
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom monomers to polymers: steric and supramolecular effects on dimensionality of coordination architectures of heteroleptic mercury(II) halogenide–tetradentate Schiff base complexes†
Ghodrat
Mahmoudi
*a,
Vladimir
Stilinović
*b,
Masoumeh Servati
Gargari
a,
Antonio
Bauzá
c,
Guillermo
Zaragoza
d,
Werner
Kaminsky
e,
Vincent
Lynch
f,
Duane
Choquesillo-Lazarte
g,
K.
Sivakumar
h,
Ali Akbar
Khandar
i and
Antonio
Frontera
 *c
*c
aYoung Researchers and Elite Club, Tabriz Branch, Islamic Azad University, Tabriz, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk
bDepartment of Chemistry, Faculty of Science, University of Zagreb, Horvatovac 102a, HR-10000 Zagreb, Croatia
cDepartamento de Química, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, Palma de Mallorca (Baleares) 07122, SPAIN. E-mail: toni.frontera@uib.es
dUnidad de RX, Edificio CACTUS, Campus Vida, Santiago Compostela, 15782, Spain
eX-ray Crystallography Laboratory, University of Washington, USA
fDepartment of Chemistry and Biochemistry University of Texas at Austin, Austin, TX 78712, USA
gLaboratorio de Estudios Cristalográficos, IACT, CSIC-Universidad de Granada, Avda. de las Palmeras 4, Armilla, Granada, 18100, Spain
hDepartment of Physics, Anna University, Chennai 600 025, India
iDepartment of Inorganic Chemistry, Faculty of Chemistry, University of Tabriz, P.O. Box 5166616471, Tabriz, Iran
First published on 25th March 2015
Abstract
In this study, neutral mercury(II) complexes of the composition [Hg(L1)(μ-Cl)2Hg3Cl6]n (1), [Hg(L1)(μ-Br)2HgBr2] (2), [Hg(L3)Br2] (2a), [Hg(L1)I2] (3), [Hg(L2)Cl2]·CH3OH (4) and [Hg(L2)(μ-Br)HgBr3]2 (5) (L1 = benzilbis((pyridin-2-yl)methylidenehydrazone); L2 = benzilbis((acetylpyridin-2-yl)methylidenehydrazone)) are described. Single-crystal X-ray crystallography showed that the molecular complexes can aggregate into larger entities depending upon the anion coordinated to the metal centre. Iodide gives discrete monomeric complexes, bromide generates a 1D coordination polymer formed through Hg–Br–Hg bridges and chloride gives rise to an inorganic–organic hybrid material. The significant differences in the reaction conditions indicate that the anions exert a substantial influence on the formation of the compounds – smaller anions show a larger potential for bridging metal ions and forming coordination polymers. A minute increase in the bulkiness of the ligand (two extra methyl substituents in L2) dramatically changes the coordination architectures, and leads to the formation of monomeric (chloride and iodide) and oligomeric (bromide) structures, rather than polymeric structures. The noncovalent C–H/π and π-hole interactions observed in the solid state architecture of some complexes have been rationalized by means of theoretical DFT calculations.
1. Introduction
Construction of coordination networks by self-assembly has attracted considerable attention in crystal engineering and supramolecular chemistry, due to their versatile structural diversity and potential applications in different areas from catalysis to nonlinear optics.1–9 Factors that play important roles in controlling the architecture of self-assembled species include: the structure of ligands, coordination geometry of metal ions, counter-anions, and supramolecular interactions of the coordination compound with its surroundings.10–16 Among these, anions play a very important role in the self-assembled construction.17–24Due to their applications in the paper industry and as preservatives, fluorescent lamps, sensors and batteries,24–29 mercury and its compounds are of immense importance in chemistry and related disciplines. The spherical d10 configuration of Hg(II) is associated with a flexible coordination environment, thus the geometries of these complexes can vary from linear to octahedral or even distorted hexagonal bipyramidal, and severe distortions from ideal coordination polyhedra occur easily. Furthermore, due to the lability of d10 metal complexes, the formation of coordination bonds is reversible, which enables metal ions and ligands to rearrange during the supramolecular assembly to allow the formation of the thermodynamically most stable structure, by varying the coordination polyhedron and coordination number of the mercury atom. Consequently, mercury(II) can readily accommodate different kinds of coordination frameworks, using a variety of organic ligands along with different inorganic/organic bridging units.30
In line with the above discussion, we recently reported on the syntheses and self-assembly of some Hg(II) compounds of the composition [Hg2(μ-L)(SCN)4]n, [Hg2(μ-L)(μ-Cl)2Cl2]n, [Hg2(μ-L)Br4]·[Hg2(μ-L)(μ-Br)2Br2]n and [Hg2(μ-L)I4] {L = N,N′(bis-(pyridin-2-yl)benzylidene)-1,2-ethanediamine}.31 Schiff base ligands are frequently used in coordination chemistry due to their significant ability to form stable complexes with metal ions.32 The Schiff base ligand (L) previously used by us31a played an important role in the formation of coordination polymers with fascinating structures. In this manuscript we investigated the effect of related azine-based Schiff base ligands (Scheme 1) on the solid state architecture of a series of Hg(II)-containing coordination compounds, namely [Hg(L1)(μ-Cl)2Hg3Cl6]n (1), [Hg(L1)(μ-Br)2HgBr2] (2), [Hg(L3)Br2] (2a), [Hg(L1)I2] (3), [Hg(L2)Cl2]·CH3OH (4) and [Hg(L2)(μ-Br)HgBr3]2 (5), prepared by the reaction of a ligand (L1 or L2) and HgX2 (X = Cl, Br, I) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio. Herein, we report the structures and topological analyses of these compounds and discuss the influence of the anions on the structures of the coordination species. Moreover, the noncovalent C–H/π and π-hole interactions observed in the solid state architecture of some complexes have been rationalized by means of theoretical DFT calculations.
:10 molar ratio. Herein, we report the structures and topological analyses of these compounds and discuss the influence of the anions on the structures of the coordination species. Moreover, the noncovalent C–H/π and π-hole interactions observed in the solid state architecture of some complexes have been rationalized by means of theoretical DFT calculations.
 | ||
| Scheme 1 Molecular diagrams of L1 and L2. | ||
2. Experimental methods
2.1. Materials and measurements
The ligands L1 and L2 were prepared following a reported method described elsewhere31b and used without further purification. All other reagents and solvents used for the synthesis and analysis were commercially available and used as received. FT-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer. Microanalyses were performed using a Heraeus CHN–O-Rapid analyzer. Melting points were measured on an Electrothermal 9100 apparatus and were uncorrected.Caution ! Mercury and its compounds are toxic.33 Only a small amount of these materials should be prepared and handled with care.
2.2. Synthesis of L1 and L2
Benzil dihydrazone (5.61 g, 23.58 mmol) was dissolved in 100 ml of anhydrous methanol. To this colourless solution, 4.50 ml (47.16 mmol) of freshly distilled 2-pyridinecarboxaldehyde was added. The resulting yellowish mixture was refluxed for 18 h, while maintaining a dry atmosphere. Then it was slowly cooled to room temperature to yield a yellowish crystalline solid, which was filtered off and dried in air. L2 was prepared using the same procedure as that for L1 except that 2-pyridinecarboxaldehyde was replaced with 2-acetylpyridine.2.3. Synthesis of [Hg(L1)(μ-Cl)2Hg3Cl6]n (1), [Hg(L1)(μ-Br)2HgBr2] (2), [Hg(L1)I2] (3), [Hg(L2)Cl2]·CH3OH (4) and [Hg(L2)(μ-Br)HgBr3]2 (5)
Mercury(II) chloride and L1 (0.021 g, 0.05 mmol; 0.135 g, 0.5 mmol) were placed in the main arm of a branched tube. Methanol (10 ml) was carefully added to fill the arms. The tube was sealed and immersed in an oil bath at 60 °C while the branched arm was kept at ambient temperature. After 2 days, crystals of 1 that were isolated in the cooler arm were filtered off, washed with acetone and ether, and dried in air. Crystals of 2 and 3 were prepared by a similar synthetic procedure to that used for 1, except that HgCl2 was replaced with HgBr2 and HgI2, respectively. For 4 and 5 a similar synthetic procedure to that used for 1 and 2 was used except that L1 was replaced with L2. It is interesting to note that in all these cases an excess amount of HgX2 (X = Cl, Br and I) was used – all attempts to prepare crystalline complexes in the branched tube from equimolar mixtures were unsuccessful. For 1: (0.157 g, yield 84%), found: C, 20.68; H, 1.43; N, 5.67%; calcd. for C26H20Cl8Hg4N6: C, 20.79 H, 1.37; N, 5.59%. IR (cm−1) selected bands: 527(w), 687(vs), 776(vs), 973(m), 1155 (w), 1185(m), 1258(s), 1298(s), 1438(s), 1477(s), 1588(s), 1617(s), 3060(w). For 2: (0.277 g, yield 74%), found: C, 27.35; H, 1.83; N, 7.49%; calcd. for C26H20Br4Hg2N6: C, 27.46 H, 1.77; N, 7.39%. IR (cm−1) selected bands: 491(w), 625(m), 685(vs), 778 (m), 977(s), 1250(m), 1435(s), 1555(vs), 1613(m), 1651(m), 3060(w). For 3: (0.295 g, yield 68%), found: C, 35.75; H, 2.37; N, 9.76%; calcd. for C26H20I2HgN6: C, 35.86 H, 2.31; N, 9.65%. IR (cm−1) selected bands: 527(w), 690(s), 776(m), 1058 (w), 1156(m), 1256(w), 1439(m), 1592(m), 1623(s), 3060(w). For 4: (0.205 g, yield 55%), found: C, 46.44; H, 3.85; N, 11.35%; calcd. for C29H28Cl2HgN6O: C, 46.56 H, 3.77; N, 11.23%. IR (cm−1) selected bands: 574(w), 694(s), 780(s), 1003 (w), 1159(m), 1250(w), 1437(m), 1576(s), 1612(s), 3060(w). For 5: (0.118 g, yield 74%), found: C, 28.75; H, 2.15; N, 7.28%; calcd. for C28H24Br4Hg2N6: C, 28.86 H, 2.08; N, 7.21%. IR (cm−1) selected bands: 566(w), 692(s), 778(m), 1010 (w), 1157(m), 1250(w), 1435(m), 1592(m), 1620(s), 3059(w).2.4. Synthesis of [Hg(L3)Br2] (2a)
To a solution of benzilbis((pyridin-2-yl)methylidenehydrazone) (0.021 g, 0.05 mmol) in methanol, (50 mL) a solution of HgBr2 (0.18 g, 0.5 mmol) in methanol (40 mL) was added dropwise under stirring, which resulted in the immediate formation of a yellow precipitate. Stirring was continued for 5 h and then the mixture was filtered. The residue was washed with methanol (3 × 10 mL) and dried under vacuum. The dried solid was dissolved by boiling in acetonitrile (45 mL) and filtered while hot. The filtrate, upon cooling to room temperature, afforded a yellow crystalline material (0.193 g, yield 44%); found: C, 46.35; H, 2.93; N, 8.09%; calcd. for C34H25Br2HgN5O: C, 46.41 H, 2.86; N, 7.96%. IR (cm−1) selected bands: 688(m), 772(m), 1011(m), 1158 (m), 1385(s), 1437(m), 1565 (m), 1588(m), 1616(m), 1672(m), 3026(w).2.5. X-ray crystallography
The diffraction data were collected using a Nonius KappaCCD (1, 100 K, λ = 0.71073 Å), a Bruker APEX II (2 and 2a, 100 K, λ = 0.71073 Å), a Bruker X8 Proteum (3, 296 K, λ = 1.54178 Å) and an Oxford Diffraction Xcalibur Kappa CCD (4 and 5, 295 K, λ = 0.71073 Å) X-ray diffractometer. Data were processed with HKL Scalepack (1),34 Apex2 (2, 2a, 3)35 and CrysAlisPro (4 and 5)36 programs and corrected for absorption using SADABS.37 The structures were solved by direct methods,38 which revealed the position of all non-hydrogen atoms. These atoms were refined on F2 by a full-matrix least-squares procedure using anisotropic displacement parameters.38 All hydrogen atoms were located in difference Fourier maps and included as fixed contributions riding on attached atoms with isotropic thermal displacement parameters 1.2 times those of the respective atom. All calculations were performed and the drawings were prepared using the WINGX crystallographic suite of programs.39 The crystal data are listed in Table 1. Further details are available from the Cambridge Crystallographic Centre – CCDC 1046009–1046016 contain the supplementary crystallographic data for 1–5, 2a, L1 and L2.| 1 | 2 | 2a | 3 | 4 | 5 | L1 | L2 | |
|---|---|---|---|---|---|---|---|---|
| Chemical formula | C26H20N6Hg4Cl8 | C26H20N6Hg2Br4 | C34H25N5OHgBr2 | C26H20N6HgI2 | C29H28N6OHgCl2 | C56H48N12Hg4Br8 | C26H20N6 | C28H24N6 |
| M r | 1502.44 | 1137.3 | 879.98 | 870.87 | 748.06 | 2330.63 | 416.48 | 444.53 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/n |
P |
P |
P |
P21/c |
P |
| a/Å | 7.6970(3) | 8.7552(8) | 9.3733(7) | 9.7571(6) | 8.7640(4) | 9.5360(4) | 10.3079(4) | 9.6825(15) |
| b/Å | 15.9190(5) | 14.1329(14) | 35.998(2) | 11.5338(8) | 11.8787(4) | 10.9904(4) | 18.8288(7) | 11.376(2) |
| c/Å | 16.0270(7) | 23.646(2) | 9.8847(6) | 14.2949(12) | 15.0315(6) | 15.8293(6) | 11.5821(5) | 12.377(2) |
| α/° | 104.8910(10) | 90 | 90 | 68.980(4) | 73.615(3) | 86.580(4) | 90 | 74.062(3) |
| β/° | 101.344(2) | 96.847(5) | 104.978(7) | 83.774(4) | 88.714(5) | 73.062(3) | 105.811(2) | 76.504(4) |
| γ/° | 99.190(4) | 90 | 90 | 68.968(3) | 88.110(6) | 78.506(6) | 90 | 66.011(3) |
| V/Å3 | 1814.35(12) | 2905.0(5) | 3222.0(4) | 1401.23(18) | 1500.34(10) | 1555.19(10) | 2162.87(15) | 1186.1(3) |
| Z | 2 | 4 | 4 | 2 | 2 | 1 | 4 | 2 |
| Z′ | 1 | 1 | 1 | 1 | 1 | 1/2 | 1 | 1 |
| ρ calc/(g cm−3) | 2.75 | 2.6 | 1.814 | 2.064 | 1.656 | 2.489 | 1.279 | 1.245 |
| μ/mm−1 | 17.489 | 16.087 | 7.292 | 27.346 | 5.340 | 15.028 | 0.079 | 0.077 |
| F[000] | 1348 | 2072 | 1688 | 808 | 732 | 1068 | 872 | 468 |
| Crystal size/mm3 | 0.30 × 0.256 × 0.1 | 0.42 × 0.37 × 0.15 | 0.21 × 0.15 × 0.07 | 0.12 × 0.10 × 0.04 | 0.57 × 0.33 × 0.09 | 0.39 × 0.35 × 0.17 | 0.35 × 0.30 × 0.25 | 0.37 × 0.22 × 0.20 |
| T/K | 100(2) | 100(2) | 295(2) | 296(2) | 295(2) | 295(2) | 295(2) | 100(2) |
| Reflections collected | 12858 |
45335 |
27751 |
16554 |
11390 |
11042 |
26908 |
21324 |
| Unique reflections | 8150 | 5885 | 5631 | 4532 | 5181 | 5845 | 6510 | 5436 |
| Observed reflections | 4352 | 4451 | 2225 | 3030 | 4110 | 4466 | 3839 | 3895 |
| Parameters | 398 | 343 | 388 | 316 | 359 | 363 | 290 | 310 |
| R 1 (obs) | 0.0787 | 0.0395 | 0.0323 | 0.0922 | 0.0558 | 0.0357 | 0.0447 | 0.0387 |
| wR2 (all) | 0.2325 | 0.0859 | 0.0456 | 0.2643 | 0.1369 | 0.0983 | 0.1168 | 0.0863 |
| S | 0.964 | 1.03 | 0.681 | 1.032 | 1.025 | 1.057 | 1.010 | 1.033 |
| Max./min. Δρ/(e Å−3) | 2.695/−3.586 | 1.169/−2.048 | 0.476/−0.465 | 2.463/−1.544 | 2.106/−2.964 | 1.363/−1.786 | 0.190/−0.138 | 0.222/−0.180 |
2.6. Theoretical methods
The geometries of the complexes included in this study were computed at the BP86-D3/def2-TZVP level of theory using the crystallographic coordinates within the TURBOMOLE program.40 This level of theory that includes the latest available dispersion correction (D3) is adequate for studying non covalent interactions dominated by dispersion effects like π-stacking. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.41 The “atoms-in-molecules” (AIM)42 analysis of the electron density has been performed at the same level of theory using the AIMAll program.433. Results and discussion
3.1. Synthesis and spectroscopic results
Ligands L1 and L2 were synthesized by refluxing benzil dihydrazone with the corresponding aldehyde/keton in alcohol, according to the well-known condensation reaction between a primary amine and a ketone. The yields were almost quantitative in all cases. The reaction of equimolar amounts of these ligands and HgX2 (X = Cl, Br and I) in methanol gave the corresponding complexes. However, single crystals could only be obtained if a tenfold excess of HgX2 was used.An original attempt to produce single crystals of 2 using an equimolar mixture of reactants failed and an attempt was made as described in section 2.4. This however yielded an unexpected product (2a). Apparently this procedure led to partial hydrolysis of the ligand (L1). This hydrolysis must have yielded several products among which probably were benzil and asymmetric benzyl-hydrazone-((pyridin-2-yl)methylidenehydrazone). The condensation of these two intermediates would then produce ligand L3 (Scheme 2) which was detected in the crystal structure of 2a.
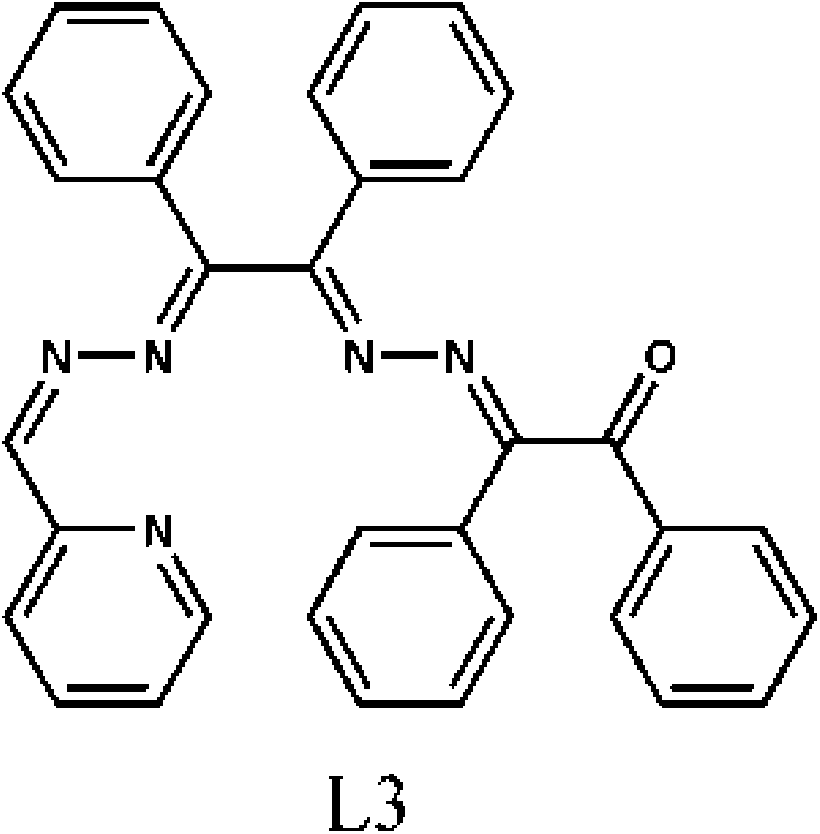 | ||
| Scheme 2 Molecular diagram of L3. | ||
The IR spectra of 1–5 exhibit ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) + ν(CC) stretching vibrations44 in the range 1650–1560 cm−1, which are characteristic of metal bound imines.
N) + ν(CC) stretching vibrations44 in the range 1650–1560 cm−1, which are characteristic of metal bound imines.
3.2. Crystal structures of 1–5
Single crystal X-ray diffraction studies of all six coordination compounds 1–5 were made. All the bond lengths and angles in the ligands have the usual values for coordinated imines.45,46 ORTEP representations of the molecules of 1–5 and perspective views of structural motifs in the crystal structures are shown in Fig. 1–8. | ||
| Fig. 1 a) ORTEP plot of a monomeric unit of 1 with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. b) A section of the polymeric double chain in the structure of 1. | ||
 | ||
| Fig. 2 a) ORTEP plot of a monomeric unit of 2 with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. b) A section of the polymeric chain in the structure of 2. | ||
 | ||
| Fig. 3 a) ORTEP plot of a molecule of 3 with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. b) A C–H⋯N hydrogen bonded dimer in the structure of 3. | ||
 | ||
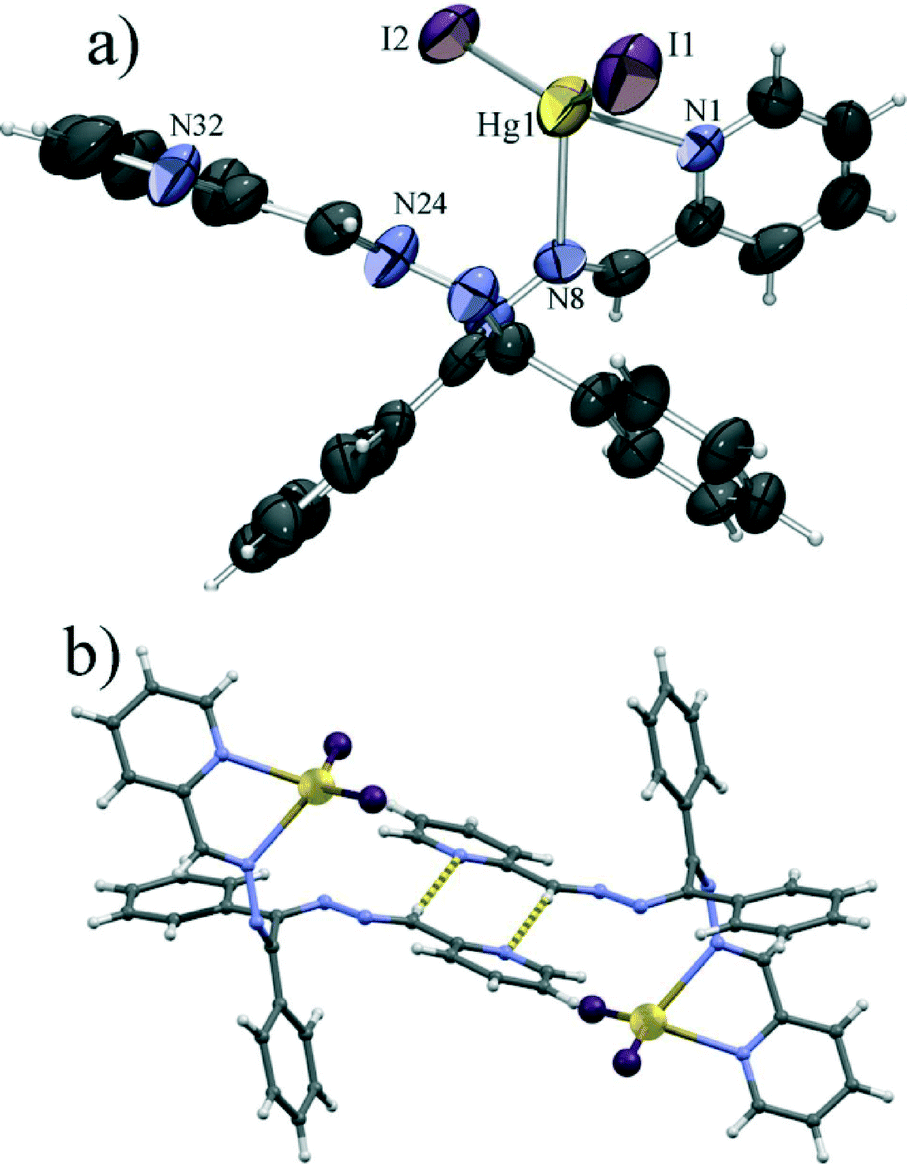
| Fig. 4 a) ORTEP plot of a molecule of 2a with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. b) Overlap of the molecules of 2a (brown), 3 (violet) and HgI2L2 (blue) showing the overall similarity of the three molecules. The chelate ring atoms have been chosen as anchor atoms for the overlap. | ||
 | ||
| Fig. 5 ORTEP plot of an asymmetric unit of 4 with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. | ||
 | ||
| Fig. 6 a) ORTEP plot of a monomeric unit of 5 with the labelling of the metal and donor atoms. Thermal ellipsoids are shown at 50% probability and hydrogen atoms are shown as small spheres of arbitrary radii. b) Tetranuclear molecules of 5 interconnecting into chains via weak Hg–Br interactions. | ||
 | ||
| Fig. 7 Overlap of Hg2Br4L units in 2 (red) and 5 (blue). Mercury atoms are shown as small spheres. The chelated mercury atom and the chelating nitrogen atoms of the organic ligand have been chosen as anchor atoms for the overlap. Mercury atoms are shown as spheres and bromides as sticks. | ||
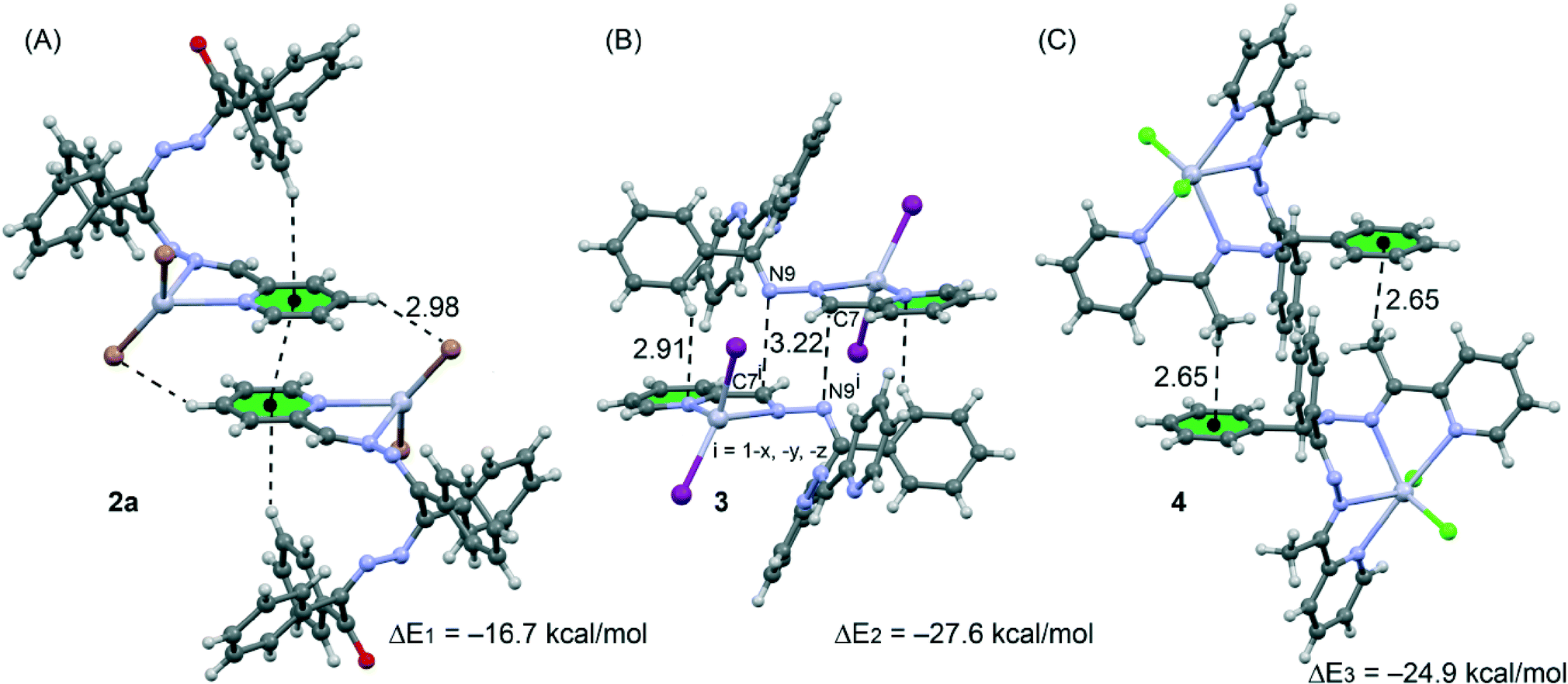 | ||
| Fig. 8 Dimeric crystal fragments of compounds 2a (A), 3 (B) and 4 (C) and their interaction energies. | ||
Single crystal diffraction experiments have shown 1 to be a 1D coordination polymer. The monomeric unit of the polymer comprises four symmetrically independent mercury(II) cations interconnected by bridging chloride anions. The polymeric structure is achieved through chains of HgCl2 units along the crystallographic a axis which are further garnished with Hg2Cl4L1 units comprising a HgCl4 tetrahedron to which a terminal HgL1 is bonded (Fig. 1a). The two symmetrically independent mercury atoms which form the backbone of the coordination polymer (Hg3 and Hg4) have a distorted octahedral coordination with two short (ca. 2.3 Å) and two long Hg–Cl bonds (ca. 3.0 Å) forming the (HgCl2)n chain, and two long Hg–Cl bonds of which one (2.82 Å and 2.95 Å) binds the Hg atoms of the chain to the HgCl4 tetrahedra, while the substantially longer other one (3.25 Å and 3.60 Å) binds the Hg atoms of one (HgCl2)n chain to bridging chlorides of a neighbouring chain (Fig. 1b). The HgCl4 tetrahedra formed about mercury atoms Hg2 are disphenoidally distorted with two shorter bonds to a terminal chloride (Hg2–Cl4 of 2.33 Å) and to a chloride bridging to the (HgCl2)n chain (Hg2–Cl3 of 2.40 Å), and two longer bonds to chlorides bridging to the terminal mercury Hg1 cation (Hg2–Cl1 of 2.70 Å and Hg2–Cl2 of 2.68 Å) of which one (Cl2) also bridges to the (HgCl2)n chain. The coordination of the terminal mercury ion can be described as a heavily distorted trigonal prism with two chloride anions and N1 of the ligand L1 as vertices of one base and N2, N3 and N4 as vertices of the other. Of the Hg–N bonds, the bonds with the pyridine nitrogen atoms are markedly shorter (Hg1–N1 of 2.21 Å and Hg1–N3 of 2.26 Å) than those with imine nitrogen atoms (Hg1–N2 of 2.60 Å and Hg1–N4 of 2.47 Å). The molecule of L1 is helically twisted to encompass the Hg1 atom so that the ligand molecule assumes a conformation of approximate C2 symmetry.
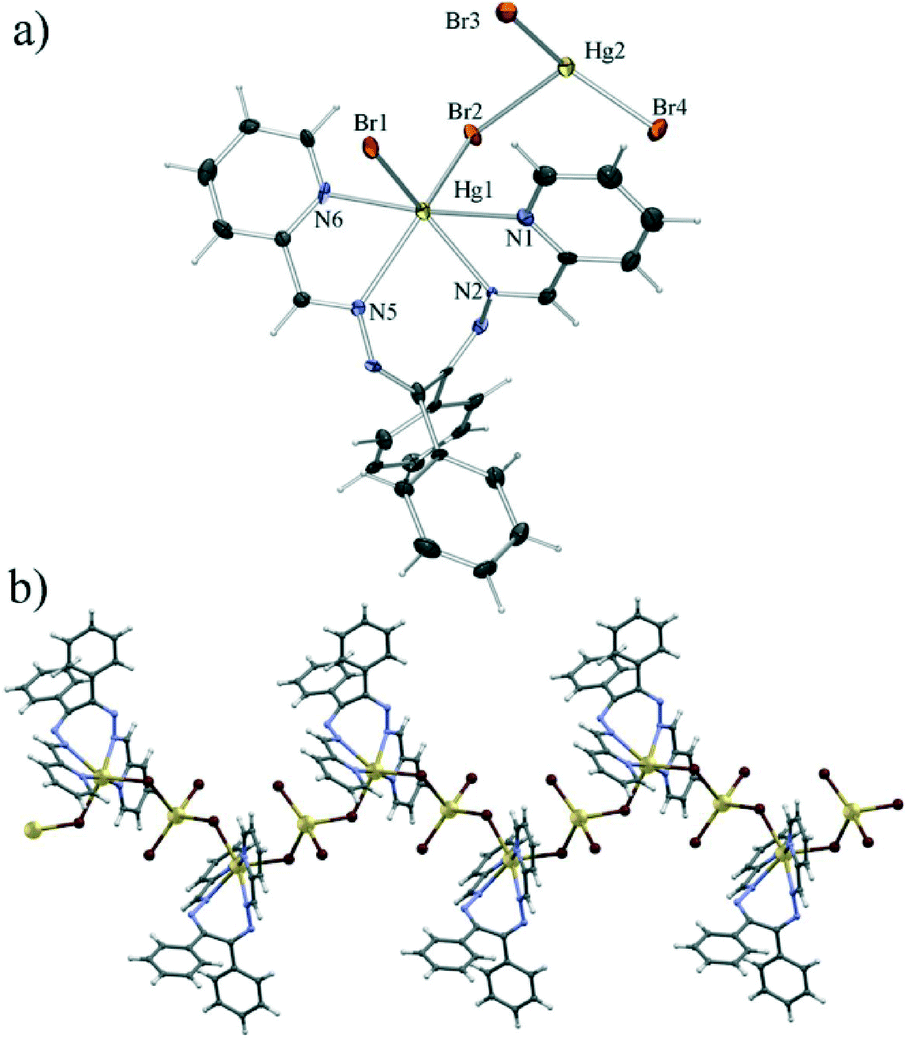
Compound 2, formed when chloride was replaced with bromide, was also found to be a coordination polymer. Here however the monomeric unit comprises only two symmetrically independent mercury(II) cations, each bonded to two bromides which acted as bridges between mercury cations forming a polymer along the crystallographic b axis (Fig. 2). One of the mercury cations (Hg2) is the centre of a HgBr4 tetrahedron, which is again disphenoidally distorted with two shorter bonds to terminal bromides (Hg2–Br3 of 2.45 Å and Hg2–Br4 of 2.46 Å) and two longer bonds to bridging bromides (Hg2–Br1 of 2.82 Å and Hg2–Br2 of 2.86 Å). The coordination of the other mercury cation (Hg1) is similar to that of Hg1 in 1 with two bridging bromides and a tetracoordinating L1. Here however all the Hg–N bonds are of similar lengths (in the range 2.42 Å to 2.55 Å), and the overall coordination polyhedron is an intermediate between a (distorted) octahedron and a (distorted) trigonal prism.
Although structurally quite different from the chloride derivative 1, the bromide derivative was found to be isostructural with a previously described chloride analogue32 (see Fig. S1†) from which it only differs due to the larger Hg–Br bond lengths as compared to the Hg–Cl bonds, which subsequently leads also to an increase in the length of the b axis, as compared to the isostructural chloride.
Introduction of iodide, in place of chloride or bromide, leads to a significant change in the molecular structure. Unlike 1 and 2, which are polymeric, the iodide derivative 3 was found to be a mononuclear complex. Also, L1 is in this case only bidentate, chelating mercury(II) with only one pyridine and one imine nitrogen atom. This makes the mercury ion tetrahedrally coordinated with the two nitrogen atoms and two iodide anions (Fig. 3a). This HgI2[N]2 tetrahedron shows even larger disphenoidal distortion, not so much due to the difference in Hg–I (Hg1–I1 of 2.64 Å and Hg1–I2 of 2.65 Å) and Hg–N bond lengths (Hg1–N1 of 2.45 Å and Hg1–N3 of 2.56 Å), but due to the large difference between the chelate bite angle (φ(N1–Hg1–N2) = 67.0°) and the I1–Hg1–I2 angle (φ(I1–Hg1–I2) = 135.4°) caused by the large van der Waals radius of iodide (non bonded I⋯I distance of 4.46 Å). The large van der Waals radius of iodide is also the probable cause of the change of the coordination mode of L1 which due to the increased size of the halogen can no longer act as a tetradentate ligand because of steric hindrance. The molecular structure is almost identical to that reported for the HgI2L2 complex,47 with minor differences in the conformation of the ligand molecule, most ostensively in the non-chelating arm of the ligand. These differences are likely caused by the different supramolecular environments – the azomethine CH group of the uncoordinated arm of L1 forms a hydrogen bond with a pyridine nitrogen of a neighbouring molecule (C26–H26⋯N32 of 3.60 Å) closing a centrosymmetric R22(8) motif (Fig. 3b). Such a C–H⋯N hydrogen bond, which is often a significant interaction in imines derived from pyridine,48 is not possible in the case of L2, as there the azomethine hydrogen has been replaced with a methyl group.
The steric effect on the coordination of the ligand to mercury(II) halogenides is also demonstrated by 2a (HgBr2L3), where mercury is coordinated by two bromides and an organic ligand derived from L1 by replacing one 2-pyridyl group with a benzyl and a phenyl ring, rendering it far bulkier (Fig. 4a). Not only is the coordination polyhedron of the cation here almost identical to that in the iodide complexes, being a disphenoidally distorted tetrahedron with similar Hg–Br (Hg1–Br1 of 2.45 Å and Hg1–Br2 of 2.49 Å) and Hg–N bond lengths (Hg1–N1 of 2.38 Å and Hg1–N2 of 2.70 Å) while the angle between terminal Hg–Br bonds (φ(Br1–Hg1–Br2) = 142.0°) is much larger than the chelate angle ((N1–Hg1–N2) = 66.7°), but the general shape of the molecule is also quite similar to the shapes of 3 and HgI2L2 molecules (Fig. 4b).
Unlike in the case of the iodide complex where the replacement of L1 with its dimethyl derivative L2 does not lead to any significant change in coordination or molecular geometry, both the mercury(II) chloride and bromide complexes obtained with L2 have been found to be quite different from the respective L1 complexes.
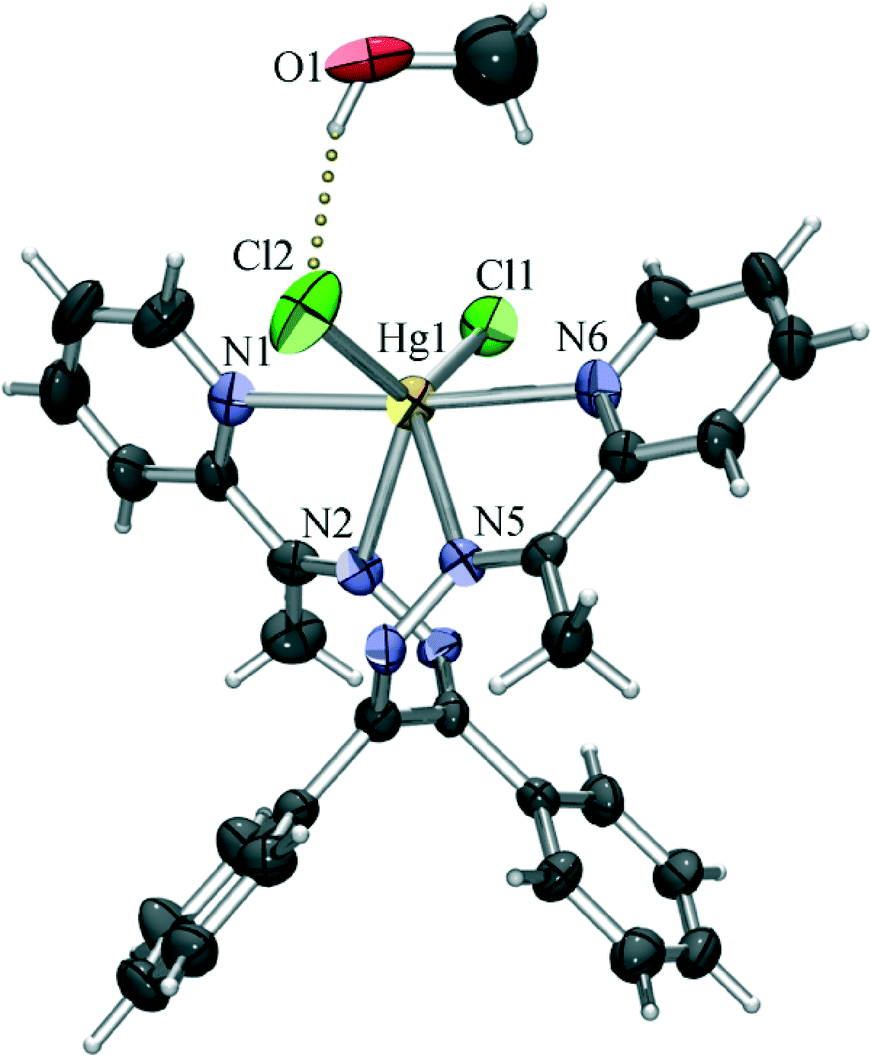
The chloride derivative (4) is a monomeric species found to crystallise as a methanol solvate. The coordination of the mercury(II) ion is similar to that of the terminal Hg1 in the structure of 1, with the four nitrogen atoms of the tetradentate L2 and two chloride anions defining the vertices of a highly irregular polyhedron (intermediate between a octahedron and a trigonal prism). Unlike in 1, however, all the Hg–N bonds are of similar lengths (in the 2.50 Å to 2.57 Å range), and the Hg–Cl bonds are considerably shorter (Hg1–Cl1 of 2.44 Å and Hg1–Cl2 of 2.47 Å) than those in 1, which is only to be expected as here the chlorides are not shared with another mercury cation. The slight difference in length of the two Hg–Cl bonds can be connected to intermolecular hydrogen binding. As noted earlier, 4 crystallises as a methanol solvate, and the methanol molecule is hydrogen bonded to one coordinated chloride (O1–H1o⋯Cl2 of 3.14 Å), which can be assigned as a reason for the slight stretching of the Hg1–Cl2 bond (Fig. 5). By binding to a coordinated chloride, the methanol molecule blocks it from forming other interactions, and thus prevents the chloride to act as a bridge towards another mercury atom. Therefore, although L2 is a slightly sterically more demanding ligand than L1, it cannot be concluded that the absence of polymerisation in this case is due to the change of the ligand. Rather, the supramolecular environment (i.e. hydrogen bonding of the coordinated chloride) which prevents the formation of chloride bridges appears to be a more likely cause of the monomeric nature of 4.
The latter conclusion is to an extent justified by the structure of the bromide derivative (5), which was found to be a tetranuclear complex with bridging bromides. This complex consists of two Hg2Br4L2 monomeric units, equivalent to those forming the polymer 2, although of a different conformation (Fig. 6a and 7). Unlike in 2, however these units bind in a head-to-head manner to a centrosymmetric dimer. The resulting complex molecule thus comprises two terminal HgBrL2 units and two HgBr4 tetrahedra with a common edge (consisting of two Hg4 atoms related by an inversion centre) which bridges them. The HgBr4 tetrahedra are extremely distorted with the Hg2 atom almost coplanar with Br4, Br5 and Br6 (elevated from the Br4–Br5–Br6 plane by only 0.13 Å) to which it binds with short bonds (Hg2–Br5 of 2.62 Å, Hg2–Br4 of 2.47 Å and Hg2–Br6 of 2.48 Å) and with a very long bond (Hg2–Br6 of 3.24 Å) to the other Br6. The coordination of the terminal Hg1 cation is quite similar to the one described in 2, with similar Hg–N bond lengths (in the range 2.40 Å to 2.58 Å), although with a large difference between the terminal (Hg1–Br3 of 2.64 Å) and the bridging Hg–Br (Hg1–Br5 of 2.86 Å) bonds. The Br3–Hg1–Br5 angle is also significantly larger (φ(Br3–Hg1–Br5) = 150.4° in 5 as opposed to 110.9° in 2), although this can be attributed to the close proximity of Br3 from a neighbouring molecule which approaches Hg1 (Hg1⋯Br3′ contact of 3.52 Å) between the two coordinated bromides spreading them apart. This weak quasi-coordinative interaction interconnects the tetranuclear molecules into chains in the crystallographic [−1 0 1] direction (Fig. 6b).
3.5 Theoretical study of the supramolecular assemblies
We have focused the theoretical study on the analysis of the interesting supramolecular assemblies observed in the solid state of complexes 2a, 3 and 4 (see Fig. 8). In complex 2a we have analysed the C–H⋯π/π⋯π/π⋯H–C supramolecular assembly (see Fig. 8A) observed in the solid state. In 3 we have analysed a self-complementary dimer that is governed by two symmetrically related C7⋯N9 π-hole interactions. That is, the lone pair of the N9 atom interacts with the positive π-hole located at the carbon C7 atom of the imidic CN bond (see Fig. 8B). This lp⋯π-hole interactions49 are attracting increasing attention from the scientific community due to their important role in crystal engineering and supramolecular chemistry.50 Finally, in compound 4 we have also analysed a self-assembled dimer where two complementary C–H⋯π interactions are established (see Fig. 8C).
The complexation energies of the selected crystallographic fragments are also included in Fig. 9. It can be observed that they are large and negative, indicating that they are strong binding motifs in the solid state structures. We have focused the theoretical study on the analysis of the influence of the complexation of the organic ligand to the Hg(II) metal centre on the strength of the noncovalent interactions. Therefore we have used several theoretical models based on the crystal structures. For compound 2a we have used two models and the binding energies (Fig. 9) are compared to the corresponding one obtained for the crystallographic dimer shown in Fig. 8A.
 | ||
| Fig. 9 Theoretical models used to evaluate the noncovalent interactions in compound 2a without (A) and with (B) the phenyl groups. | ||
We have first analysed the influence of the intramolecular C–H⋯π interaction on the binding energy of the dimer. Therefore we have used a model where the phenyl groups that participate in the C–H⋯π interactions have been replaced with hydrogen atoms (see arrows in Fig. 9A). As a result the binding energy (ΔE4 = −16.4 kcal mol−1) was slightly reduced compared to ΔE1 = −16.7 kcal mol−1 (see Fig. 8A), therefore the influence of the C–H⋯π interaction on the strength of the π⋯π interaction is almost negligible. Secondly, we have analyzed the influence of the metal complexation on the binding energy by eliminating the HgBr2 part of the molecule. By doing so, the interaction energy is significantly reduced to ΔE5 = −7.0 kcal mol−1 which is the contribution of the π⋯π interaction to the supramolecular assembly. The difference between ΔE1 and ΔE5 corresponds to the contribution of both C–H⋯Br hydrogen bonds, that is ΔE1 − ΔE5 = −9.7 kcal mol−1.
The theoretical models used to analyse the contribution of the different noncovalent interactions in complexes 3 and 4 are shown in Fig. 10. For complex 3 we have used two models. In the first one we have replaced the phenyl groups that establish the C–H⋯π interaction with hydrogen atoms (see small arrows in Fig. 10A). As a result the interaction energy is reduced from ΔE2 = −27.6 kcal mol−1 (see Fig. 8B) to ΔE6 = −18.8 kcal mol−1 which corresponds to the interaction energy associated with the double π-hole interaction. Therefore each C–H⋯π interaction can be estimated as (ΔE2 − ΔE6)/2 = −4.3 kcal mol−1. In order to analyze the influence of the metal coordination on the π-hole interaction, we have used another theoretical model (see Fig. 10B) where the HgI2 moieties have been eliminated. In this model the interaction energy is further reduced to ΔE6 = −5.8 kcal mol−1, indicating that the complexation of the imidic nitrogen atom to the Hg metal centre has a strong influence on the magnitude of the π-hole at the carbon atom. To corroborate this explanation, we have computed the atomic Mulliken charge at C7 carbon in the presence and absence of the HgI2 moiety. As a result, the charge at the imidic carbon atom becomes 0.16 e more positive in the presence of the metal centre.
Finally, in complex 3 we have used a theoretical model to evaluate the influence of the metal complexation on the self-complementary CH3⋯π interaction (see Fig. 10C). In this case the interaction energy in the absence of HgCl2 is considerably reduced from ΔE2 = −24.9 kcal mol−1 (see Fig. 8C) to ΔE6 = −11.3 kcal mol−1 which corresponds to the interaction energy associated with the double CH3⋯π interaction. A likely explanation for this significant reduction is that the acidity of the methyl hydrogen atom increases as a consequence of the complexation of the organic ligand to the Hg(II) ion.
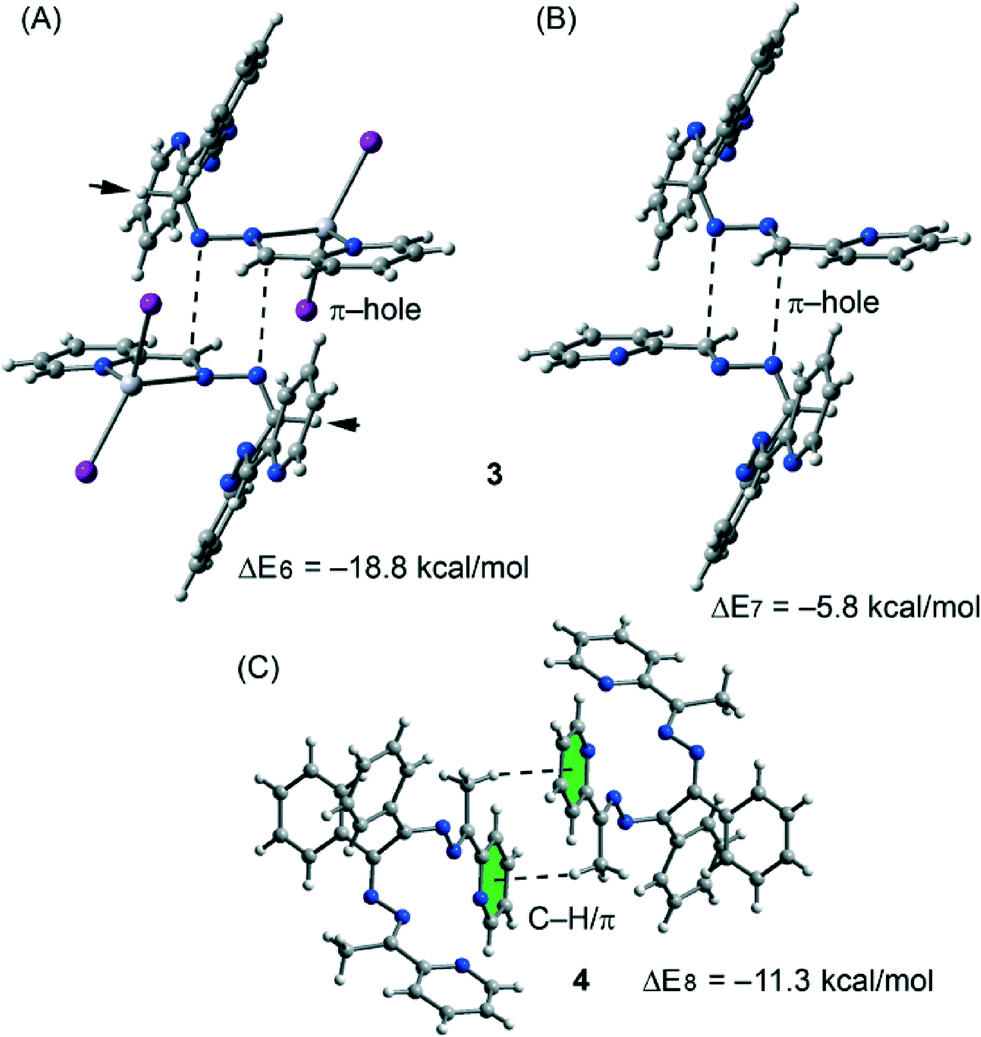 | ||
| Fig. 10 Theoretical models used to evaluate the noncovalent interactions in compounds 3 (A and B) and 4 (C). | ||
In order to characterize the noncovalent interactions explained above, we have used Bader's “atoms-in-molecules” methodology42 which provides an unambiguous definition of chemical bonding. The existence of a bond critical point and a bond path connecting two atoms can be used as a confirmation of covalent/noncovalent bonding. AIM analyses of the theoretical models corresponding to compounds 2a, 3 and 4 are shown in Fig. S2 and S3.† All noncovalent interaction described above have been confirmed by the existence of several bond critical points and bond paths connecting the interacting atoms (see the ESI† for details).
4. Conclusions
In conclusion, we herein reported the syntheses and structural characterization of six new mercury(II) complexes 1–5 with halogenide and organic ligands, where a comparison between structurally similar ligands, L1 and L2, was made. Although the difference between the two ligands is minute (they differ only in the presence/absence of two methyl groups) the choice of ligand has an immense effect on the coordination behaviour of mercury when bromide or chloride was the anionic ligand present. This is related to the differences in the supramolecular assemblies formed by the complexes, which in turn affect the molecular packing. The simple addition of a methyl group to the ligand controls on one hand the formation of a dimer instead of a polymer (as in compound 5) and, on the other hand, the formation of supramolecular assemblies due to the participation in CH3⋯π interactions, as demonstrated by DFT analysis.Acknowledgements
We are grateful to the University of Tabriz Research Council for the financial support to this research. The project “Factoría de Cristalización, CONSOLIDER INGENIO-2010” provided the X-ray structural facilities used for this work. AF and AB thank MINECO of Spain (project CONSOLIDER INGENIO 2010 CSD2010-00065, FEDER funds) and CTI (UIB) for free allocation of computer time.Notes and references
- K. Y. Wang, L. J. Zhou, M. L. Feng and X. Y. Huang, Dalton Trans., 2012, 41, 6689–6695 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- C. S. Liu, X. S. Shi, J. R. Li, J. J. Wang and X. H. Bu, Cryst. Growth Des., 2006, 6, 656–663 CAS.
- S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33–46 CrossRef CAS.
- S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145–168 CrossRef CAS.
- C. D. Wu, A. G. Hu, L. Zhang and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- L. C. Tabares, J. A. R. Navarro and J. M. Salas, J. Am. Chem. Soc., 2001, 123, 383–387 CrossRef CAS.
- Y. Zhao, Z. Lin, H. Wu and C. Duan, Inorg. Chem., 2006, 45, 10013–10015 CrossRef CAS PubMed.
- H. R. Khavasi, A. R. Salimi, H. Eshtiagh-Hosseini and M. M. Amini, CrystEngComm, 2011, 13, 3710–3717 RSC.
- I. Kuzu, I. Krummenacher, J. Meyer, F. Armbruster and F. Breher, Dalton Trans., 2008, 5836–5865 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- (a) S. Re and S. Nagase, Chem. Commun., 2004, 658–659 RSC; (b) B. H. Northrop, Y. R. Zheng, K. W. Chi and P. J. Stang, Acc. Chem. Res., 2009, 42, 1554–1563 CrossRef CAS PubMed.
- C. J. Elsevier, J. Reedijk, P. H. Walton and M. D. Ward, Dalton Trans., 2003, 1869–1880 RSC.
- B. Olenyuk, A. Fechtenkötter and P. J. Stang, J. Chem. Soc., Dalton Trans., 1998, 1707–1728 RSC.
- G. S. Papaefstathiou and L. R. MacGillivray, Coord. Chem. Rev., 2003, 246, 169–184 CrossRef CAS.
- H.-P. Zhou, X.-P. Gan, X.-L. Li, Z.-D. Liu, W.-Q. Geng, F.-X. Zhou, W.-Z. Ke, P. Wang, L. Kong, F.-Y. Hao, J.-Y. Wu and Y.-P. Tian, Cryst. Growth Des., 2010, 10, 1767–1776 CAS.
- H.-P. Zhou, J.-H. Yin, L.-X. Zheng, P. Wang, F.-Y. Hao, W.-Q. Geng, X.-P. Gan, G.-Y. Xu, J.-Y. Wu, Y.-P. Tian, X.-T. Tao, M.-H. Jiang and Y.-H. Kan, Cryst. Growth Des., 2009, 9, 3789–3798 CAS.
- S. Y. Lee, S. Park, H. J. Kim, J. H. Jung and S. S. Lee, Inorg. Chem., 2008, 47, 1913–1915 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides, M. Shatruk, A. Chouai, L. M. Perez, J. Bacsa and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 5895–5912 CrossRef CAS PubMed.
- C. S. Campos-Fernandez, B. L. Schottel, H. T. Chifotides, J. K. Bera, J. Bacsa, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 12909–12923 CrossRef CAS PubMed.
- F. Zeng, J. Ni, Q. Wang, Y. Ding, S. W. Ng, W. Zhu and Y. Xie, Cryst. Growth Des., 2010, 10, 1611–1622 CAS.
- P. Manna, S. K. Seth, A. Das, J. Hemming, R. Prendergast, M. Helliwell, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, Inorg. Chem., 2012, 51, 3557–3571 CrossRef CAS PubMed.
- V. Stilinović, K. Užarević, I. Cvrtila and B. Kaitner, CrystEngComm, 2012, 14, 7493–7501 RSC.
- K. Huber, Wisconsin Mercury Source Book, Wisconsin Department of Natural Resources, Bureau of Watershed Management, Madison, WI, 1997 Search PubMed.
- M. F. Hawthorne and Z. Zheng, Acc. Chem. Res., 1997, 30, 267–276 CrossRef CAS.
- A. Tamayo, B. Pedras, C. Lodeiro, L. Escriche, J. Casabo, J. L. Capelo, B. Covelo, R. Kivekas and R. Sillanpaa, Inorg. Chem., 2007, 46, 7818–7826 CrossRef CAS PubMed.
- J. G. Melnick, K. Yurkerwich, D. Buccella, W. Sattler and G. Parkin, Inorg. Chem., 2008, 47, 6421–6426 CrossRef CAS PubMed.
- S. Park, S. Y. Lee and S. S. Lee, Inorg. Chem., 2010, 49, 1238–1244 CrossRef CAS PubMed.
- A. Morsali and M. Y. Masoomi, Coord. Chem. Rev., 2009, 253, 1882–1905 CrossRef CAS PubMed.
- (a) A. A. Khandar, V. T. Yilmaz, F. Costantino, S. Gumus, S. A. Hosseini-Yazdia and G. Mahmoudi, Inorg. Chim. Acta, 2013, 394, 36–44 CrossRef CAS PubMed; (b) M. Akkurt, A. A. Khandar, M. N. Tahir, S. A. Hosseini-Yazdi and G. Mahmoudi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 62, m903–m904 Search PubMed.
- (a) J. Lewiński, J. Zachara, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185–1199 CrossRef PubMed; (b) M. Sakamoto, K. Manseki and H. Okawa, Coord. Chem. Rev., 2001, 219–221, 379–414 CrossRef CAS; (c) H. Wang, W. Cao, T. Liu, C. Duan and J. Jiang, Chem. – Eur. J., 2013, 19, 2266–2270 CrossRef CAS PubMed; (d) Q. Ma, X. Feng, W. Cao, H. Wang and J. Jiang, CrystEngComm, 2013, 15, 10383–10388 RSC; (e) H. Wang, D. Zhang, Z.-H. Ni, X. Li, L. Tian and J. Jiang, Inorg. Chem., 2009, 48, 5946–5956 CrossRef CAS PubMed.
- A. J. Bloodworth, J. Organomet. Chem., 1970, 23, 27–30 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Macromolecular Crystallography, Part A, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- Bruker, APEX2, Bruker AXS Inc., Madison, WI, USA, 2004 Search PubMed.
- Oxford Diffraction, CrysAlis CCD and CrysAlis RED. Version 1.170. Oxford Diffraction Ltd, Wroclaw, Poland 2003 Search PubMed.
- G. M. Sheldrick, SADABS, Programs for Scaling and Correction of Area detection Data, University of Göttingen, Göttingen (Germany), 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Haser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- AIMAll (Version 13.11.04), T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33–46 CrossRef CAS.
- A. Blagus, D. Cinčić, T. Friščić, B. Kaitner and V. Stilinović, Maced. J. Chem. Chem. Eng., 2010, 29, 117–138 CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer and A. G. Orpen, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- M. G. B. Drew, S. De and D. Datta, Inorg. Chim. Acta, 2009, 362, 2487–2491 CrossRef CAS PubMed.
- V. Stilinović, D. Cinčić and B. Kaitner, Acta Chim. Slov., 2008, 55, 874–879 Search PubMed.
- (a) H. B. Burgi, J. D. Dunitz and E. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef CAS; (b) J. Lewiński, W. Bury and I. Justyniak, Eur. J. Inorg. Chem., 2005, 4490–4492 CrossRef.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2015, 51, 1491–1493 RSC; (b) A. Bauzá, R. Ramis and A. Frontera, J. Phys. Chem. A, 2014, 118, 2827–2834 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S3 and results and discussion of AIM analysis. CCDC 1046009–1046016 contain the supplementary crystallographic data for 1–5, L1 and L2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00382b |
| This journal is © The Royal Society of Chemistry 2015 |