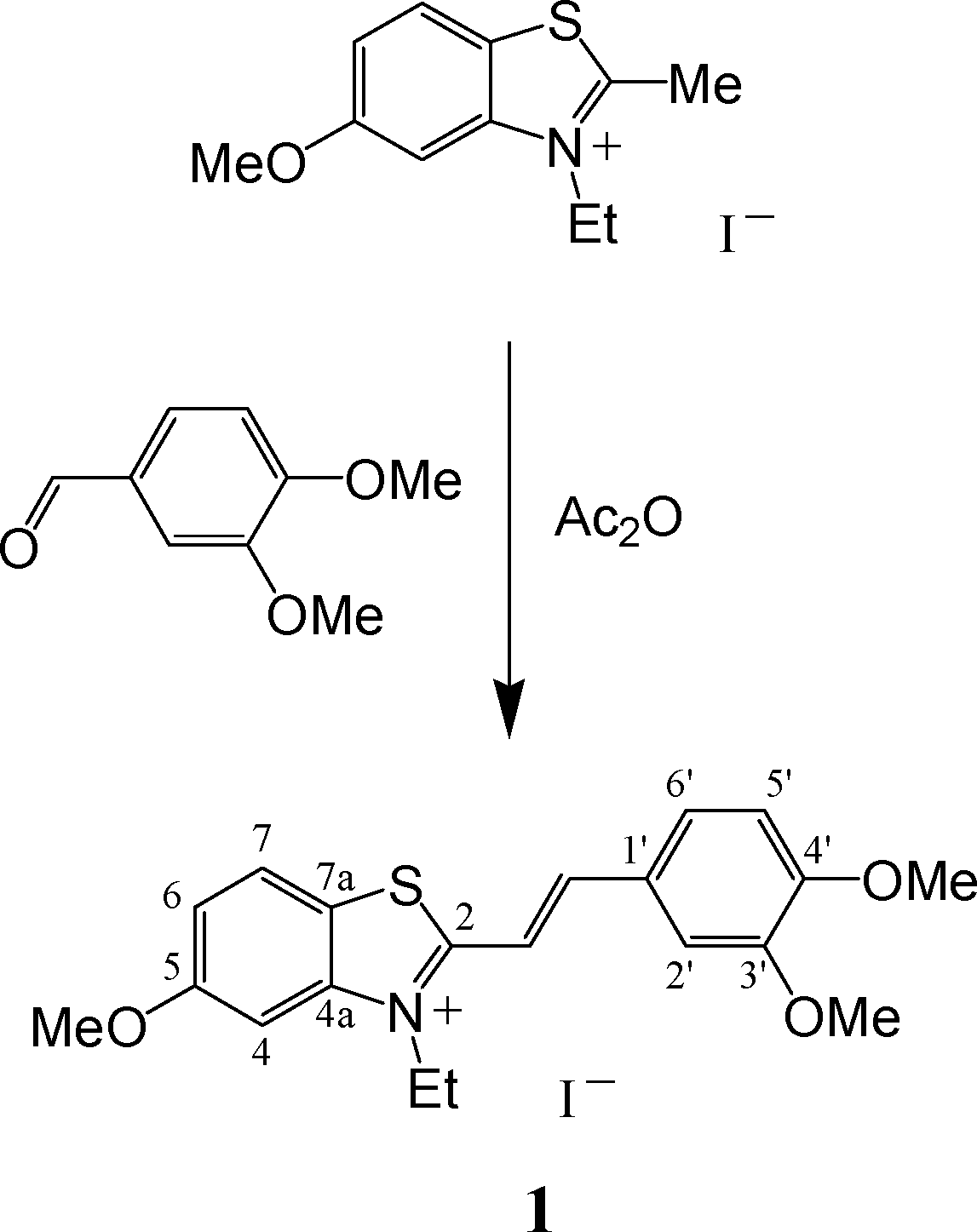 | ||
| Scheme 2 Synthesis of dye 1. | ||
Samples of dye 1 in the form of thin polycrystalline film on a glass support and single crystals were investigated by the 1H NMR and X-ray diffraction methods before and after their irradiation with visible light. The irradiation of the film for 40 hours did not lead to the formation of new compounds. Different behavior was demonstrated by single crystals of this compound.
In the same crystalline sample two types of single crystals were observed – thin shapeless plates (2) and well-shaped prisms (3a and 3b). The vast majority of the crystals were of the type 2. The volume of the crystal unit cell of 3a is twice as large as that for 3b. We have established that unit cell of 3b may be a sub-cell of that of 3a, with the transformation matrix (−1 0 0, 0.5 0.5 0.5, 0.5 0.5 −0.5). A result of structure solution for transformed, reduced unit cell 3a′, resulted in the same structure as that of 3b. However, this transformation is accompanied by omitting some (about 80) strong reflection and significantly more, about 200 of medium intensity reflections from the experimental data. Moreover, it was found that final refinement of 3b resulted in the site occupation factor (s.o.f.) of solvate water molecule O1W equal to 0.4, whereas, this is equal to 0.5 in 3a′.
Structure 3a contains one water molecule per two formula units of the dye. It should be noted that in all the structures studied, water molecules occupy cavities in the crystal packing and they are weakly bound to I− anions. Therefore, we cannot exclude that the crystal partly looses water during storage. Interestingly, we have managed to find only two crystals of type 3b among about ten prismatic crystals and there were exclusively in the sample that had been stored for more than two months. In the freshly prepared samples, there appears to be no crystals with the unit cell dimensions of 3b. It should be also noted that in the experimental data of 3b there are no reflections that would allow us to double the unit cell. We give data for both forms, 3a and 3b in this article.
Single crystals of 2, 3a, and 3b were subjected to irradiation with visible light from an incandescent lamp (60 W). Crystals 2 and also a polycrystalline film of dye 1 turned out to be stable to irradiation for a very long time (about a year for 2) and these did not show any sign of decomposition. On the contrary, prismatic crystals 3a and 3b lost their colour under irradiation, from bright-yellow to near colourless and split into fragments of thin needle-like crystals as it shown schematically in Fig. 2.
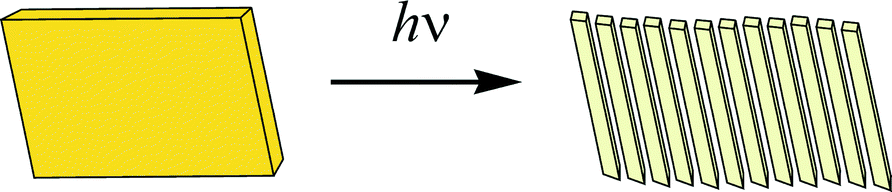 | ||
| Fig. 2 Splitting of a single crystal 3a (3b) under expose to visible light. | ||
These small needle-like crystals conserved their shape under the further irradiation with visible light. For two of these needle-like crystals (4a and 4b) we performed single crystal X-ray structure determinations. These showed that the unit cell volumes are similar to that of crystal 3b. A single crystal of 4a was investigated using a Bruker-D8 Venture diffractometer using both Mo and Cu X-radiation since the crystal gave very weak reflections. Table 1 shows only the results from the data collection using Cu X-radiation although the results of structure solution and refinement from both experiments were similar. Crystal of 4b, which gave very weak reflections, was investigated using synchrotron X-irradiation with λ = 0.68890 Å.
| Compound | 2 | 3a | 3b | 4a | 4b |
|---|---|---|---|---|---|
| Empirical formula | C20H24INO4S | C20H24INO3.5S | C20H22.8INO3.4S | C40H46I2N2O7S2 | C20H23INO3S |
| M | 499.37 | 491.37 | 489.77 | 982.74 | 484.35 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
| a/Å | 7.7237(5) | 7.4692(4) | 7.484(5) | 7.808(4) | 7.564(4) |
| b/Å | 10.2576(6) | 15.9898(9) | 10.197(7) | 10.354(6) | 10.338(5) |
| c/Å | 13.6801(8) | 18.4114(11) | 14.512(10) | 13.780(7) | 14.496(7) |
| α/° | 78.320(2) | 110.533(2) | 94.449(10) | 93.69(2) | 93.619(5) |
| β/° | 84.144(2) | 91.966(2) | 103.752(11) | 103.61(2) | 104.077(5) |
| γ/° | 76.520(2) | 94.712(2) | 105.623(10) | 105.59(2) | 105.686(5) |
| V/Å3 | 1030.43(11) | 2047.5(2) | 1024.1(12) | 1033.3(10) | 1048.4(9) |
| Z | 2 | 4 | 2 | 1 | 2 |
| μ/mm−1 | 1.680 | 1.688 | 1.687 | 13.304 | 1.524 |
| Crystal size/mm3 | 0.21 × 0.14 × 0.005 | 0.26 × 0.12 × 0.04 | 0.42 × 0.34 × 0.02 | 0.11 × 0.01 × 0.005 | 0.08 × 0.01 × 0.005 |
| Radiation | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) | MoKα (λ = 0.71073 Å) | CuKα (λ = 1.54178 Å) | Synchrotron (λ = 0.68890 Å) |
| T/K | 123 | 123 | 173 | 103 | 100 |
| Data collected | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 967 967 |
31145 |
9357 | 2646 | 8031 |
| Unique data (Rint) | 6250 (0.0661) | 10716 (0.0554) |
4331 (0.0224) | 2045 (0.0846) | 4543 (0.0558) |
| 2θ range/° | 4.68–61.20 | 4.56–58.00 | 4.74–54.00 | 19.98–119.68 | 4.00–55.08 |
| No. of variables | 244 | 485 | 241 | 238 | 242 |
| R 1 [I > 2σ(I)] | 0.0637 | 0.0582 | 0.0319 | 0.0871 | 0.0679 |
| wR2 (all data) | 0.1615 | 0.1366 | 0.0783 | 0.2280 | 0.1643 |
| GOF | 0.989 | 1.010 | 1.031 | 1.116 | 1.065 |
| Δρmax,min/ē Å−3 | 2.88/−1.01 | 4.15/−0.83 | 0.81/−0.96 | 1.93/−1.76 | 2.03/−2.04 |
Structures of the formula units in crystals 2, 3a, and 3b are shown in Fig. 3.
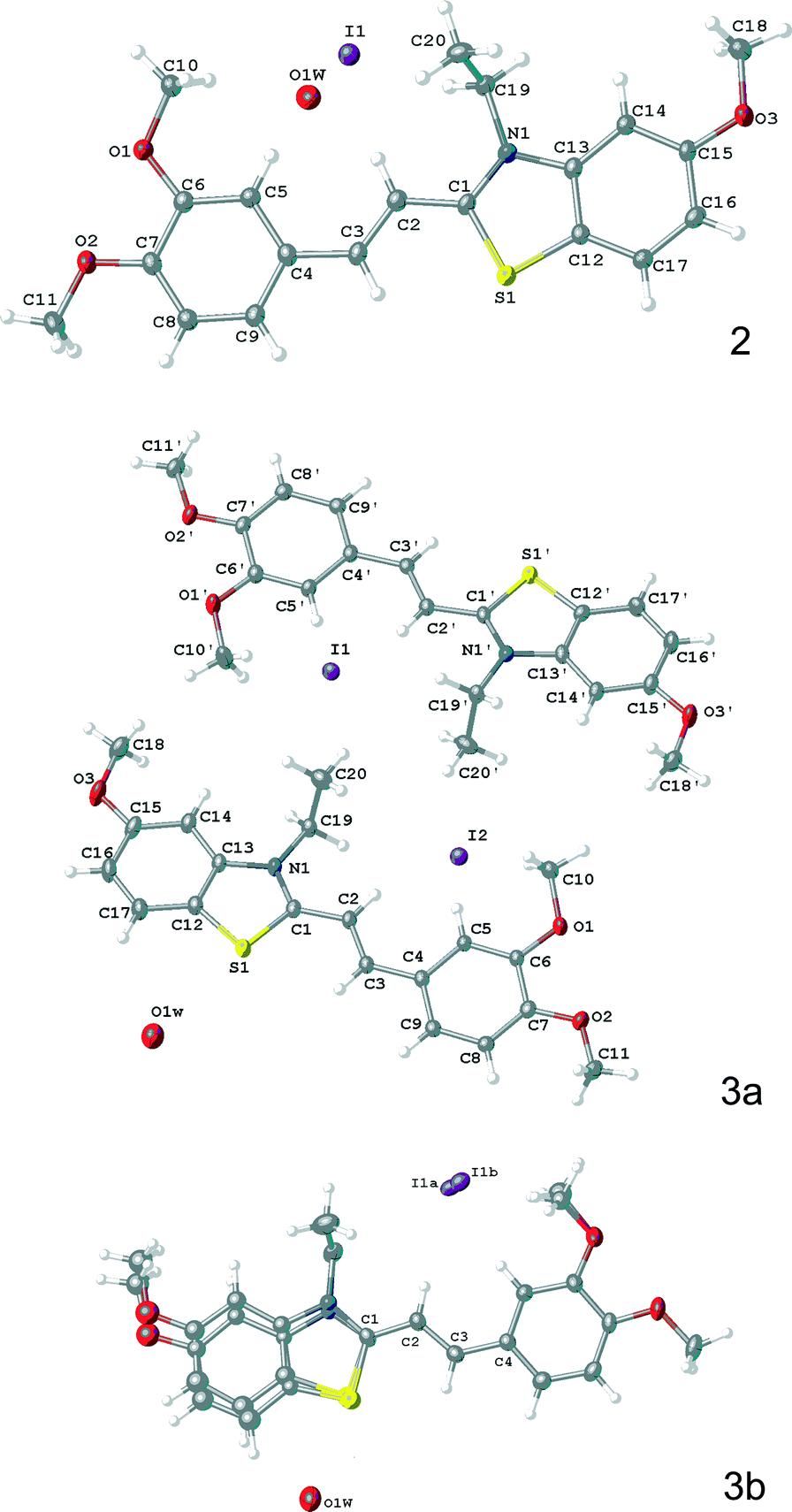 | ||
| Fig. 3 The structures of formula units determined for crystals of 2, 3a, and 3b. | ||
In structure 3b, the cation exhibits a disorder in benzothiazole fragment and one of methyl group of dimethoxybenzene residue. The I− anion is also disordered over two positions with ratio 50:50.
In all of three structures the conjugated fragment of the cation is almost planar. The bond length distribution in the ethylene fragments C(Het)–CC–C(Ar) was shown to be 1.436(5), 1.349(5), 1.449(5) Å in 2, 1.434(3), 1.358(3), 1.449(3) and 1.439(3), 1.347(3), 1.456(3) Å in 3a, 1.416(4), 1.345(4), 1.445(4) Å in 3b, which suggests a localization of the double bond. The C–C distribution in these fragments are remarkably similar, within error, however, the photochemical reaction proceeds only in crystals 3a and 3b to result in the same rctt isomer of cyclobutane 5 (see Scheme 3), whereas 2 is stable to light irradiation. The most likely reason for this difference in reaction to light results from subtle, but important variation in the crystal packing.
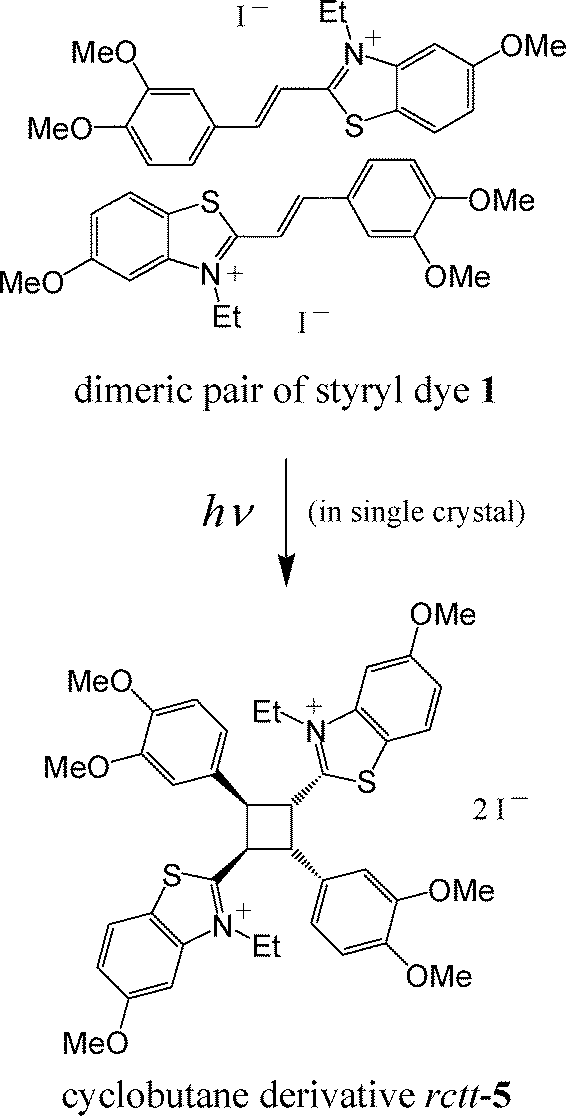 | ||
| Scheme 3 PCA of styryl dye 1 that takes place in the single crystals 3a and 3b. The rctt isomer of cyclobutane means that the initial dimeric pair is centrosymmetric. | ||
Fragment of crystal packing 2 is shown in Fig. 4.
 | ||
| Fig. 4 Fragment of the crystal packing 2 showing the stacks of cations; in a stack each molecule is related with the adjacent one through symmetry centre. | ||
In 2, cations form centrosymmetrically related stacks (see type 2 in Fig. 1), with the syn-“head-to-tail” mutual arrangement of the structural units. As is common for a stack with such a geometry,6,45 the d1 (within dimeric pair) and d2 (between adjacent dimeric pairs) distances are different. First of these distances (d1 = 3.75 Å), satisfies Schmidt's criterion on the PCA reaction feasibility, and the second one is much longer (d2 = 4.68 Å). These parameters should indicate that the PCA reaction is feasible in these crystals, however, the PCA reaction in crystals 2 does not take place.
Similar results have been obtained by us for crystals of one of several solvate forms of styryl dye 6 with TsO− counterion28 (Fig. 5), where a parallel-dimeric cation packing motif was observed (see type 4 in Fig. 1) and Schmidt's criterion for distance d is satisfied. For these two cases one common peculiarity is found, namely – the ethyl substituents at the nitrogen atoms (N1, Fig. 4) are oriented towards the centre of dimeric pair. In the other crystal forms of dye 6, the ethyl substituents are oriented outward from the centre of the dimeric pair and the PCA reaction proceeds by a SCSC process.
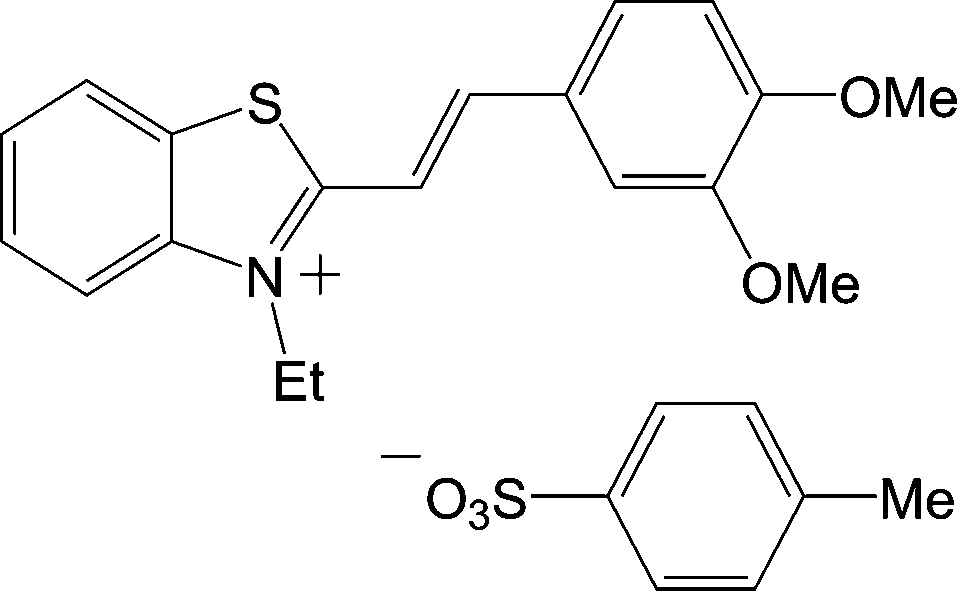 | ||
| Fig. 5 Molecular structure of cation and anion in dye 6. | ||
It should be noted that for crystals of styryl dyes of the 4-pyridine and 4-quinoline series that were investigated earlier and where Schmidt's criterion is satisfied, the orientation of N-substituent does not influence the feasibility of the PCA reaction. We may suppose that in the case of the 2-benzothiazole series styryl dyes, a steric interaction of “inside” oriented ethyl groups prevents the ethylene fragments from close approach, which is essential for the feasibility of the PCA reaction. Apparently, this is the reason for stability of crystal 2 towards light irradiation. Since a polycrystalline film of compound 1 is also stable to light, it is logically to suggest that only crystal modification 2 forms during a fast crystallization during film formation. This is also in agreement with the fact that in crystal sample crystal form 2 predominates.
Crystal forms 3a and 3b are characterized in the near the identical centrosymmetrically related stacking packing motif (Fig. 6) analogous to that observed in form 2. In crystals 3a, two types of stacks occur, formed of crystallographically independent cations.
 | ||
| Fig. 6 Fragment of one of two crystallographically independent stacks of cations in crystal 3a; the same fragment of the second independent stack has an analogous structure; in a stack each molecule is related with the adjacent one through symmetry centre. | ||
In the stacks of 3a distances d1 and d2 comprise 3.57 and 4.64 Å in one independent stack and 3.54 and 4.61 Å, in another one. In 3b these distances are equal to 3.55 and 4.61 Å. In pairs with shorter distances (d1), the ethyl substituents at nitrogen atoms are oriented outward from the centre of these pairs. This orientation of the ethyl substituents differs from that observed in 2 and this cannot prevent the ethylene fragments from approaching one another in the course of PCA.
Since distances d2 are longer than upper limit for the PCA feasibility, in a cation stack with letter symbols …–A–B–C–D–… (see Fig. 6), only in crystallographically equivalent pairs A–B, C–D, … is PCA feasible. In non-equivalent to them pairs B–C, D–E, … (E is the next molecule in a stack) the PCA reaction is not possible. If both distances were shorter than 4.2 Å, the PCA reaction would be accompanied by crystal degradation because in the course of PCA general symmetry of crystal would be violated. This situation is often observed in crystals of styryl dyes, where simultaneous accomplishing PCA in crystallographically non-equivalent cation pairs results in general crystal symmetry violation, that is, crystal degradation.5–7,45
Fig. 7 shows structures of structural units in crystals 4a and 4b, obtained on light irradiation of crystal forms 3a and/or 3b.
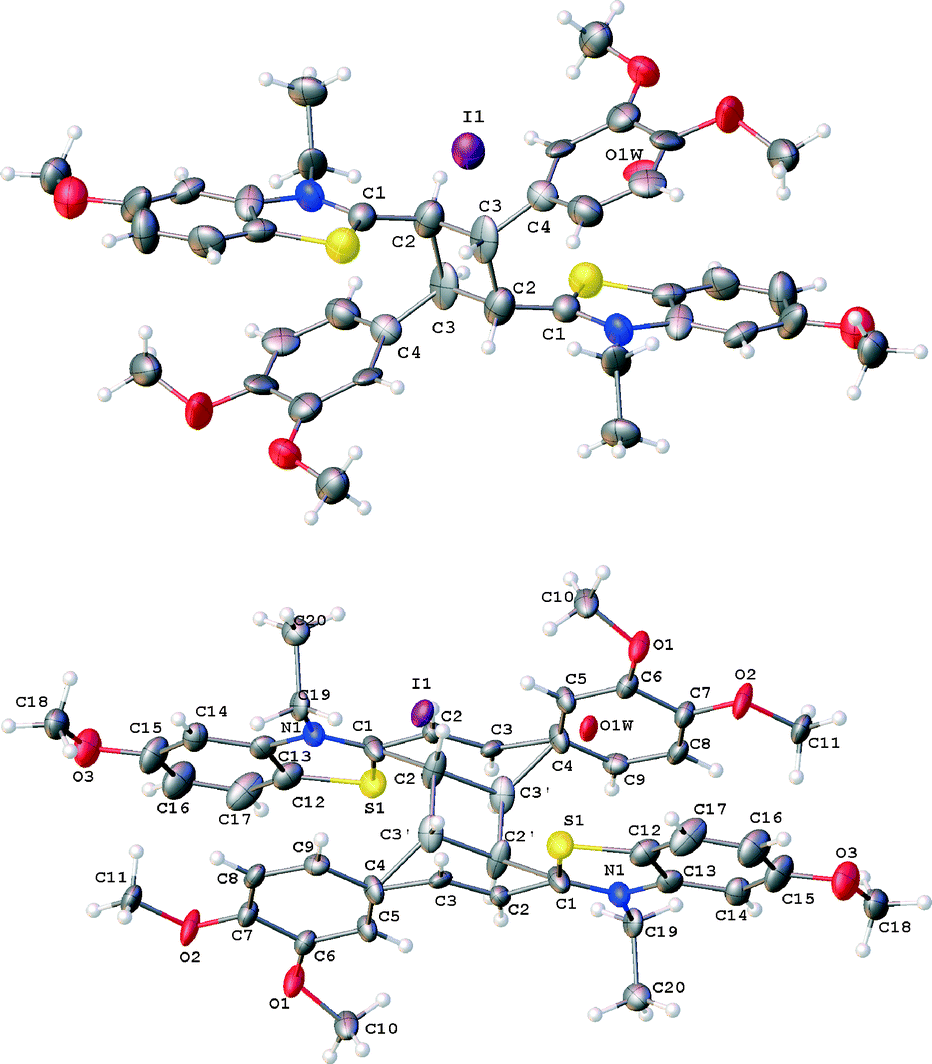 | ||
| Fig. 7 Structure of crystals 4a (top) and 4b (bottom). | ||
Crystals 4a represent a product of PCA if the reaction full accomplished (cyclobutane derivative rctt-5), whereas, in crystal 4b the PCA was only partly achieved (30%). In these crystals the site occupation of the solvate water molecules are close to 0.4–0.5. We cannot determine the exact values of these parameters because of rather low accuracy of the experiments where data were measured from very small weakly diffracting crystals.
Earlier, for styryl dyes and protonated styrylpyridines, significant role of a “soft” environment of the “pre-organized” pairs in crystals for accomplishing the PCA reaction with crystal retention was established. In these compounds this role was performed by flexible crown-ether fragments or translationally and rotationally mobile anions and solvate molecules that are not prone to form secondary interactions, such as hydrogen bonds and interactions of the S⋯X (X = I, O, S) type.3–7,17,18,28,45,46 An occurrence of such “soft” shells provides mutual tuning of the structural units changing their shape in the course of PCA.
In the systems investigated here, almost all changes in shape take place inside the region of the crystal that is determined by the “pre-organized” pair. A superposition of “pre-organized” pair in 3a and cyclobutane 4a in two projections is shown in Fig. 8.
 | ||
| Fig. 8 Superposition of “pre-organized” pair in 3a and cyclobutane product in 4a in two projections. | ||
The benzene ring and its MeO groups only are the most significantly displaced from the initial arrangement of the “pre-organized” dimeric pair.
An analysis of crystal packings of 3a,b shows that short contacts between these groups and adjacent cations are lacking. All intermolecular contacts between the adjacent stacks occur through the I− anions. Fig. 9 illustrates short contacts of I1 and I2 anions with the adjacent dimeric pairs in structure 3a.
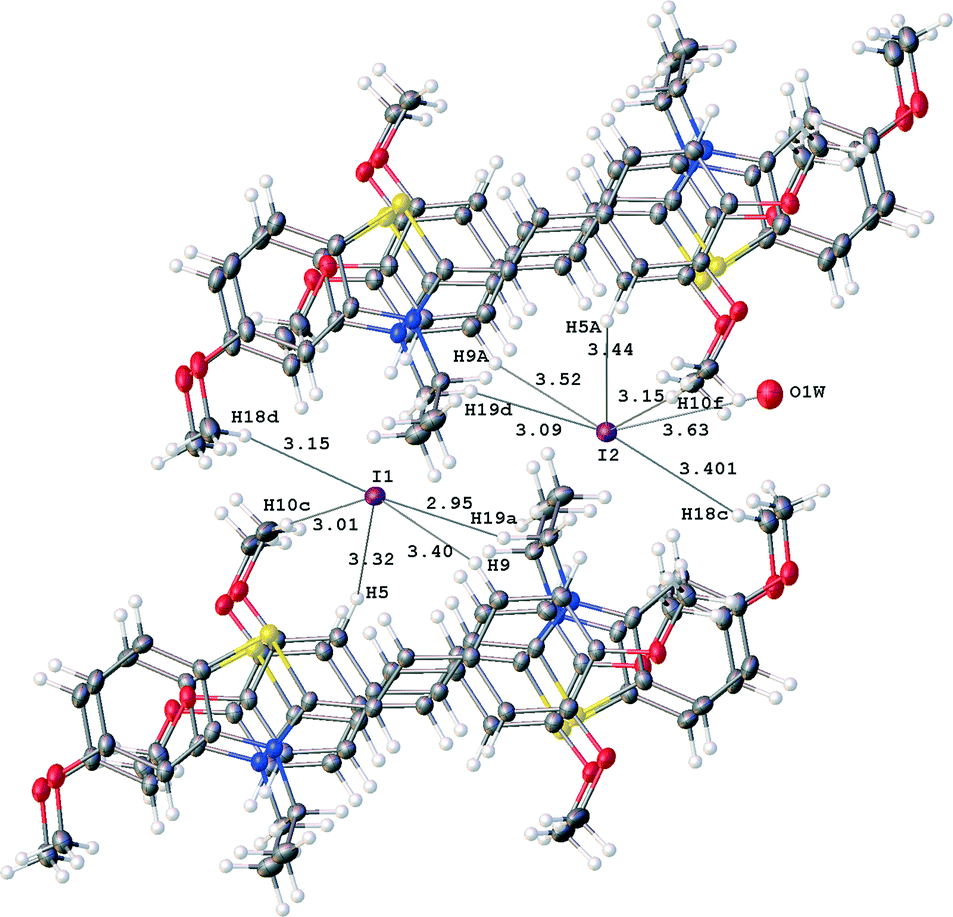 | ||
| Fig. 9 Short contacts of I1 and I2 anions in structure 3a; distances I⋯H and I2⋯O1W are given in Å. | ||
Both anions are surrounded by C–H fragments of the adjacent cations. The environments of I1 and I2 are similar, with the exception of one additional contact of I2 anion with the solvate water molecule. Distances from the iodide anions to other C–H fragments are longer than 3.9 Å. The short contacts form non-spherical environment of the anions. This means that the anions are situated in large cavities, in which they may be distributed loosely within the vicinity of the final coordinates of the iodide anions. This explains the appearance of rather high residual electron density peaks near the iodide anions at a distance from them equal to ~0.14 Å (see Table 1).
Interestingly, only one of anions, I2, retains the water molecule H2O1W in crystal. The O1W⋯I2 distance is equal to 3.63 Å and this may correspond to a weak hydrogen bond if un-located hydrogen atom of the water molecule is oriented towards the anion. If this hydrogen is oriented differently, this contact may correspond to another type of weak interaction.
Conclusions
Conducted investigation made it possible to explain why styryl dye of the 2-benzothiazole series is subjected to PCA transformation under irradiation with visible light in one crystal modification and is stable under the same conditions in another crystal modification, in spite of the fact that their crystal packings relate to the same type, namely – centrosymmetrically related stacks, in which Schmidt's criterion is performed.Experimental section
General
The melting point (uncorrected) was measured in a capillary on a Mel-Temp II apparatus. 1H and 13C NMR spectra were recorded on a Bruker DRX500 instrument (500.13 and 125.76 MHz, respectively) in DMSO-d6 at 25 °C using the solvent as an internal standard (δH 2.50 ppm and δC 39.43 ppm). 2D NOESY and heteronuclear correlation (HSQC and HMBC) spectra were used to assign the proton and carbon signals (atom numbering is shown in Scheme 2). The absorption spectrum of a solution of dye 1 (C = 2 × 10−5 M) in acetonitrile was measured on a Cary 4000 spectrophotometer (Agilent) in a range of 230–600 nm (at an interval of 1 nm) in a 1 cm quartz cell. The elemental analysis was performed in the Laboratory of Microanalysis of A. N. Nesmeyanov Institute of Elementoorganic Compounds of the Russian Academy of Sciences (Moscow, Russian Federation). The sample for elemental analysis was dried at 80 °C in vacuo.Preparations
3,4-Dimethoxybenzaldehyde was purchased from “Aldrich”. 3-Ethyl-5-methoxy-2-methyl-1,3-benzothiazol-3-ium iodide was prepared according to published procedure.53CHHet), 8.14 (d, J = 15.7 Hz, 1H, CHCHHet), 8.28 (d, J = 9.0 Hz, 1H, 7-H) ppm. 13C NMR: δ = 14.05 (MeCH2), 44.10 (CH2N), 55.77 (4′-MeO), 55.97 (3′-MeO), 56.42 (5-MeO), 99.69 (4-C), 110.61 (CHCHHet), 111.56 (2′-C), 111.64 (5′-C), 117.61 (6-C), 119.60 (7a-C), 124.92 (7-C), 125.55 (6′-C), 126.82 (1′-C), 142.29 (4a-C), 148.55 (CHCHHet), 149.03 (3′-C), 152.80 (4′-C), 160.72 (5-C), 171.75 (2-C) ppm. UV-vis (MeCN): λmax = 437 nm (ε = 33500 M−1 cm−1).
X-ray diffractometry
Crystals 2 and 3a, b were grown by slow saturation of acetonitrile solution of dye 1 with benzene vapors. Crystals 2 and 3a, b were mounted on a CCD area SMART-Apex-II diffractometer under a stream of cooled nitrogen where their crystallographic parameters and diffraction data were measured (graphite monochromator, λ(MoKα), ω-scan). Crystal 4a was investigated on a Bruker-D8 Venture diffractometer (graphite monochromator, λ(CuKα), ω-scan). The data for crystal 4b were collected on a Rigaku Saturn 724+ diffractometer at Station 119 of the Diamond Light Source synchrotron (undulator, λ = 0.68890 Å, ω-scan, 1.0° per frame). All experiments were processed using Bruker Apex-II software. Absorption correction was applied using SADABS program.54Structures 2–4 were solved by direct methods and refined by least squares in an anisotropic approximation for all non-hydrogen atoms in structures 2 and 3a. In structures 3b and 4b, only atoms of the ordered fragments were refined in an anisotropic approximation.
The atomic positions of hydrogen atoms were calculated geometrically. Their refinement was performed using a “riding” model. Solvate water molecule H atoms were not located. The refinement of water molecule oxygen atom in structure 3b gave better result with site occupation factor equal to 0.4. The refinement of structure 3b with the s.o.f. of O1W equal to 0.5 resulted in somewhat higher values of R and U(eq) of the oxygen atom, R = 3.42, U(eq) = 0.07 instead of 3.41 and 0.05 in the case when s.o.f is equal to 0.4, respectively.
In experiment for 3a rather high values for residuals are obtained, which may be associated with somewhat statistical distribution of the anions over several close positions. Our attempts to include these peaks as iodide anions in least squares refinement with a low occupancies failed since the refinement became unstable.
For 4a and 4b X-ray experiments were collected from very small crystals. Such single crystals gave weak X-ray reflections at Cu and sinchrothrone radiation, respectively. Nevertheless, we have managed to collect a rather big number of weak experimental reflections in the near area on θ. The structures have been successfully solved and refined in the anisotropic or anisotropic-isotropic approximations, but with a rather high standard deviations in geometric parameters. However, taking into account that only general view of the structure, that is, the connectivity schemes were required for our discussion and also special importance of this data we included them in the article.
All the calculations were performed using SHELXTL-Plus55 and Olex-2 (ref. 56) software.
A summary of the crystallographic data and structure determination parameters is provided in Table 1.
The experimental data for all structures are deposited in the Cambridge Crystallographic Data Centre (CCDC registration numbers are 1056057 (2), 1056058 (3a), 1056059 (3b), 1056060 (4a), 1056061 (4b)).
Acknowledgements
The Diamond Light Source is thanked for the award of instrument time on Station 119 (MT 8682) and the instrument scientists for their kind support. This work was also supported by the Russian Foundation for Basic Research (project no. 14-03-00012) and Photochemistry Center of the Russian Academy of Sciences. L.G.K. thanks the Royal Society of Chemistry for International Author Grant.Notes and references
- G. J. M. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS.
- Photochemistry in Organized and Constrained Media, ed. V. Ramamurthy, VCH, New York, 1991 Search PubMed.
- A. I. Vedernikov, S. P. Gromov, N. A. Lobova, L. G. Kuz'mina, Yu. A. Strelenko, J. A. K. Howard and M. V. Alfimov, Russ. Chem. Bull., Int. Ed., 2005, 54, 1954 CrossRef CAS.
- L. G. Kuz'mina, A. I. Vedernikov, N. A. Lobova, A. V. Churakov, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, New J. Chem., 2007, 31, 980 RSC.
- A. I. Vedernikov, L. G. Kuz'mina, S. K. Sazonov, N. A. Lobova, P. S. Loginov, A. V. Churakov, Yu. A. Strelenko, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Russ. Chem. Bull., Int. Ed., 2007, 56, 1860 CrossRef CAS PubMed.
- L. G. Kuz'mina, A. I. Vedernikov, S. K. Sazonov, N. A. Lobova, P. S. Loginov, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Crystallogr. Rep., 2008, 53, 428 CrossRef.
- L. G. Kuz'mina, A. I. Vedernikov, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Nanotechnol. Russ., 2008, 3, 408 Search PubMed.
- F. Li, J. Zhuang, G. Jiang, H. Tang, A. Xia, L. Jiang, Y. Song, Y. Li and D. Zhu, Chem. Mater., 2008, 20, 1194 CrossRef CAS.
- L. R. MacGillivray, J. Org. Chem., 2008, 73, 3311 CrossRef CAS PubMed.
- G. K. Kole, G. K. Tan and J. J. Vittal, Org. Lett., 2010, 12, 128 CrossRef CAS PubMed.
- X.-H. Miao and L.-G. Zhu, Dalton Trans., 2010, 39, 1457 RSC.
- B. R. Bhogala, B. Captain, A. Parthasarathy and V. Ramamurthy, J. Am. Chem. Soc., 2010, 132, 13434 CrossRef CAS PubMed.
- S. Clément, F. Meyer, J. De Winter, O. Coulembier, C. M. L. Vande Velde, M. Zeller, P. Gerbaux, J.-Y. Balandier, S. Sergeyev, R. Lazzaroni, Y. Geerts and P. Dubois, J. Org. Chem., 2010, 75, 1561 CrossRef PubMed.
- M. Linares and A. Briceño, New J. Chem., 2010, 34, 587 RSC.
- T. D. Hamilton, D.-K. Bučar and L. R. MacGillivray, New J. Chem., 2010, 34, 2400 RSC.
- A. Papagni, P. Del Buttero, C. Bertarelli, L. Miozzo, M. Moret, M. T. Pryce and S. Rizzato, New J. Chem., 2010, 34, 2612 RSC.
- S. P. Gromov, A. I. Vedernikov, L. G. Kuz'mina, D. V. Kondratuk, S. K. Sazonov, Yu. A. Strelenko, M. V. Alfimov and J. A. K. Howard, Eur. J. Org. Chem., 2010, 2587 CrossRef CAS PubMed.
- L. G. Kuz'mina, A. I. Vedernikov, S. K. Sazonov, N. A. Lobova, A. V. Churakov, E. Kh. Lermontova, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Russ. Chem. Bull., Int. Ed., 2011, 60, 1734 CrossRef.
- Y. Sonoda, Molecules, 2011, 16, 119 CrossRef CAS PubMed.
- C. Karunatilaka, D.-K. Bučar, L. R. Ditzler, T. Friščić, D. C. Swenson, L. R. MacGillivray and A. V. Tivanski, Angew. Chem., Int. Ed., 2011, 50, 8642 CrossRef CAS PubMed.
- G. K. Kole, G. K. Tan and J. J. Vittal, J. Org. Chem., 2011, 76, 7860 CrossRef CAS PubMed.
- I. G. Ovchinnikova, D. K. Nikulov, E. V. Bartashevich, E. G. Matochkina, M. I. Kodess, P. A. Slepukhin, A. V. Druzhinin, O. V. Fedorova, G. L. Rusinov and V. N. Charushin, Russ. Chem. Bull., Int. Ed., 2011, 60, 824 CrossRef CAS.
- D.-K. Bučar, A. Sen, S. V. S. Mariappan and L. R. MacGillivray, Chem. Commun., 2012, 48, 1790 RSC.
- G. K. Kole, G. K. Tan and J. J. Vittal, CrystEngComm, 2012, 14, 7438 RSC.
- E. Elacqua, P. Kaushik, R. H. Groeneman, J. C. Sumrak, D.-K. Bučar and L. R. MacGillivray, Angew. Chem., Int. Ed., 2012, 51, 1037 CrossRef CAS PubMed.
- P. Zhang, Y. Wang, R. Bao, T. Luo, Z. Yang and Y. Tang, Org. Lett., 2012, 14, 162 CrossRef CAS PubMed.
- X.-M. Chen, M. Chen, Z.-T. Huang and Q.-Y. Zheng, Synthesis, 2012, 44, 3693 CrossRef PubMed.
- L. G. Kuz'mina, A. I. Vedernikov, E. Kh. Lermontova, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, Russ. Chem. Bull., Int. Ed., 2013, 62, 1726 CrossRef PubMed.
- S. Bhattacharya, J. Stojaković, B. K. Saha and L. R. MacGillivray, Org. Lett., 2013, 15, 744 CrossRef CAS PubMed.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950 RSC.
- J.-K. Sun, W. Li, C. Chen, C.-X. Ren, D.-M. Pan and J. Zhang, Angew. Chem., Int. Ed., 2013, 52, 6653 CrossRef CAS PubMed.
- R. Santra, M. Garai, D. Mondal and K. Biradha, Chem. – Eur. J., 2013, 19, 489 CrossRef CAS PubMed.
- H. Kashida, T. Doi, T. Sakakibara, T. Hayashi and H. Asanuma, J. Am. Chem. Soc., 2013, 135, 7960 CrossRef CAS PubMed.
- Y.-F. Han, G.-X. Jin and F. E. Hahn, J. Am. Chem. Soc., 2013, 135, 9263 CrossRef CAS PubMed.
- B. C. Pemberton, A. Ugrinov and J. Sivaguru, J. Photochem. Photobiol., A, 2013, 255, 10 CrossRef CAS PubMed.
- H. Asahara, T. Iwamoto, T. Kida and M. Akashi, Tetrahedron Lett., 2013, 54, 688 CrossRef CAS PubMed.
- S. Yamada, M. Kusafuka and M. Sugawara, Tetrahedron Lett., 2013, 54, 3997 CrossRef CAS PubMed.
- R. Medishetty, T. T. S. Yap, L. L. Koh and J. J. Vittal, Chem. Commun., 2013, 49, 9567 RSC.
- N. Vallavoju and J. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084 RSC.
- B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179 RSC.
- K. M. Hutchins, J. C. Sumrak and L. R. MacGillivray, Org. Lett., 2014, 16, 1052 CrossRef CAS PubMed.
- A. A. Parent, D. H. Ess and J. A. Katzenellenbogen, J. Org. Chem., 2014, 79, 5448 CrossRef CAS PubMed.
- K. M. Hutchins, T. P. Rupasinghe, L. R. Ditzler, D. C. Swenson, J. R. G. Sander, J. Baltrusaitis, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2014, 136, 6778 CrossRef CAS PubMed.
- T. Runčevski, M. Blanco-Lomas, M. Marazzi, M. Cejuela, D. Sampedro and R. E. Dinnebier, Angew. Chem., Int. Ed., 2014, 53, 6738 CrossRef PubMed.
- L. G. Kuz'mina, A. I. Vedernikov, A. V. Churakov, E. Kh. Lermontova, J. A. K. Howard, M. V. Alfimov and S. P. Gromov, CrystEngComm, 2014, 16, 5364 RSC.
- S. P. Gromov, A. I. Vedernikov, N. A. Lobova, L. G. Kuz'mina, S. N. Dmitrieva, Yu. A. Strelenko and J. A. K. Howard, J. Org. Chem., 2014, 79, 11416 CrossRef CAS PubMed.
- R. Jasiński, Monatsh. Chem., 2015, 146, 591 CrossRef PubMed.
- R. Jasiński, Tetrahedron, 2013, 69, 927 CrossRef PubMed.
- R. Jasiński, M. Kubik, A. Łapczuk-Krygier, A. Kącka, E. Dresler and A. Boguszewska-Czubara, React. Kinet., Mech. Catal., 2014, 113, 333 Search PubMed.
- S. P. Gromov, Rev. J. Chem., 2011, 1, 1 CrossRef.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed.
- S. P. Gromov, O. A. Fedorova, M. V. Alfimov, S. I. Druzhinin, M. V. Rusalov and B. M. Uzhinov, Russ. Chem. Bull., 1995, 44, 1922 CrossRef.
- SMART and SADABS Software. Reference Manuals, Bruker AXS Inc., Madison, Wisconsin (USA), 1998 Search PubMed.
- SHELXTL-Plus, Version 5.10, Bruker AXS Inc., Madison, Wisconsin (USA), 1997 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Pushman, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
Footnote |
| † CCDC 1056057–1056061. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00653h |
| This journal is © The Royal Society of Chemistry 2015 |