DOI:
10.1039/C5CE00329F
(Paper)
CrystEngComm, 2015,
17, 3701-3707
Photoelectrochemical epitaxy of silver oxide clathrate Ag7O8M (M = NO3, HSO4) on rutile-type Nb-doped TiO2 single crystals
Received
13th February 2015
, Accepted 9th April 2015
First published on 24th April 2015
Abstract
Silver oxide clathrate Ag7O8M (M = NO3, HSO4) compounds were synthesized photoelectrochemically on rutile-type Nb-doped TiO2 single-crystal substrates. Epitaxial crystal growth was achieved for some clathrate compositions and substrate surface orientations, where commensurate growth is possible due to lattice matching between the pseudo lattice of the clathrate Ag6O8 cages and the TiO2 surface, similar to the well-known case of epitaxial C60 growth on single-crystal substrates. Particularly for the growth of Ag7O8NO3 on Nb-doped TiO2(110), fully (111)-oriented epitaxial crystallites without any other orientations were obtained. The selectivity for Ag7O8NO3 growth and the suppression of the formation of by-products, such as O2, were found to depend on the electrode potential. The highest selectivity was obtained at +0.2 V vs. Ag in a 0.01 M AgNO3 solution. An investigation of Ag7O8(MM′) (M = NO3, M′ = HSO4) depositions from solutions with different AgNO3 and Ag2SO4 mixing ratios showed that the growth of Ag7O8HSO4 is much faster than that of Ag7O8NO3. The process of incorporating monovalent M anions into the clathrate Ag6O8 cages was identified as the rate-limiting step for the growth of silver oxide clathrate compounds.
1. Introduction
Silver oxide clathrates with a general formula of Ag7O8M are a class of clathrate compounds where M is a monovalent molecular anion that is surrounded by Ag6O8 cages.1 The possible molecular anions in silver oxide clathrates include NO3, HSO4, ClO4, HF2, BF4, PF6, and HCO3,2–4 with some of the compounds exhibiting superconductivity at low temperatures, such as Ag7O8NO3 that has a Tc of 1.04 K.5,6 As is evident from the chemical formula, a Ag6O8 cage contains Ag ions in multiple valence states, including Ag+, Ag2+, and Ag3+. Silver oxide clathrate compounds can therefore be synthesized only under strongly oxidizing conditions; as a result, they are thermodynamically unstable and are likely to undergo self-decomposition into AgOx and other derivatives in air even at room temperature.7
So far, several different methods of synthesizing Ag7O8M have been reported, but depending on the concentration of Ag+, the pH, and the choice of counter anion, most methods lead to the formation of various simple binary silver oxides, such as Ag3O4, Ag2O4, Ag2O, and Ag2O2, in addition to the desired clathrate phase.7–11 Electrochemical oxidation in a 5 M AgNO3 aqueous solution with pH = 1.45 is usually preferred for the synthesis of Ag7O8NO3, using a Pt electrode that is biased beyond the Ag7O8NO3 equilibrium potential of +1.59 V vs. the standard hydrogen electrode potential (SHE).9 We have shown earlier that Ag7O8NO3 films can be synthesized photocatalytically under ultraviolet (UV) light irradiation in an aqueous AgNO3 solution either directly on Nb- or La-doped SrTiO3(001) single crystals.12 Ag7O8NO3 is metallic among many photo-oxidized products, and thus the photo-deposition process of Ag7O8NO3 on substrates that have been coated with an epitaxial ferroelectric PbTiO3 thin film can be used for an efficient domain switching to pole a large area because the metallic Ag7O8NO3 can stabilize the resultant polar surface.13–15 A common feature of all these surfaces is the presence of valence band levels that are derived predominantly from O 2p states and are thus more positive than the equilibrium potential. Similar light-induced deposition of Ag7O8NO3, though not photocatalytically, has been observed on GaAs substrates when synchrotron X-rays were used as a light source.16 More recently, we also demonstrated photoelectrochemical synthesis of Ag7O8NO3 on Nb-doped SrTiO3(001) single-crystal electrodes in a AgNO3 solution and discussed the role of the electrode potential in this photochemical process.17
Despite the availability of different synthesis methods, the crystal growth mechanism of silver oxide clathrate compounds is still very poorly understood. In particular, it is not clear if silver oxide clathrates can be epitaxially grown on appropriate lattice-matched single-crystal substrates. There is a growing interest in developing electrochemical room-temperature epitaxy techniques, often termed “electrochemical epitaxy”.18 However, the growth of silver oxide clathrate compounds is usually not epitaxial, even when single-crystal electrodes, like Pt(100), Pt(111), Pt(110)10 and Nb-doped SrTiO3(001) are used, probably due to the large difference in the crystal structures of the clathrate films and the single-crystal substrate electrodes. So far, only out-of-plane orientation control has been achieved in the growth of Ag7O8NO3 on Pb–Tl–O buffer layers11 but there is no conclusive data on epitaxial growth of any silver oxide clathrate compounds. If epitaxy for these materials can be achieved, a new guiding principle may be developed for epitaxial growth of materials that have very different crystal structures from the substrate.
In the present work, we therefore examine the possibility of epitaxial photoelectrochemical growth of Ag7O8NO3 and Ag7O8HSO4 on Nb-doped TiO2 single crystals under UV irradiation at a well-controlled electrode potential and compare the results with earlier studies that used Nb-doped SrTiO3(001) as a substrate. Since the clathrate formation reaction competes with water photo-oxidation, we take a careful look at the electrode potential dependence of the reaction selectivity against the formation of by-products, such as O2. The crystal growth mechanism of silver oxide clathrates is studied by investigating the growth behaviour of Ag7O8(MM′) (M = NO3, M′ = HSO4) in mixtures of AgNO3 and Ag2SO4 solutions at different mixing ratios. We identify the rate-limiting step for the growth of silver oxide Ag7O8M clathrate compounds and show that single-phase Ag7O8NO3 can be epitaxially grown on Nb-doped TiO2(110) surfaces.
2. Experimental section
We used single-crystal n-type 0.5 wt% Nb-doped rutile TiO2 substrates with (110), (101), (100), and (001) surface cuts from Shinkosha.19 Ohmic substrate contacts were made by coating the backside of the crystals with an In–Ga alloy and attaching copper wires with silver paste. All parts of the samples, except for the middle part of the front surface, were covered with epoxy resin to avoid leak currents in the electrochemical cell. The photoelectrochemical synthesis was performed at room temperature in a three-electrode electrochemical cell, using a Pt plate counter electrode and a silver wire reference electrode. The electrolytes used in this study included 0.01 M AgNO3 and 0.01 M Ag2SO4 aqueous solutions, and mixtures of AgNO3 and Ag2SO4 solutions with different mixing ratios. The photoelectrochemical reaction was carried out under UV light illumination from a high-pressure Hg lamp (Ushio, USH-150SC) at a constant flux of 28 mW cm−2 and the photocurrent was recorded at a sampling rate of 5 Hz. The light intensity was measured with an integrating light counter at 254 nm (Ushio, UIT-150-A). The electrode potential was set in the range between −0.5 and +1 V vs. Ag. After photoelectrochemical treatment, the deposited materials were first identified by θ–2θ X-ray diffraction (XRD) analysis (MAC Science SRA M18XHF22), followed by, if needed, X-ray Φ scan analysis (PANalytical X'Pert PRO MRD) in order to investigate the epitaxial in-plane orientation of the Ag7O8NO3 and Ag7O8HSO4 crystals with respect to the TiO2 substrate lattice. The morphology of the Ag7O8NO3 and Ag7O8HSO4 crystals was observed by scanning electron microscopy (SEM) and electron probe microanalysis (EPMA) was used to estimate the total volume and the composition of the deposition products (Hitachi S-4000). The photocatalytic reaction kinetics on Nb-doped TiO2 was investigated by measuring the relative selectivity σ between Ag7O8NO3 and other by-products, such as O2. The selectivity is defined as| |  | (1) |
where I(Ag/Ti) is the measured EPMA intensity ratio of Ag and Ti, and Qphoto is the total electrical charge conveyed per unit surface area by the photocurrent during the photoelectrochemical reaction.
3. Results and discussion
3.1. Growth of Ag7O8NO3 on Nb-doped TiO2 single crystals
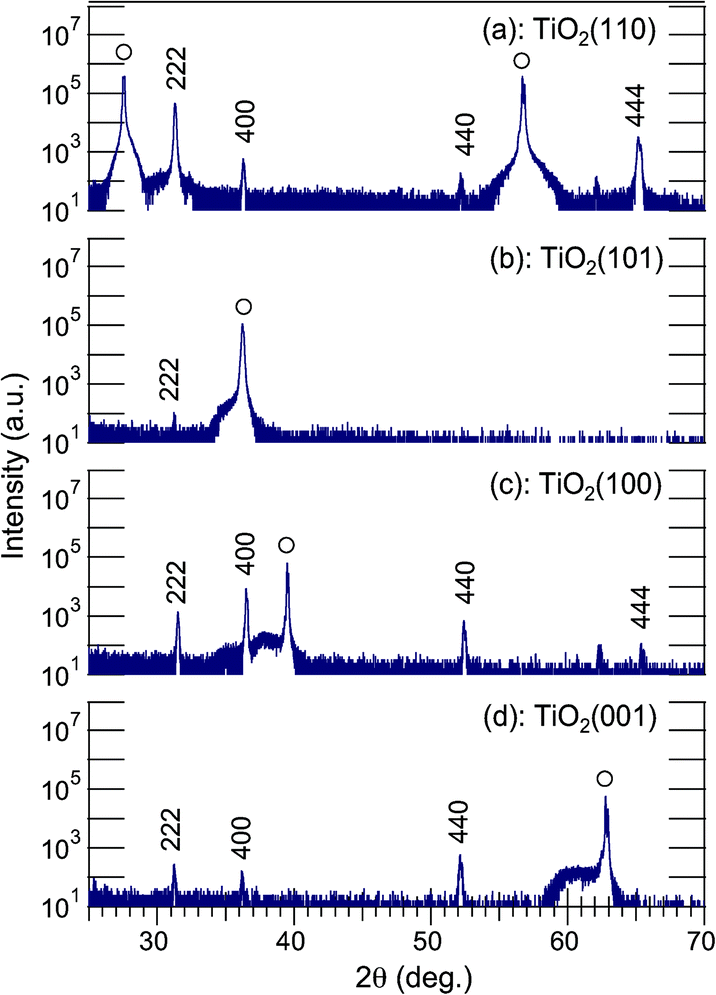
Fig. 1 shows θ–2θ XRD patterns of Ag7O8NO3 crystals that were photoelectrochemically deposited in 2 hours on Nb-doped TiO2 (110), (101), (100), and (001) single-crystal substrates in 0.01 M AgNO3 at an electrode potential of +0.3 V vs. Ag. Diffraction peaks were observed at 2θ = 31.28°, 36.27°, 52.26°, and 65.56°,20 revealing that only Ag7O8NO3 was deposited on the TiO2 substrates, irrespective of the crystal face. No other peaks that could be assigned to binary silver oxides were detected. All XRD patterns in Fig. 1 show reflections of (111)-oriented Ag7O8NO3 crystals. In particular, Fig. 1(a) shows that the 222 peak for the sample grown on TiO2(110) has the highest intensity, even though the deposition times are identical. As the FWHM values for the θ–2θ scans are not so different, the highest intensity of Ag7O8NO3 on TiO2(110) is due to its unique out-of-plane orientation.
 |
| | Fig. 1 A set of θ–2θ XRD patterns of photoelectrochemically deposited Ag7O8NO3 grown on Nb-doped TiO2 (a) (110), (b) (101), (c) (100), and (d) (001) single-crystal substrates in a 0.01 M AgNO3 solution at an electrode potential of +0.3 V vs. Ag. The deposition time was 2 h. All plots are shown on a logarithmic scale. | |
The crystal quality of Ag7O8NO3 was further analyzed by visualizing the grain structure by SEM, as shown in Fig. 2. It is clear that a few large faceted crystals and many small grains grew on TiO2(101), (100), and (001) substrates, suggestive of a polycrystalline nature of Ag7O8NO3 on these surfaces. Only Fig. 2(a) shows large hexagonal crystal platelets with a unique in-plane orientation growing on the TiO2(110) substrate. This implies epitaxial growth of (111)-oriented Ag7O8NO3 crystals on the TiO2(110) substrate. The thickness of each Ag7O8NO3 platelet could be estimated to be in a range of a few hundred nanometers from a cross-sectional SEM image (not shown). The epitaxial relationship was determined from the Φ scans recorded for the TiO2(111) and Ag7O8NO3(400) reflections, as shown in Fig. 3. There are six Ag7O8NO3(400) peaks and two TiO2(111) reflection peaks, which means that (111)-oriented Ag7O8NO3 grew epitaxially on TiO2(110) in two equivalent domains that are rotated by 180° with respect to each other. The in-plane relationship between the Ag7O8NO3 crystals and the TiO2(110) substrate is Ag7O8NO3(111)[1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0]//TiO2(110)[001], Ag7O8NO3(111)[112]//TiO2(110)[10] or Ag7O8NO3(111)[11
0]//TiO2(110)[001], Ag7O8NO3(111)[112]//TiO2(110)[10] or Ag7O8NO3(111)[11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ]//TiO2(110)[10].
]//TiO2(110)[10].
 |
| | Fig. 2 SEM images of the same set of Ag7O8NO3 samples for (a) (110), (b) (101), (c) (100), and (d) (001) single-crystal TiO2 substrates that were used for the XRD analysis in Fig. 1. | |
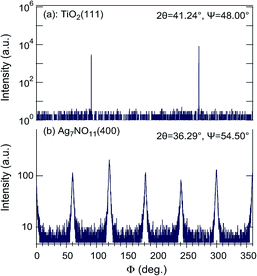 |
| | Fig. 3 X-ray Φ scans of (a) TiO2(111) and (b) Ag7O8NO3(400) reflections for a (111)-oriented Ag7O8NO3 sample epitaxially grown on a TiO2(110) substrate. Six clathrate peaks were observed from two equivalent domains rotated by 180° relative to each other. | |
The FWHM value of the (111)-oriented epitaxial Ag7O8NO3 film on TiO2(110), estimated from the Φ scan, is as large as 1.815°, suggesting that the crystallinity and mosaic spread, though these two parameters cannot be independently discussed here, are significant the in-plane direction. Nevertheless, the X-ray analysis results are consistent with the SEM observation of a unique in-plane orientation of the large crystallites in Fig. 2(a). The analysis of the Φ scan data, summarized in Table 1, revealed that some Ag7O8NO3 crystals grew epitaxially on other TiO2 crystal planes as well. For example, the data in Fig. 1 shows that (111)-oriented Ag7O8NO3 crystals were always present, regardless of the TiO2 substrate orientation. These photoelectrochemical deposition results are consistent with our earlier work on electrochemical deposition of clathrates on Nb:SrTiO3(001) substrates,12 and photodeposition on Nb- or La-doped SrTiO3(001)17 and on PbTiO3/Nb:SrTiO3(001).13–15 Ag7O8NO3 appears to have a crystal habit of preferentially growing along the (111) direction, but the selectivity for pure (111)-oriented growth is the highest for photoelectrochemical deposition on TiO2(110) substrates.
Table 1 Epitaxial relationship between TiO2 substrates and photoelectrochemically deposited Ag7O8NO3 crystals
| Substrate |
Ag7O8NO3 |
Ag7O8NO3 |
Ag7O8NO3 |
TiO2 |
|
|
Epitaxial orientation |
Non-epitaxial orientation |
In-plane direction |
In-plane direction |
| TiO2(110) |
(111) |
× |
[11] |
[10] |
|
|
|
|
[10] |
[001] |
| TiO2(101) |
(100) |
(111) |
[001] |
[010] |
|
|
|
|
[010] |
[10] |
| TiO2(100) |
(110) |
(111) |
[001] |
[001] |
|
|
|
(100) |
[10] |
[010] |
| TiO2(001) |
(110) |
(111) |
[001] |
[100] |
|
|
|
(100) |
[10] |
[010] |
3.2. Epitaxial growth model of Ag7O8NO3/Nb-doped TiO2 interfaces
We follow earlier work by Breyfogle et al.,11 who found that the growth orientation of Ag7O8NO3 crystals is tunable by selecting a suitable lattice-matched and textured Pb–Tl–O bottom layer, and consider the lattice mismatch between Ag7O8NO3 crystals and TiO2 substrates. Fig. 4(a) schematically illustrates the Ag7O8NO3 crystal structure,1 showing a network composed of large face-sharing rhombicuboctahedral Ag6O8 cages. Silver ions are located at the centre points of the shared cage faces and in the middle of the small cubic voids in between the cages. As is illustrated in Fig. 4(b), the dominant factor determining whether commensurate epitaxial Ag7O8NO3 crystal growth can occur on a substrate is the Ag6O8 cage size, which determines the lattice mismatch between the TiO2 surface and the clathrate cage pseudo lattice. This is analogous to the epitaxial growth of C60, which is another well-known molecular cage compound that can be epitaxially grown on single-crystal substrates.21,22 The in-plane distances of the cage pseudo lattice viewed along the (111) direction are 6.993 Å and 6.056 Å. We have taken a closer look at the structure of the unreconstructed TiO2(110) surface to understand the reason for the epitaxial growth of (111)-oriented Ag7O8NO3 crystals on TiO2(110) substrates. This surface consists of 5-fold coordinated titanium (Ti5c) atoms, in-plane oxygen atoms, and bridging oxygen atoms (Ob), as shown in Fig. 4(c). The bridging oxygen atoms are known to have a strong effect on the chemical reaction at the interface between the TiO2(110) surface and an electrolyte.23 The distance between adjacent bridging oxygen lines is 6.495 Å. Fig. 4(d) and (e) show two possible in-plane alignments of (111)-oriented Ag7O8NO3 crystals on the TiO2(110) surface. The black closed and open circles mark the positions of Ti atoms along the Ob rows and at the Ti5c sites, respectively, as illustrated in Fig. 4(f). The model in Fig. 4(d) clearly shows a smaller lattice mismatch than that in Fig. 4(e), and corresponds to the experimentally observed in-plane orientation as shown by the Φ scan in Fig. 3. In general, it is difficult to stabilize a triangular lattice, such as that of the (111)-oriented Ag7O8NO3 crystals, on a rectangular lattice. However, the tetragonal TiO2(110) surface has strong in-plane anisotropy, allowing the (111)-oriented Ag7O8NO3 crystals to grow epitaxially with only moderate in-plane mismatch.
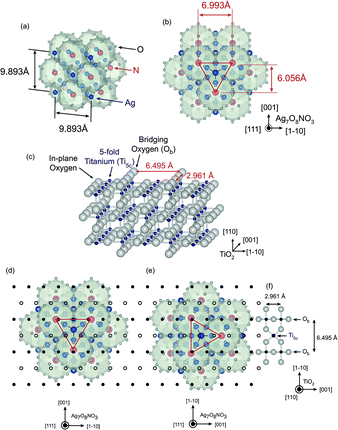 |
| | Fig. 4 (a) Schematic illustration of the Ag7O8NO3 crystal structure in a cubic system. Blue, red, and gray spheres denote Ag, N, and O, respectively. (b) A view of the (111) surface of a Ag7O8NO3 crystal. The red lines mark the outlines of the surface unit cell of the rhombicuboctahedral pseudo lattice. (c) Illustration of the TiO2(110) surface structure. The surface layer consists of bridging oxygens (Ob), 5-fold coordinated titanium atoms (Ti5c), and in-plane oxygens. (d, e) Two possible in-plane alignments of Ag7O8NO3 crystals on a TiO2(110) surface. The black open and closed circles mark the positions of the Ti5c and bridging titanium sites, respectively, on the underlying TiO2(110) surface. The configuration in (d) corresponds to the experimental results in Fig. 3, showing only a small lattice mismatch between the rhombicuboctahedral cage lattice and the bridging oxygens on the TiO2(110) surface. | |
3.3. Reaction selectivity against O2 by-products
The effect of the electrode potential on the growth of Ag7O8NO3 crystals on TiO2(110) was investigated. Fig. 5 shows the relative selectivity for the formation of Ag7O8NO3 (filled circles) as a function of the electrode potential, together with SEM images of crystals grown at electrode potentials of −0.15, +0.2, and +0.45 V. In principle, no deposition of Ag7O8NO3 (relative selectivity = 0) should occur at negative electrode potentials vs. Ag and, as expected, the inset SEM image for the −0.15 V sample shows almost no growth. It is thermodynamically more likely for Ag ions to be reduced to Ag metal at negative electrode potentials and this behaviour was experimentally observed for electrode potentials <−0.3 V, with Ag metal deposits growing on the TiO2 electrode instead of a clathrate phase. These data points are marked with open circles in Fig. 5. On the other hand, for positive electrode potentials, a gradual increase in selectivity was found up to a potential of ca. +0.2 V, at which point the selectivity reached a maximum and decreased again for higher potentials. The inset SEM images of the +0.2 V and +0.45 V samples show that the selectivity changes are clearly reflected in the morphology of the crystals. Based on our earlier work on the growth selectivity of Ag7O8NO3 on Nb-doped SrTiO3(001),17 it appears that the selectivity maximum at around +0.2 V is a common feature of silver oxide clathrates. A notable difference, however, is that on SrTiO3(001) substrates, the clathrate formation selectivity actually increased at negative electrode potentials vs. Ag, although the absolute amount of deposited Ag7O8NO3, of course, dropped in accordance with the decreasing total photocurrent. A strong electric field, on the order of 1 MV cm−1, has been shown to occur at the interface between an electrode and an electrolyte.24 Hot carriers that can be injected through quantized interface levels due to the strong electric fields25 may be partly responsible for the observed electrode potential dependence.
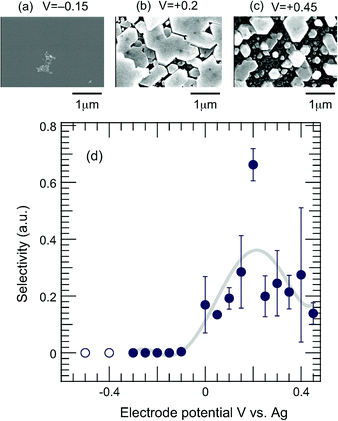 |
| | Fig. 5 (a) SEM images taken for samples grown at electrode potentials of (a) −0.15, (b) +0.2 and (c) +0.45 V, respectively. (d) Relative selectivity of the formation of Ag7O8NO3 (filled circles) as a function of electrode potential. Open circles correspond to the formation of Ag metal particles. | |
3.4. Comparison of the growth behaviours of Ag7O8NO3 and Ag7O8HSO4 on Nb-doped TiO2 single crystals
The photoelectrochemical synthesis of other types of silver oxide clathrates on Nb-doped TiO2 single crystals was also studied under experimental conditions that were identical except for the composition of the electrolyte. Ag7O8HSO4 crystals were successfully obtained in a 0.01 M Ag2SO4 solution, while the synthesis of Ag7O8ClO4 in a 0.01 M AgClO4 solution was possible, but the absolute amount was too small to be fully characterized. We therefore compare the growth of Ag7O8HSO4 with Ag7O8NO3. Fig. 6 shows a set of θ–2θ XRD patterns of Ag7O8HSO4 layers that were photoelectrochemically deposited in 2 hours on Nb-doped TiO2 (110), (101), (100), and (001) single-crystal substrates in 0.01 M Ag2SO4 at an electrode potential of +0.3 V vs. Ag. The diffraction peak positions were very close to those of Ag7O8NO3 because of the similar lattice constants (Ag7O8NO3: 0.989 nm, Ag7O8HSO4: 0.992 nm).7 No other peaks belonging to binary silver oxides or any other secondary phases could be detected. According to a Φ scan analysis (not shown), some Ag7O8HSO4 crystals always grew epitaxially, regardless of the TiO2 substrate orientation. The relative intensities of the Ag7O8HSO4 XRD peaks were different from those of Ag7O8NO3, which reflects a different out-of-plane orientation preference. The main difference was the presence of (100)-oriented grains in addition to the (111)-orientation, even on TiO2(110). A set of SEM images corresponding to the different substrate orientations are shown in Fig. 7. A comparison with the SEM images of Ag7O8NO3 crystals in Fig. 2 clearly shows that the size and areal density of Ag7O8HSO4 crystals are considerably larger than those of Ag7O8NO3 on all TiO2 crystal faces, but most remarkably on TiO2(110). However, it should be pointed out that since the concentration of Ag+ ions in a 0.01 M Ag2SO4 solution is two times higher than that in a 0.01 M AgNO3 solution, the growth behaviours of Ag7O8NO3 and Ag7O8HSO4 cannot be directly compared based on these two independent experiments if the growth rate depends on the concentration of Ag+ in the solution.
 |
| | Fig. 6 A set of θ−2θ XRD patterns of photoelectrochemically deposited Ag7O8HSO4 grown on Nb-doped TiO2 (a) (110), (b) (101), (c) (100), and (d) (001) single-crystal in a 0.01 M Ag7O8HSO4 solution at an electrode potential of +0.3 V vs. Ag in 2 h. All plots are shown on a logarithmic scale. | |
 |
| | Fig. 7 SEM images of the same set of Ag7O8HSO4 samples for (a) (110), (b) (101), (c) (100), and (d) (001) single-crystal TiO2 substrates that were used for the XRD analysis in Fig. 6. | |
In order to rule out this possibility, simultaneous photodeposition of Ag7O8(MM′) (M = NO3, M′ = HSO4) on TiO2(110) was tested by using mixtures of AgNO3 and Ag2SO4 solutions with different mixing ratios at an electrode potential of +0.3 V vs. Ag, where any unexpected effects that would appear even when all the experimental conditions are equal will be minimized. As shown by the series of SEM images in Fig. 8, no large crystallites were formed in a pure 0.01 M AgNO3 solution after a 2-hour deposition. As the concentration of Ag2SO4, cAg2SO4, in the mixed solution increased, the number of large crystals also increased, reaching a maximum size for cAg2SO4 = 0.01 and decreasing for higher Ag2SO4 concentrations. It is noted that a very small, e.g. 20%, increase of Ag+ in the solution by adding Ag2SO4 (cAg2SO4 is 0.001 M) is enough to cause a big change in the morphology and growth rate of the silver oxide clathrate compounds. According to our previous work, the concentration dependence of the growth rate and morphology is not very strong in the photocatalytic synthesis of Ag7O8NO3 on Nb:SrTiO3(001);12 the observed changes may not be simply due to the Ag+ concentration effect. As most of the volume of the deposited clathrate is contained in the large crystallites, the areal density of such crystals and their volume as a function of cAg2SO4 should be proportional to the total amount of photodeposited silver-oxide clathrate compounds, which can be measured from the EPMA Ag/Ti intensity ratio as shown in Fig. 9(a). On the other hand, the intensity ratio of the 400 and 222 XRD peaks, plotted as a function of cAg2SO4 in Fig. 9(b), shows a similar tendency to the Ag/Ti intensity ratio. The large crystals can thus be identified as (100)-oriented clathrate crystals. The large number of smaller plate-like crystals that can be seen in the inset of Fig. 7(a) is spread under and around the large (100) crystals and appears to have formed at the initial growth stage. These small crystallites are responsible for the weaker 222 and 444 XRD peaks in Fig. 6 and these crystallites are thus oriented along the (111) direction. As was pointed out earlier, among the silver oxide clathrates studied here, only Ag7O8HSO4 crystals can grow along the (100) direction on TiO2(110). We can therefore conclude that when mixtures of AgNO3 and Ag2SO4 solutions are used under the present experimental conditions, the dominant (100)-oriented Ag7O8(MM′) phase is either pure Ag7O8HSO4 or at least a Ag7O8HSO4-rich mixed clathrate. The preferential formation of Ag7O8HSO4 clathrate is probably caused by the faster growth rate of Ag7O8HSO4 crystals when compared to Ag7O8NO3 for both (100) and (111) orientations. As can be seen in the inset of Fig. 7(a), the average size of the (111)-oriented crystals of Ag7O8HSO4 is much smaller than that of Ag7O8NO3 crystals in Fig. 2(a). Both images were taken after a 2-hour photodeposition run under otherwise identical conditions. This size difference can be understood in terms of the kinetic effect, i.e., a higher growth rate leads to the formation of a larger number of smaller crystals. The small (111)-oriented crystallites grown from a mixed solution may contain Ag7O8NO3 or Ag7O8NO3-rich mixed clathrates as well as Ag7O8HSO4, but the volume fraction of the former must be very small due to the lower growth rate. This assumption is supported by the observation that, as shown in Fig. 9(c), the EPMA intensity ratio of S to Ag appears to be nearly constant, irrespective of the mixing ratio of the growth solution. Furthermore, the decrease of the EPMA Ag/Ti intensity ratio, i.e., the amount of Ag7O8HSO4 or Ag7O8HSO4-rich mixed clathrates deposited as a major product, at cAg2SO4 higher than 0.01 M, can be understood when considering the very narrow Ag+ ion concentration window of less than 0.005 M where Ag7O8HSO4 can be synthesized by electrochemical oxidation.2
 |
| | Fig. 8 A series of SEM images of silver oxide clathrate compounds photodeposited in solutions of AgNO3 and Ag2SO4 with different mixing ratios at an electrode potential of +0.3 V vs. Ag in 1 h. | |
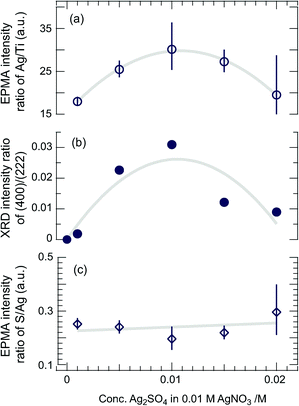 |
| | Fig. 9 (a) EPMA intensity ratio of Ag/Ti as a function of the Ag2SO4 concentration in the photodeposition solution. The Ag/Ti ratio is proportional to the total amount of photodeposited silver oxide clathrate compounds. (b) Intensity ratio of the (400) and (222) XRD peaks. (c) EPMA S/Ag intensity ratio as a function of cAg2SO4. | |
In simultaneous photodeposition of Ag7O8(MM′) (M = NO3, M′ = HSO4) the process that forms Ag6O8 cages through the oxidation of Ag+ ions is common for both materials and the rate-limiting step for the growth of silver oxide clathrate Ag7O8M compounds must therefore be a process that involves the monovalent anions. It is possible, for example, that the different incorporation kinetics of the anions inside the Ag6O8 cages are responsible for the observed differences in the Ag7O8NO3 and Ag7O8HSO4 growth rates.
4. Conclusion
We have succeeded in photoelectrochemically synthesizing silver oxide clathrate Ag7O8M (M = NO3, HSO4) compounds on Nb-doped rutile TiO2 single-crystal substrates. An epitaxial relationship between the clathrate crystals and TiO2 substrates was found for some combinations of the clathrate anions and substrate orientations, with a particularly clear case being the fully (111)-oriented growth of Ag7O8NO3 occurring on TiO2(110). This result has confirmed that commensurate, epitaxial lattice matching can occur between a single-crystal substrate lattice and the pseudo lattice of cages in an inorganic compound, such as the Ag7O8NO3 clathrate used in this work. This work shows that such a matching is not unique to organic cage compounds like C60. A certain electrode potential of ca. +0.2 V vs. Ag resulted in the preferential deposition of Ag7O8NO3 over other by-products. It was found that the process of incorporating monovalent anions, such as NO3− or HSO4−, into the Ag6O8 cages is the likely rate-limiting step in this photoelectrochemical silver oxide clathrate synthesis, leading to different growth rates of Ag7O8NO3 and Ag7O8HSO4.
Acknowledgements
This work was partially supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (no. 20360294, 25706022, 26105002) and the Asahi Glass Foundation. Some of the experiments were performed using the joint-use facilities of the Institute for Solid State Physics, the University of Tokyo. Crystal structure illustrations were generated with VESTA.26
Notes and references
- I. Náray-Szabó, G. Argay and P. Szabó, Acta Crystallogr., 1965, 19, 180–184 CrossRef.
- B. Standke and M. Jansen, J. Solid State Chem., 1987, 67, 278–284 CrossRef CAS.
- A. Parkin, S. F. Johnstone, A. R. Mount, S. Parsons, C. R. Pulham and D. Sanders, J. Appl. Crystallogr., 2004, 37, 312–318 CAS.
- M. Jansen and S. Vensky, Z. Naturforsch. B Chem. Sci., 2000, 55, 882–886 CAS.
- M. B. Robin, K. Andres, T. H. Geballe, N. A. Kuebler and D. B. McWhan, Phys. Rev. Lett., 1996, 17, 917–919 CrossRef.
- K. Kawashima, M. Ishii, H. Okabe, J. Akimitsu, M. Kriener, H. Takatsu, S. Yonezawa and Y. Maeno, J. Phys. Soc. Jpn., 2008, 77, 024707 CrossRef.
- G. I. N. Waterhouse, J. B. Metson and G. A. Bowmaker, Polyhedron, 2007, 26, 3310–3322 CrossRef CAS.
- C. H. Wong, T. H. Lu, C. N. Chen and T. J. Lee, J. Inorg. Nucl. Chem., 1972, 34, 3253–3257 CrossRef CAS.
- S. S. Djokic, J. Electrochem. Soc., 2004, 151, C359–364 CrossRef CAS.
- E. Michailova and A. Milchev, J. Appl. Electrochem., 1988, 18, 614–618 CrossRef.
- B. E. Breyfogle, R. J. Phillips and J. A. Switzer, Chem. Mater., 1992, 4, 1356–1360 CrossRef CAS.
- R. Tanaka, S. Takata, M. Katayama, R. Takahashi, J. K. Grepstad, T. Tybell and Y. Matsumoto, J. Electrochem. Soc., 2010, 157, E181–183 CrossRef CAS.
- R. Takahashi, J. K. Grepstad, T. Tybell and Y. Matsumoto, Appl. Phys. Lett., 2008, 92, 112901 CrossRef.
- R. Takahashi, M. Katayama, Ø. Dahl, J. K. Grepstad, Y. Matsumoto and T. Tybell, Appl. Phys. Lett., 2009, 94, 232901 CrossRef.
- R. Takahashi, M. Katayama, Ø. Dahl, J. K. Grepstad, Y. Matsumoto and T. Tybell, Appl. Phys. Lett., 2010, 97, 059903 CrossRef.
- Y. Sun, Y. Ren, D. R. Haeffner, J. D. Almer, J. D. Wang, W. Yang and T. T. Truong, Nano Lett., 2010, 10, 3747–3753 CrossRef CAS PubMed.
- R. Tanaka, S. Takata, R. Takahashi, J. K. Grepstad, T. Tybell and Y. Matsumoto, Electrochem. Solid-State Lett., 2012, 15, E19–22 CrossRef CAS.
- J. A. Switzer and G. Hodes, MRS Bull., 2010, 35, 743–752 CAS.
- Y. Yamamoto, K. Nakajima, T. Ohsawa, Y. Matsumoto and H. Koinuma, Jpn. J. Appl. Phys., Part 1, 2005, 40, L511–514 CrossRef.
- JCPDS Card no. 006–0434.
- M. Sakurai, H. Tada, K. Saiki and A. Koma, Jpn. J. Appl. Phys., 1991, 30, L1892–1894 CrossRef CAS.
- S. Yaginuma, K. Itaka, M. Haemori, M. Katayama, K. Ueno, T. Ohnishi, M. Lippmaa, Y. Matsumoto and H. Koinuma, Appl. Phys. Express, 2008, 1, 015005 CrossRef.
- C. L. Pang, R. Lindsay and G. Thornton, Chem. Soc. Rev., 2008, 37, 2328–2353 RSC.
- Y. Matsumoto, S. Takata, R. Tanaka and A. Hachiya, J. Appl. Phys., 2011, 109, 014112 CrossRef.
- D. S. Boudreaux, F. Williams and A. Nozik, J. Appl. Phys., 1980, 51, 2158–2163 CrossRef CAS.
- K Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence