
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The N-atom in [N(PR3)2]+ cations (R = Ph, Me) can act as electron donor for (pseudo) anti-electrostatic interactions†
Antonio
Bauzá
a,
Antonio
Frontera
 *a,
Tiddo J.
Mooibroek
*b and
Jan
Reedijk
cd
*a,
Tiddo J.
Mooibroek
*b and
Jan
Reedijk
cd
aDepartment of Chemistry Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma (Baleares), Spain. E-mail: toni.frontera@uib.es; Fax: (+) 34 971 173426
bSchool of Chemistry of the University of Bristol, Cantock's Close, BS8 1TS, Bristol, UK. E-mail: chtjm@bristol.ac.uk
cLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300 RA Leiden, The Netherlands
dDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 15th April 2015
Abstract
A CSD analysis and DFT study reveal that the nitrogen lone-pair in [N(PPh3)2]+ is partially intact and involved in intramolecular hydrogen bonding. Computations with the model cation [N(PMe3)2]+ indicate that intermolecular hydrogen bonding with this ion's N-atom is possible (−8.3 kcal mol−1 for H2O as hydrogen bond donor), presenting a rare example of pseudo anti electrostatic hydrogen bonding.
Non-covalent interactions are known to be of great importance for many chemical and biological sciences.1–4 For example, both intra- and intermolecular hydrogen bonds are significant structural determinants in biological systems.5,6 While intramolecular interactions may determine what molecular conformer is most stable, intermolecular interactions govern molecular aggregation and crystallization. Hence, using entities known for their intermolecular interactions can be exploited in crystal engineering.
For example, the bis(triphenylphosphane)iminium cation ([PPN]+, 1, Scheme 1) is a popular counter cation‡,8–13 to crystallize anionic species; its ability to form π–π stacking networks could be an important factor for the ease with which [PPN]+ containing compounds crystallize.13 Despite the widespread use of 1 as counter ion, surprisingly little information is available concerning the inter- and intramolecular interactions of this ion.13
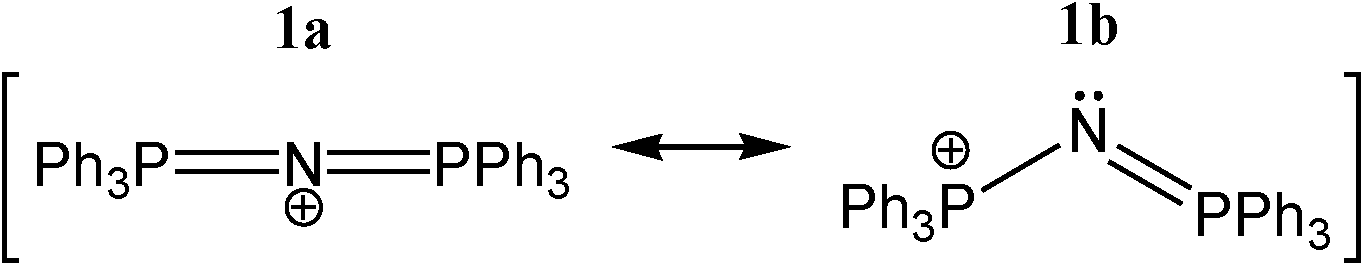 | ||
| Scheme 1 Common linear and bent resonance structures of the [N(PPh3)2]+ cation, typically abbreviated as [PPN]+. | ||
One particular question is whether the N-atom in 1 could act as an electron donor. While the most widely used notation (1a)§ would predict that the N-atom is positively charged and linear, resonance structure 1b would predict that the lone-pair is intact and that the P–N–P angle is non-linear. Naturally, the conjugated nature of 1 will spread out the positive charge, but it is not a priori obvious what repercussions this would have exactly on the lone-pair electrons of N. If some electron density remains on N, could [PPN]+ still act as hydrogen bond acceptor for cationic protons, despite it being a cation itself? If so, this would be a rare case of ‘anti-electrostatic hydrogen bonding’ (AEHB).14 That is, hydrogen bonding between two entities that are formally of the same charge, in this case positive. Such AEHB can in principle take two forms (where D = ‘donor’ and A = ‘acceptor’):
(A1) [D–Hδ+]+⋯[Aδ−]+ (cationic donor/acceptor)
(A2) [D–Hδ+]−⋯[A]− (anionic donor/acceptor)
If the interacting partner lacks a formal charge but is positively or negatively polarized (e.g. O–Hδ+, N–Hδ+; R2Oδ−, R3Nδ−), one could speak of a ‘pseudo anti-electrostatic hydrogen bond,’ which one can write in two general forms:
(B1) D–Hδ+⋯[Aδ−]+ (cationic acceptor)
(B2) [D–Hδ+]−⋯Aδ− (anionic donor)
Both these scenarios (A and B) are markedly different from conventional hydrogen bonding (C) where both interacting partners are charge-neutral or where the charge helps to make the bond stronger:
(C1) D–Hδ+⋯Aδ− (neutral donor/acceptor)
(C2) [D–Hδ+]+⋯Aδ− (cationic donor)
(C3) D–Hδ+⋯[A]− (anionic acceptor)
(C4) [D–Hδ+]+⋯[A]− (charge complementary)
We herein report on a combined DFT study and Cambridge Crystallographic Database (CSD)15 analysis that confirm the non-linearity of 1, and the ability of the N-atom in 1 (and in [N(PMe3)2]+) to act as electron donor to establish (pseudo-) AEHB of the form A1 and B1.
Shown in Fig. 1a and b are perspective views of a very recent [PPN]+ containing crystal structure, [PPN]+[Au(CN)(CCPh)]−.7 As is indicated in the figures, short intramolecular C–H⋯N contact distances (2.55–2.63 Å) are present that lie within the sum of the van der Waals radii of H (1.09 Å) and N (1.55 Å).16 Such short contacts are indicative of hydrogen bonding and seem to have been largely overlooked in the past.8,9,13,17,18 These four apparent H-bonds are characterized by PNCC torsion angles close to 0°, in this example ranging from 1.31° (H23) to 22.3° (H11). Two of them (belonging to R1 and R1′, in red, see Fig. 1a) can be distinguished as perpendicular to the plane running through the PNP core. The other two hydrogen atoms (belonging to R2 and R2′, in blue, see Fig. 1b) are more parallel to this plane. The aryl rings R3 and R3′ (green) seem to be π–π stacking and their o-H atoms do not display short C–H⋯N distances (3.32–3.95 Å).
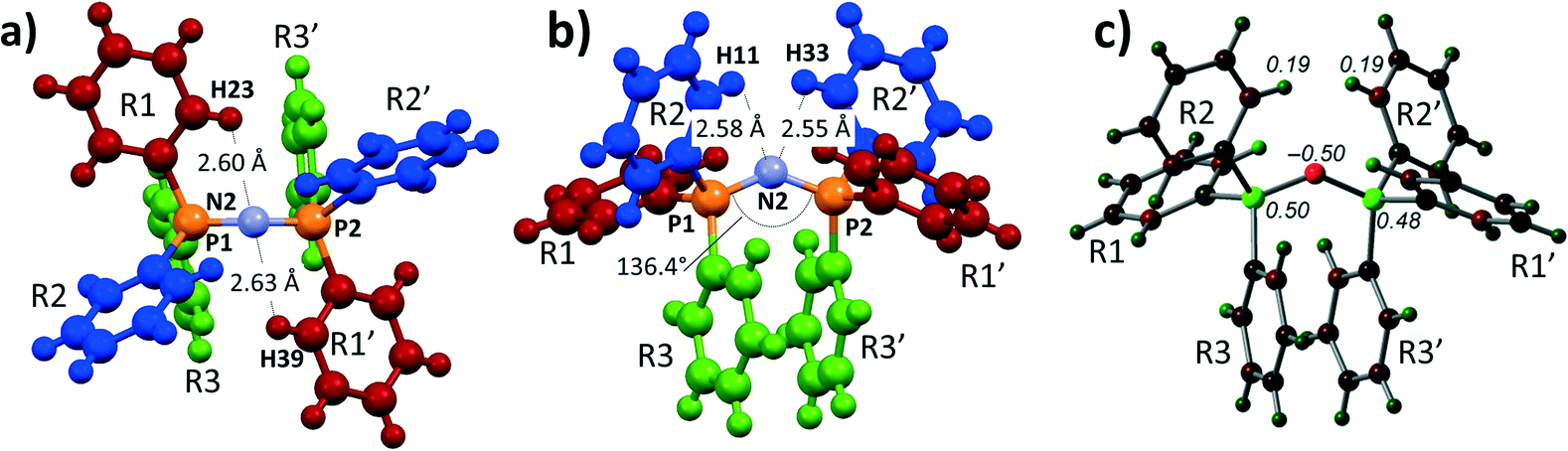 | ||
| Fig. 1 a) and b) Perspective views and numbering scheme of 1 as determined by X-ray crystallography of [PPN]+[Au(CN)(CCPh)]−·H2O·Acetone.7 Solvent molecules and anion are omitted for clarity; c) DFT reproduction of 1 at the BP86-D3/def2-TZVP level of theory, showing the Mulliken charge distribution (in e). Atoms are colour coded based on charge, ranging from −0.5 (red) to +0.5 (green). | ||
To obtain more insight into the physical origins of these intramolecular features, the geometry of 1 as found7 in [PPN]+[Au(CN)(CCPh)]− was subjected to DFT computations on the BP86-D3/def2-TZVP level of theory (see ESI† for details). Shown in Fig. 1c is an analysis of the Mulliken charge distribution of 1, revealing that the central nitrogen atom has a partial negative charge (−0.50 e) and is thus suited to act as potential electron donor for (pseudo-)AEHB of general notations A1 or B1. The non-covalent index analysis of 1 (Fig. S1†) indeed confirmed the presence of the four intramolecular C–H⋯N hydrogen bonds, while simultaneously revealing intramolecular π–π stacking and C–H⋯π interactions.
In order to ascertain the general nature of these intramolecular H-bonding features, the CSD was evaluated.¶ Shown in Fig. 2a is a colour-coded contour density plot of the intramolecular orthoC–H⋯N distances vs. the absolute N–P–C–C torsion angles found within 1315 crystalline [PPN]+ salts deposited in the CSD.
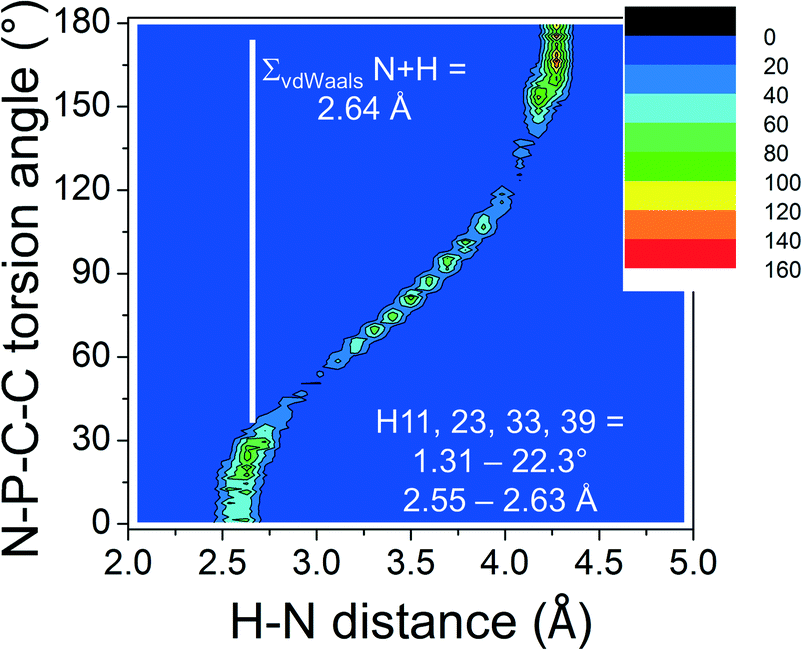 | ||
| Fig. 2 Plot of the absolute N–P–C–C torsion angle vs. the orthoC–H⋯N distances found in 1315 [PPN]+ structures found within 1120 CIFs. The numbers indicated in white are taken from the X-ray structure of 1 highlighted in this work. | ||
The feature around [15°; 2.6 Å] clearly reveals that short intramolecular C–H⋯N distances and small N–P–C–C torsion angles are normal within crystalline [PPN]+ salts. The ortho-H atoms opposite those involved in short H⋯N distances are characterized by the feature around [165°; 4.3 Å], while the widespread intermediate feature belongs to the ortho-H atoms of aryls like R3 and R3′. Fig. 3 shows a plot of P–N–P angles vs. P–N distances of [PPN]+ structures. Numerical analysis of these data showed that the average P–N distances are 1.58 ± 0.02 Å and that the average P–N–P angle is 142.6 ± 8.8°.
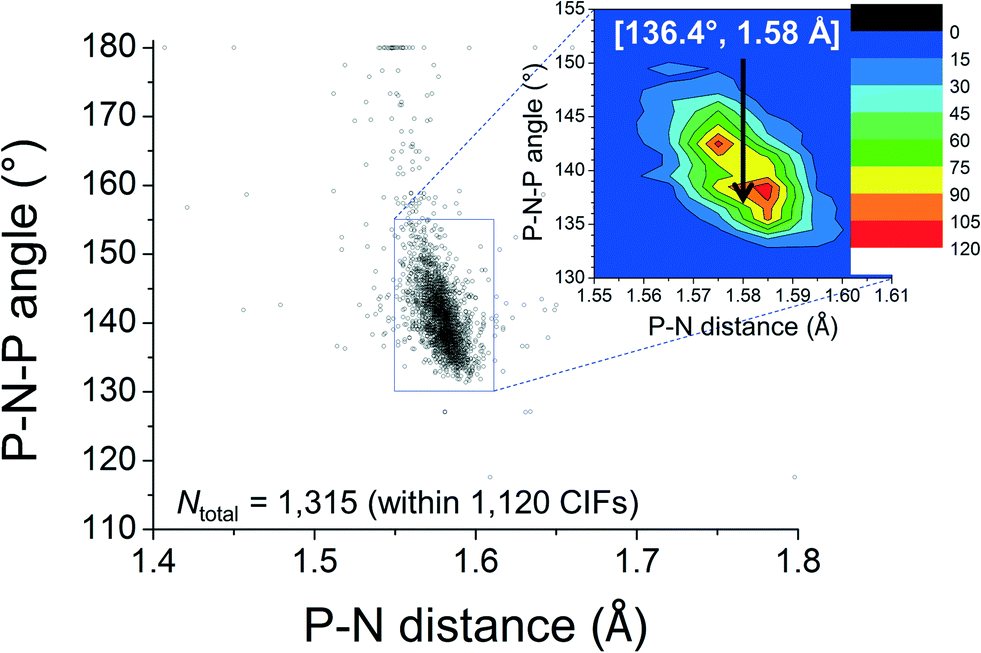 | ||
| Fig. 3 Plot of P–N–P angles vs. P–N distances found in 1315 [PPN]+ structures found within 1120 CIFs. The insert is a colour coded magnification of the densest region. The values indicated in white belong to the X-ray structure of 1. | ||
The above data clearly indicate that the cation 1 is best represented by resonance structure 1b (at least in the crystalline state), and that the lone-pair on N is normally involved in intramolecular hydrogen bonding. These interactions can be seen as intramolecular pseudo anti-electrostatic hydrogen bonds, i.e. [C–Hδ+⋯[Nδ–P2]+]+, much like in general notation B1.
We wondered if perhaps the N-atom is also capable of engaging in intermolecular interactions, to which end we conducted some additional DFT studies and performed a detailed survey of the CSD. We choose [N(PMe3)2]+ as a simplified model compound and calculated its complexation energies with several neutral (2–6) and cationic (7–11) species at the BP86-D3/def2-TZVP level of theory (see ESI† for details). The results are summarized in Table 1. The interacting energies of 1 with neutral guests are significant and negative (up to −8.3 kcal mol−1 for 2), while for cationic species the interaction is unfavourable, apparently due to the strong electrostatic repulsion.
| Complex (guest) | E BSSE | R e | ρ (a.u.) |
|---|---|---|---|
| a The H⋯O distance is 1.690 Å with ρ = 0.0465 a.u. | |||
| 2 (H2O) | −8.3 | 2.190 | 0.0187 |
| 3 (HF) | −7.4 | 1.672 | 0.0553 |
| 4 (HCN) | −0.4 | 2.128 | 0.0200 |
| 5 (HCCH) | −3.5 | 2.254 | 0.0153 |
| 6 (C6H6) | −4.4 | 2.397 | 0.0124 |
| 7 (NH4+) | 41.0 | 1.619 | 0.0673 |
| 8 (Li+) | 34.3 | 1.967 | 0.0335 |
| 9 (Na+) | 40.7 | 2.505 | 0.0165 |
| 10 (CH3Hg+) | 15.7 | 2.247 | 0.0785 |
| 11 (H3O+, H+ transferred)a | 10.2 | 1.056 | 0.2887 |
| 12 (H+) | — | 1.022 | 0.3213 |
As an example, Fig. 4 shows the supramolecular geometry and the distribution of bond critical points according to an ‘atoms in molecules’ (AIM) analysis of complexes 2, 5, and 9 (see Table 1 for the values of ρ(r) at the bond critical points that emerge upon complexation). In complex 2 (Fig. 4a), the bond-critical point between N and one of the H atoms of water confirms that the N lone-pair acts as electron donor. The water molecule is furthermore held in place by two C–H⋯O hydrogen bonds, which might explain why the EBSSE is largest for 2. Such additional intermolecular forces are absent in 5 (Fig. 4b), and the ethyne molecule is held in place by a single C–H⋯N hydrogen bond. Both these complexes can be seen as examples of intermolecular pseudo anti-electrostatic hydrogen bonding of notation D–Hδ+⋯[Nδ–P2]+.
 | ||
| Fig. 4 Computer aided renderings of 2, 5, and 9, computed at the BP86-D3/def2-TZVP level of theory. The small red dots denote the bond-critical points according to an AIM analysis. | ||
The truly anti-electrostatic interaction in complex 9 (i.e. Na+⋯[Nδ–P2]+) is characterized by a single bond-critical point, despite the large positive EBSSE of +40.7 kcal mol−1. A survey of the potential energy curve of complex 9 as a function of N⋯Na+ distance (see Fig. S2a†) revealed a small energy well at the optimized distance of 2.51 Å, with a transition state at 4.49 Å (barrier of 5.1 kcal mol−1). Analysis of the HOMO and HOMO-1 orbitals of compound 9 (Fig. S2b†) shows a large bonding orbital between the cation and the N atom in the HOMO-1, in agreement with the large covalent character of anti-electrostatic interactions.14 The large positive energies of 7–11 make it unlikely that such species could be stable, while the presence of a well indicates that such species might be kinetically stable enough to allow detection/characterization.
It is worth noting that complex 11 is least unstable of those with a cationic guest and that the proton from H3O+ actually appears to be transferred to N with a N–H distance of 1.056 Å. The bond critical point between N and H in 11 is much denser (ρ = 0.29 a.u.) compared to complexes 2–10 (ρ = 0.01–0.08 a.u.). Without the water molecule competing for the proton, i.e. in 12, the N–H distance is even shorter (1.022 Å) and the bond critical point even denser (ρ = 0.32 a.u.). This suggests that [PPN]+-like cations could be protonated to form the dicationic species [PPNH]2+ (when the acid is completely dissociated), and could thus be non-innocent in proton-transfer reactions. Three such examples are indeed known in the solid state, where a [PPNH]2+ crystallized with large non-coordinating anions (CSD recodes BIKYEB, GIXWIT and ZAVDIJ).
Finally, the CSD was inspected for intermolecular N(1)⋯X interactions in [PPN]+ compounds, where X can be any atom and the N⋯X distance was set to the sum of the van der Waals radii of interacting atoms + 1.0 Å. While a plot of the amount of hits as a function of the van der Waals corrected N⋯X distance (see Fig. S3†) indicates some van der Waals overlap, manual inspection revealed that these hits are mainly due to disordered structures. The crystal structure with CSD refcode WOLDUW appears to be an exception with a N(1)⋯H distance of 2.633 Å. A possible explanation for the lack of data in support of intermolecular interactions with the N-atom of 1 is the steric and electronic shielding provided by the above-mentioned four intramolecular hydrogen bonds.
Conclusions
The presented DFT calculations clearly indicate that the lone-pair at N in the [PPN]+ cation is partially intact and can thus act as an electron donor, be it for intramolecular or intermolecular interactions. This possibility is confirmed by the CSD analysis of over 1300 crystal structures containing [PPN]+: four intramolecular H-bonds are normally present and the PNP angle is not linear (~140°). Computations on the model compound [N(PMe2)2]+ suggest that intermolecular hydrogen bonding complexes with neutral guests – forming pseudo-AEHB of type B1 – is thermodynamically viable (from −0.5 to −8.3 kcal mol−1). The interaction with small cations – forming AEHB of type A1 – could be kinetically possible (from +10 to +40 kcal mol−1 with a ~5 kcal mol−1 barrier for 9). The CSD so far appears fairly void of examples of such intermolecular (pseudo) anti-electrostatic interactions, possibly due to steric and electronic shielding provided by the intramolecular hydrogen bonds. One wonders, however, what other cations exist or can be synthesized that contain localized electron densities capable of establishing pseudo anti electrostatic interactions. We would predict that more such examples will be found in the future.Notes and references
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- H. J. Schneider and A. Yatsimirski, Principles and methods in supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- P. D. Beer, P. A. Gale and D. K. Smith, Supramolecular chemistry, Oxford University Press, Oxford, 1999 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- B. Kuhn, P. Mohr and M. Stahl, J. Med. Chem., 2010, 53, 2601–2611 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- A. Alsalme, M. Jaafar, X. Liu, F. Dielmann, F. Ekkehardt Hahn, K. Al-farhan and J. Reedijk, Polyhedron, 2015, 88, 1–5 CrossRef CAS PubMed.
- A. Martinsen and J. Songstad, Acta Chem. Scand., Ser. A, 1977, 31, 645–650 CrossRef CAS PubMed.
- W. E. Swartz, J. K. Ruff and D. M. Hercules, J. Am. Chem. Soc., 1972, 94, 5227–5229 CrossRef CAS.
- F. J. Lalor and S. Chaona, J. Organomet. Chem., 1988, 344, 163–165 CrossRef CAS.
- V. J. Sussman and J. E. Ellis, Chem. Commun., 2008, 5642–5644 RSC.
- W. N. Sit, S. M. Ng, K. Y. Kwong and C. P. Lau, J. Org. Chem., 2005, 70, 8583–8586 CrossRef CAS PubMed.
- G. R. Lewis and I. Dance, Dalton, 2000, 299–306 RSC.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- I. A. Guzei, J. S. Dougan and P. M. Treichel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1060–1061 CAS.
- C. Knapp and R. Uzun, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, O3186–U3794 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S3, computational methods and Cartesian coordinates of computed structures. See DOI: 10.1039/c5ce00352k |
| ‡ To underline the popularity of 1, one may note that at date the CSD lists some 1500 crystal structures that contain a [PPN]+ cation, and that the Reaxys database lists 4500 unique [PPN]+ containing substances. The popularity of 1 furthermore derives from its (synthetic) accessibility, its desirable solubility properties (soluble in most organic solvents and in hot water), its fairly hydrophobic character, and because [PPN]+ is chemically stable at ambient or slightly forcing conditions (see ref. 8–13). |
| § Notation 1a is used in the CSD, Reaxys, chemexper.com, emolecules.com and the websites of companies that sell [PPN]+ salts (Sigma-Aldrich and Alfa-Aeser). |
| ¶ The CSD (version 5.36, November 2014, including 1 update) contained 1457 CIFs with a [PPN]+ cation. When retrieving geometric parameters, data could be retrieved of only 1120 CIFs, containing a total of 1,315 [PPN]+ cations. |
| This journal is © The Royal Society of Chemistry 2015 |