
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The solid state, surface and morphological properties of p-aminobenzoic acid in terms of the strength and directionality of its intermolecular synthons†
I.
Rosbottom
a,
K. J.
Roberts
*a and
R.
Docherty
b
aInstitute of Particle Science and Engineering Institute for Process, Research and Development, School of Chemical and Process Engineering, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, England. E-mail: k.j.roberts@leeds.ac.uk
bPfizer Global Research & Development, Pharmaceutical R & D (IPC 612), Sandwich, Kent CT13 9NJ, England
First published on 18th June 2015
Abstract
Empirical force-field calculations utilising the atom–atom method were used to examine the strength, directionality and chemical state of the intermolecular interactions (synthons) present in the polymorphic forms (α and β) of p-aminobenzoic acid (pABA). This is set within the context of predicting the morphology of both forms in terms of the unsatisfied synthons at each growth surface. The α lattice energy was calculated to be −24.54 kcal mol−1 with the dominant intermolecular interactions found to consist of OH⋯O carboxylic acid H-bonding dimers and head to head π–π stacking interactions. The β lattice energy was calculated to be −22.73 kcal mol−1 and the dominant interactions found to consist of a 4-membered H-bonding ring made up of two identical NH⋯O and OH⋯N interactions, plus strong head to tail π–π stacking interactions. The NH2 group was calculated to contribute more to the β lattice energy than to the α, as it acts as a H-bonding donor and acceptor in the β structure, whilst acting solely as a donor in α. Conversely, the COOH group was found to contribute more strongly to the α lattice energy due to the formation of the OH⋯O H-bonds and also NH⋯O H-bonds, while the COOH group in the β structure forms only weaker O⋯HN and OH⋯N interactions. Morphological prediction of the β form gave greater resemblance to the experimental morphology compared to α. Surface chemistry analysis revealed that the strength, character and directionality of the synthons present varies in terms of their anisotropy between these two polymorphs. The strength and character of the unsaturated synthons exposed at the major surfaces of the α crystal were found to significantly vary, which results in a needle-like morphology. In contrast, the strength and character of the synthons exposed at the major surfaces of the β morphology were found to be much more similar, which results in the more equant morphology. Overall, this paper presents a synthonic, analytical approach which holistically links the molecular properties with the bulk and surface synthons, and through this rationalises their contributions to the growth and morphology of this organic crystalline system.
1. Introduction
The study of crystal surfaces from a structural perspective can be a powerful tool in predicting the optimum conditions for solution crystallisation. Understanding and controlling the shape of crystals can be a critical quality attribute in terms of enabling an active ingredient to be processed to a viable product.1 Morphology prediction and screening is therefore very important for industry to obtain desirable crystalline shapes for filtering and downstream processing. These predictions can further the knowledge of the growth mechanisms of a crystal surface and through this direct the final morphology of a crystalline particle. In addition, knowledge of the surface chemistry of crystals, as derived from morphological simulation, can provide a vital insight into the materials crystal/crystal aggregation properties and hence their formulation.2–4Early relationships of interplanar spacing to morphological importance, linked with lattice geometry, lead to the Bravais, Friedel, Donnay and Harker model (BFDH).5–9 This model is still used to identify the morphologically dominant faces (hkl). However, this model neglects the chemistry of the interactions present within the crystal, which for a molecular crystal are often dominated by isotropic van der Waals (vdW) interactions coupled with more directive hydrogen bonds (H-bonds). In particular, the BFDH approach doesn't effectively deal with these directional H-bonding interactions, and this has been demonstrated in the prediction of the morphology of β-succinic acid.10
In 1954, Hartmann and Perdok11 expanded Born's assumption that surface energy is directly related to chemical bond energies12 through the periodic bond chain (PBC) theory and introduced the term ‘attachment energy’ (Eatt). PBC's are strong stoichiometric intermolecular interactions that run in-plane with respect to a growing face and any face containing at least two of these can be assumed to facilitate stable, slow growth and therefore be present at the surface of an experimentally grown crystal.11 In turn, it is then assumed that the faces with a low attachment energy grow slowly and are therefore morphologically important. This idea was expanded for inorganic materials by Dowty with the use of the term ‘template fraction’, which describes the fraction of energy holding growth layers to substrates.13 Hartmann and Bennema showed that assuming the relative attachment energies are proportional to relative face specific crystal growth rates is a valid approximation for faces growing by either a Burton, Carbera and Frank (BCF) mechanism14 or birth and spread mechanism15 below the roughening transition temperature,16 and a robust method for deriving the morphology of molecular crystals from their internal structure and symmetry was demonstrated by Berkovich-Yellin.17 Building on this, computational methods for the routine prediction of the strength, directivity and dispersive nature of intermolecular interactions, together with their summation for predicting crystal lattice and surface attachment energies for morphological prediction were developed through the HABIT programme18 by Roberts and co-workers.19 In parallel to this, within the crystallographic, solid-state and supra-molecular chemistry community, the importance of hydrogen bonding interactions and graph theory20,21 was recognised, in particular their potential importance for understanding polymorphism22 and for crystal engineering the design of materials.23 More recently, the concepts as to how the shape of molecules, together with the directionality and strength of their interactions, can strongly influence the physical properties of crystalline materials have been reviewed by Desiraju.24
The attachment energy model relies, to some extent, on the interactions between the molecules interacting at the crystal surface and the solution being almost the same as the bulk interactions of the crystal, and this proportionality concept has been proved to be a good approximation for a variety of studies.18,25–27 Calculating the relative strength of the intermolecular interactions using atomistic force-fields derived from empirical data, through the atom–atom method,28 can provide good prediction of the physical properties of molecular crystals.29–33 However, more recent publications highlighted the option to optimise these potentials against ab initio data and crystal structures to create an interatomic interaction potential which is specific to each crystalline system.34–36
The most significant drawback of the attachment energy model is that it fails to take into account external conditions such as temperature and surrounding solvent interactions with a crystal surface. More recent models attempt to account for the effect as to how a surface de-solvates prior to solute incorporation by calculating solvent binding energies and applying models that considers the size of the surface and the concentration of the solution.37–39 Further models have also attempted to predict the effect of different growth mechanisms on the attachment energy of a given surface,40 but these models have yet to be proven effective over a required number of crystalline systems and environments. Molecular dynamics simulations can provide valuable information in predicting the solvent adsorption at a surface and how this affects crystal growth41–43 but the downside of this approach is that these calculations are often time consuming and require a high amount of molecular modelling expertise.
In this study, a holistic method is presented, which can be relatively easily reproduced by less specialised computational scientists, for examining crystal morphology by analysing the molecular, crystal structure and morphological properties of a model organic system, i.e. the α and β forms of p-amino benzoic acid (pABA). To achieve this, the conformational space of the molecule and the intermolecular hydrogen bonding lengths are compared to similar crystal structures present in the Cambridge Structural Database (CSD).44 The lattice energies are calculated and the individual atom and functional group contributions to the lattice energy are compared between the α and β polymorphs. The bulk intermolecular interaction strengths are calculated and ranked, and the dominating interatomic interaction type established. The morphology of the two polymorphs is predicted assuming monomer attachment to each crystal surface. In addition, the morphology of α is predicted assuming a carboxylic acid dimer is the attaching crystal growth unit. Finally, the surface chemistry of both forms is analysed by establishing the key intermolecular interactions that contribute to the attachment energies of the morphologically important surfaces. This is summarised in Scheme 1.
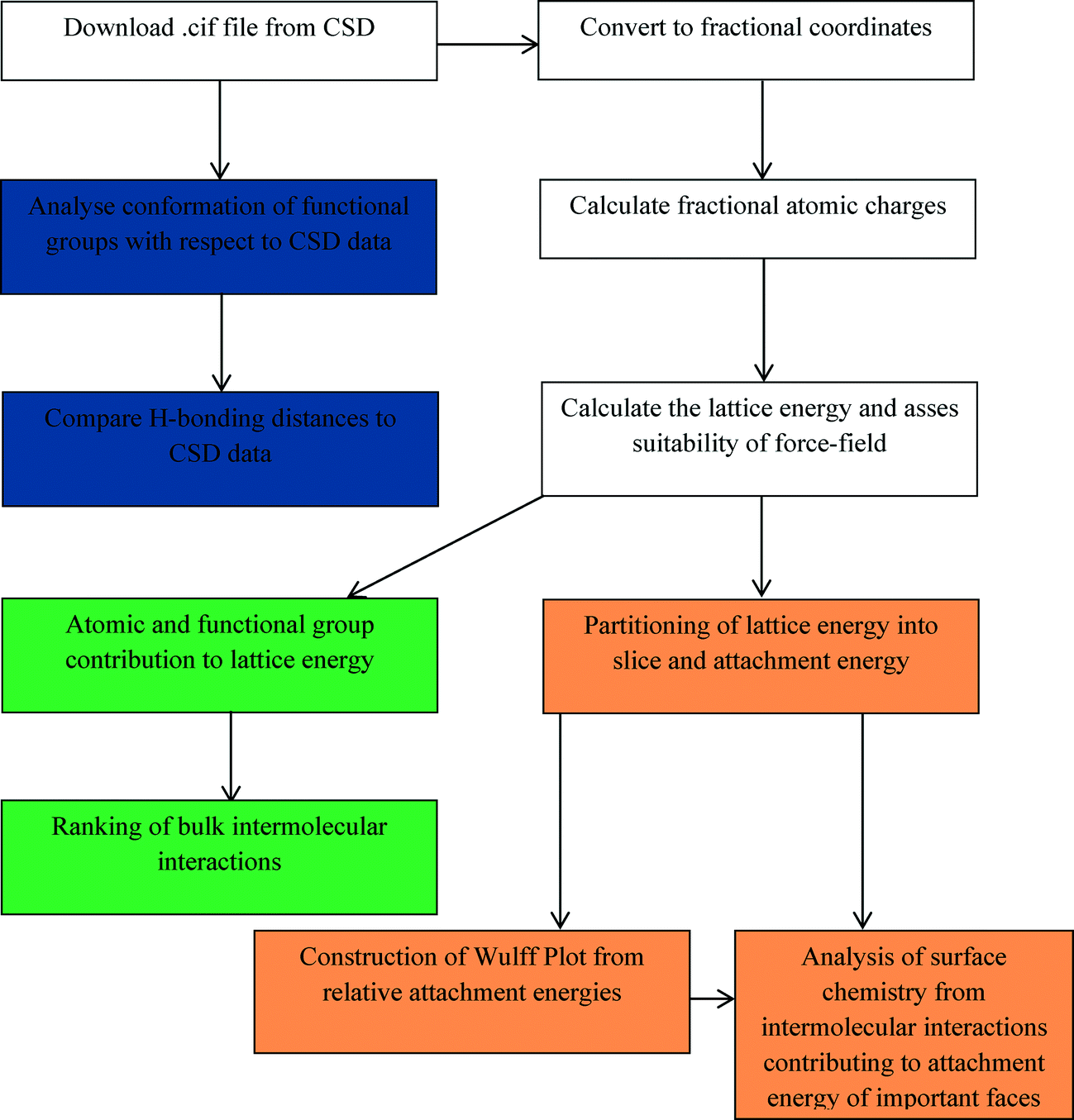 | ||
| Scheme 1 Flow diagram of how the data was obtained for each stage of the morphological analysis. Structure file preparation, charge and initial calculations shown in white. CSD data analysis shown in blue. Bulk intermolecular interaction data shown in green. Surface and morphological data shown in yellow. | ||
2. Synthonic modelling
Synthonic modelling draws upon the molecular and crystallographic structure of a material and involves the calculation of the strength, directionality and chemical state associated with pairwise intermolecular interactions (synthons) within a crystal structure using the atom–atom approach.45 This information can be used to predict physical and chemical properties of the crystal such as shape, cluster stability, mechanical properties etc.2.1. Bulk intrinsic synthons
Summation of intermolecular interactions can be used to calculate a molar lattice energy. The intermolecular interactions can be ranked by strength or distance and outputted for analysis, along with the atom by atom contribution to the lattice energy summed over the asymmetric unit. Further analysis of the lattice energy as a function of limiting radius can be utilised to reveal information on the initial coordination sphere of a crystal structure involved in nucleation and the early stages of the growth process. In turn, this reveals the intermolecular interactions that need to be saturated in the bulk crystal chemistry for lattice energy convergence. The bulk saturated intermolecular interactions can be referred to as ‘intrinsic synthons’.2.2. Surface extrinsic synthons
The lattice energy can be partitioned into slice and attachment energy per surface as defined by specified Miller planes (hkl). The magnitude of the attachment energy per face can be taken to predict the relative growth rate and hence morphological importance of the surface. Face-specific information, such as which of the bulk intrinsic synthons are unsaturated (broken) due to surface termination can be outputted for analysis. These unsaturated interactions are known as ‘extrinsic synthons’. The nature and strength of these interactions, combined with molecular scale modelling of the predicted surfaces using molecular visualisation software, can be used to reveal detailed information on the surface chemistry of the important faces and how e.g. the solute and solvent molecules potentially bind and incorporate into the lattice. This information can then be directly related to the relative growth rate and size of the crystal face.3. Materials and methods
3.1. Materials
This study focuses on the α and β forms of pABA. The crystal structures of these forms (AMBNAC01 and AMBNAC06) are taken from the CSD.44The molecular structure of pABA consists of a phenyl ring with a carboxylic acid group and amino group in the para position (Fig. 1).
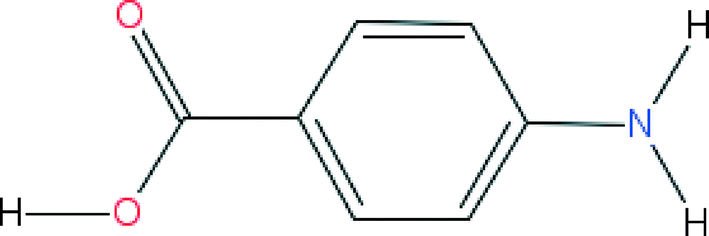 | ||
| Fig. 1 The molecular structure of pABA. Functionality consists of three hydrogen bonding donors (amino hydrogens and carboxylic acid hydrogen) and three hydrogen bonding acceptors (amino nitrogen and carboxylic acid oxygens). | ||
pABA is known to crystallise in two well-characterised polymorphs, α46 and β.47 A recent study has revealed a third polymorph, this has an orthorhombic crystal structure, which was found by crystallising from aqueous solutions containing pABA and selenous acid,48 but this latter structure was not considered here. Both the α and β crystal structures are monoclinic with a P21/n space group. The α form crystallises with two molecules in the asymmetric unit and eight molecules in the unit cell with dimensions a = 18.55 Å, b = 3.86 Å, c = 18.64 Å and β = 93.56°. The β form crystallises with one molecule in the asymmetric unit and four molecules in the unit cell with dimensions a = 6.27 Å, b = 8.58 Å, c = 12.36 Å and β = 100.13°.
Fig. 2a shows that the packing of the α form is found to be dominated by the formation of non-equivalent OH⋯O H-bonding dimers between neighbouring carboxylic acid groups. In addition, the pABA molecules are found to form a head to head stacking motif in the b direction creating π–π stacking interactions. Fig. 2b shows that the packing of the β form is found to be dominated by a 4 membered H-bonding ring motif consisting of alternating OH⋯N and NH⋯O H-bonds. In addition, the pABA molecules are also found to form head to tail stacking motifs creating π–π stacking interactions.
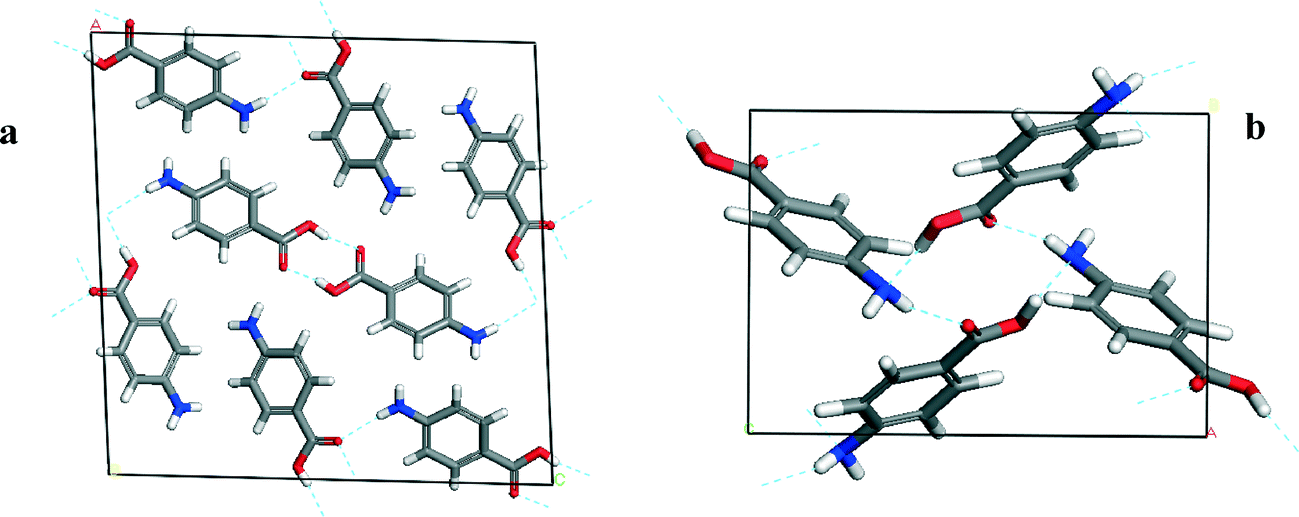 | ||
| Fig. 2 Details of unit cells of α-pABA (a) and β-pABA (b) displaying their associated packing motifs. α packing consists of COOH⋯HOOC H-bonding dimers and NH⋯O H-bonds. β packing consists of a 4 membered H-bonding ring with identical OH⋯N and NH⋯O interactions. | ||
The α form of pABA is observed to crystallise in a needle-like morphology, while the β form has a more equant morphology.49 The α morphology is of particular significance due to the associated issues with controlling the chemical process behaviour of needle-like crystals in pharmaceutical and fine chemical industries. Hence, there is a desire to control the shape of crystalline particles and recent studies have highlighted the challenge of predicting and experimentally controlling the morphology of needle-like crystals.39,50,51 Therefore there is a clear need to better understand the growth of these crystals from a molecular standpoint.
3.2. Computational methods
The intermolecular interactions were calculated using the Momany force-field29 containing a Lennard-Jones potential for the vdW interactions, a specific 10–12 H-bonding potential and a Coulombic term with respect to the electrostatic interactions. This force-field has previously shown good correlation of calculated and experimental lattice energies of crystalline materials containing both H-bonding and π-orbital functionality.5,10,55
For the calculations of the electrostatic interactions, the Restrained Electrostatic Potential (RESP) charges based on ab initio MP2/aug-cc-pvtz theory derived from the Antechamber within Ambertools were calculated.56 The single molecule of pABA was optimized at the MP2/aug-cc-pvtz level and the optimized structure's electrostatic potential was calculated with Gaussian09.57 The ESP data created from Gaussian is converted into a RESP format in Antechamber and finally the RESP fit is applied with Ambechamber to calculate the actual RESP charges.
From the intermolecular energy calculations, the lattice energy was calculated (Ecr). The suitability of the potential was evaluated by comparison with the sublimation enthalpy (ΔHs), given by eqn (1):
| Ecr = ΔHs − 2RT | (1) |
| Ecr = Esl + Eatt | (2) |
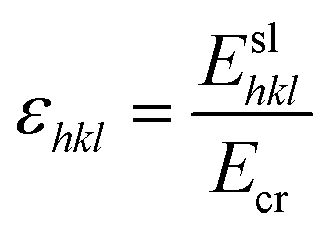 | (3) |
The nomenclature used to label the interactions identified the strongest interaction as capital A (i.e. alphabetically), with α or β referring to the polymorphic form and 1 or 2 relating to the different crystallographically independent molecules within the asymmetric unit (α-structure). The packing diagrams were annotated to show some of the strongest interactions with two labels on, e.g. Dα1/Dα2, indicates the intermolecular interactions between the two molecules within the asymmetric unit.
This basic nomenclature was also used to characterise the surface–specific interactions at a given surface (hkl).
3.3 Experimental methods
4. Results and discussion
Table 1 summarises the examination of the molecular structure, crystal chemistry, CSD analysis and key intermolecular interactions, highlighting how they contribute to the lattice energy. The detailed analysis of these results is presented in sections 4.1–4.4.| α | Attribute | β |
|---|---|---|
| Crystallographic data | ||
| 18.55 | a (Å) | 6.27 |
| 3.86 | b (Å) | 8.58 |
| 18.64 | c (Å) | 12.36 |
| 93.56 | β (°) | 100.13 |
| P21/n | Space group | P21/n |
| 4, 2 | Z, Z′ | 4, 1 |
| 1332.319/166.54 | Cell/molecular volume (Å3) | 655.907/163.98 |
| 1.373 | Density (g Å−3) | 1.389 |
| Solid form informatics | ||
| Pyramidal | NH2 geometry | Pyramidal |
| OH⋯O dimers and NH⋯O | H-bonding network | OH⋯N and NH⋯O4 membered ring |
| Head to head ~3.38 | π–π stacking interaction (Å) | Head to tail ~4.0 |
| 1.99 & 2.00 | OH⋯O H-bonding distance (Å) | N/A |
| N/A | OH⋯N H-bonding distance (Å) | 2.06 |
| 2.05 | NH⋯O H-bonding distance (Å) | 2.19 |
| Lattice energy contributions | ||
| −24.51 | Lattice energy (kcal mol−1) | −22.73 |
| 15.33%/16.8% | NH2 (monomer attachment)/NH2 (carboxylic acid dimer attachment) | 23.8% |
| 39.81%/59.9% | C6H4 (monomer attachment)/C6H4 (carboxylic acid dimer attachment) | 42.5% |
| 44.86%/23.3% | COOH (monomer attachment)/COOH(carboxylic acid dimer attachment) | 33.7% |
| 7 | Number of key interactions (above 0.9 kcal mol−1) | 8 |
| 71 | Percentage of lattice energy from key interactions | 75 |
| 30 | Molecular cluster size for lattice energy convergence | 35 |
4.1 Conformational analysis
The torsion angles of the functional groups of published structures of both polymorphs are shown in Table 2.| COOH torsion angle (°) | Polymorph | Ref. code | Lead author | Year published | C–C–N–H torsion angle (°) |
|---|---|---|---|---|---|
| 2.865, 1.172 | α | AMBNAC 01 | Lai | 1967 | 12.03, 11.17 |
| 0.866, 0.852 | α | AMBNAC 06 | Athimoolan | 2007 | 0.024, 0.008 |
| 10.397 | β | AMBNAC 04 | Gracin | 2005 | 26.844 |
Table 2 shows the COOH group of the α structures were found to be almost completely planar with respect to the phenyl ring, while the β structure was found to have a torsion angle of around 10°. The formation of the OH⋯O H-bonding dimers that run planar to the phenyl ring appears to hold the COOH planar with respect to the phenyl ring, while the NH⋯O and OH⋯N interactions in the β form are not directed planar to the ring and hence the torsion angle is around 10° away from the plane of the ring. Fig. 3 reveals that the majority of crystal structures with a COOH group attached to a phenyl ring in the CSD are close to planar.
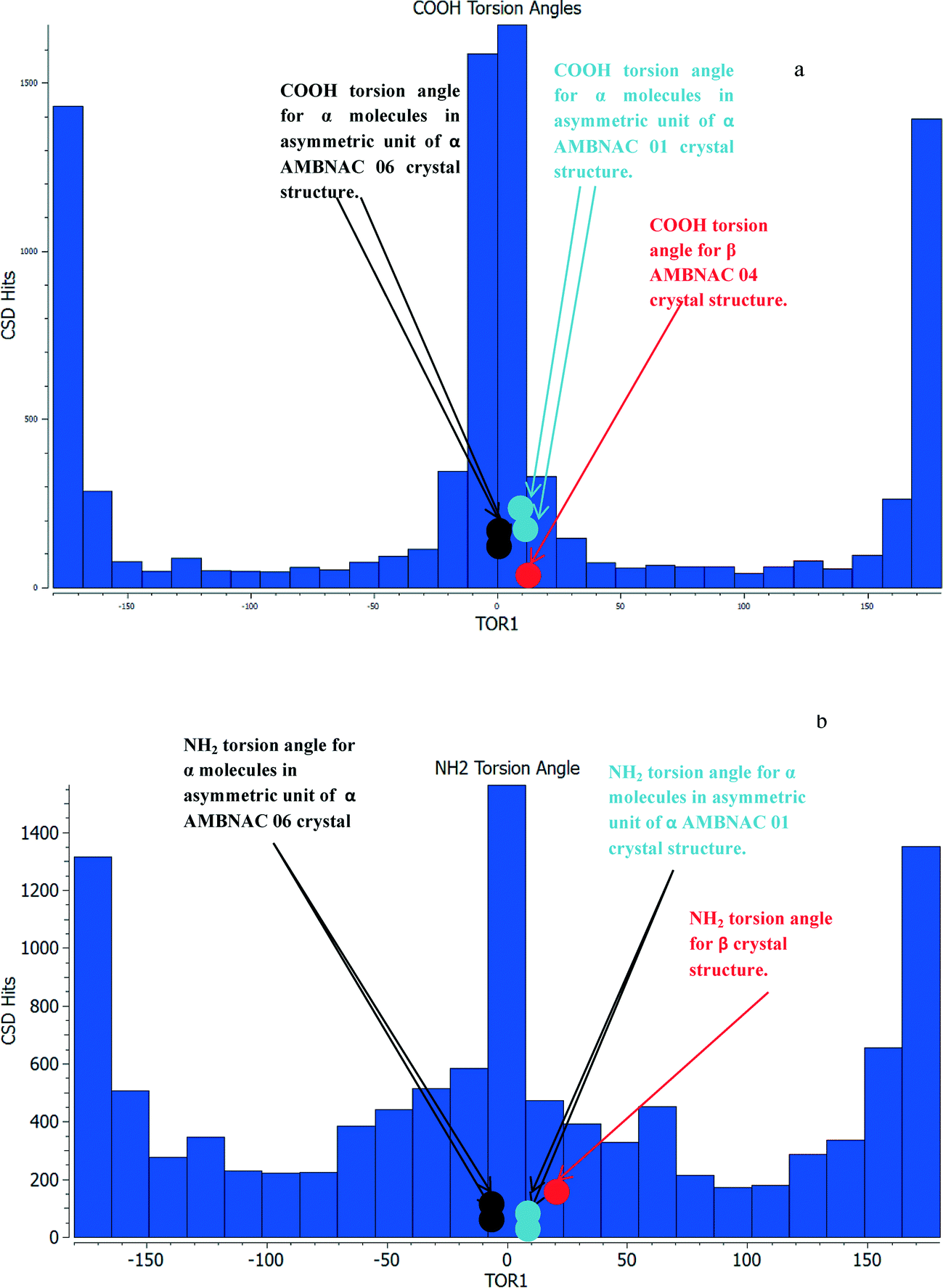 | ||
| Fig. 3 (a) Torsion angles of COOH groups attached to a phenyl ring found in the CSD. Reveals that vast majority of the groups are planar or very close to planar with respect to the phenyl ring. (b) Histogram of the amount of hits from the CSD as a function of torsion angles of NH2 hydrogens from the phenyl ring from planar to 45°. Majority of hits have a planar NH2 similar to that of the AMBNAC06 α structure, spread of hits up to around 40° torsion broadly similar. | ||
The conformation of the NH2 group is of some interest as the two structures published in the CSD have different conformations, the structure published by Lai in 1967 showing a torsion angle of around 12° from the plane of the phenyl ring, while the more recent structure from Athimoolan suggests that it is planar.
Fig. 3b shows the majority of structures were found to have a close to planar NH2. The spread of hits at more pyramidal angles was found to be fairly level all the way up to 45°. Comparison of the calculated lattice energies, ranking of intermolecular interactions and attachment energies showed little difference between the planar and pyramidal structures for the major interactions of α-pABA (section S4, ESI†). This analysis, together with recently published work by Schroeder et al.61 suggesting that the NH2 in the α structure may be pyramidal, resulted in the crystal structure with the pyramidal NH2 group published by Lai et al. (AMBNAC01) being chosen for this study.
4.2 Lattice energy calculations
The lattice energy for each structure was calculated and compared to experimentally measured sublimation enthalpies. The experimental lattice energy, as calculated from eqn (4) and based on published sublimation enthalpy data for α-pABA was found to be between 26.77 kcal mol−1 (ref. 62) measured at 373 K using a torsion effusion method, and 27.25 kcal mol−1 (ref. 63) also measured at 373 K using a calorimetric method. The calculated lattice energy for the α-form was found to provide a good match to sublimation enthalpy data, hence suggesting that the Momany force-field was a sensible choice for calculating the strength of the intermolecular interactions within the crystal structures of pABA. There are no known published values for the sublimation enthalpies of β-pABA.Table 1 demonstrates how the intermolecular packing for each polymorph affects the respective contribution of the functional groups to the lattice energy. In this, the NH2 group was found to contribute significantly more to the lattice energy of the β structure than α, as in the β structure the NH2 acts as a H-bonding donor and acceptor, while in the α structure the NH2 acts only as a donor. The strong H-bonds formed between the COOH groups in the α structure consequently give a larger contribution from the COOH group in α than β. Table 1 also compares the functional group contribution to the lattice energy of the α structure based on both monomeric and dimeric building blocks. The loss of the intermolecular energy from the carboxylic acid group was found to result in the major contributor to the lattice energy becoming the phenyl ring group, with the π–π stacking interactions becoming, in terms of interaction energy, the most important synthons in the crystal structure.
The individual atom–atom contributions to the lattice energy are given in Fig. 4.
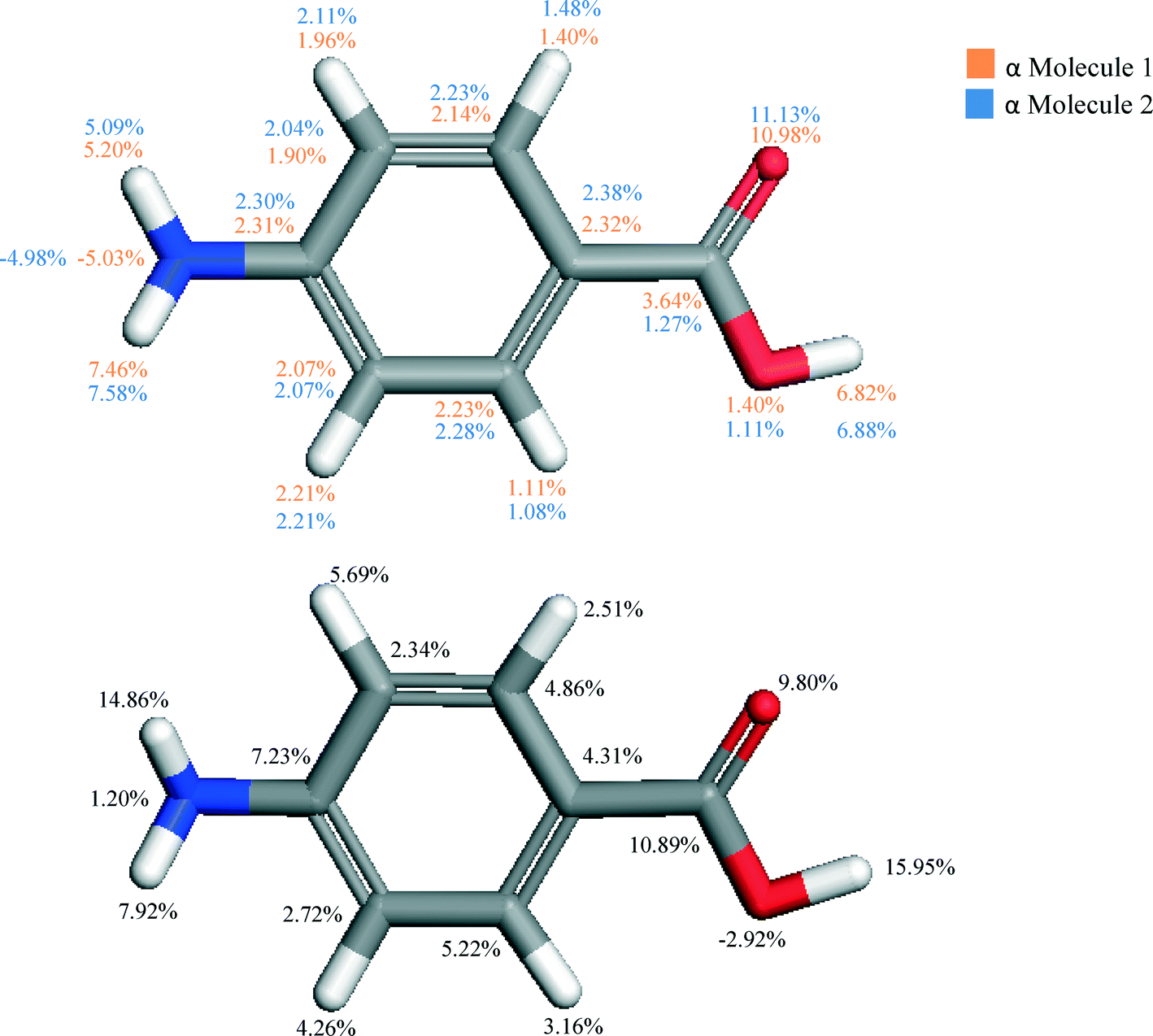 | ||
| Fig. 4 Molecular structures of pABA highlighting the percentage contribution of the lattice energy of α (top) and β (bottom) per atom. Contributions of the two molecules of α broadly similar due to the similar environments, whereas the β form shows increased importance of the amino hydrogens and hydroxyl hydrogen, and decrease in contribution from carbonyl oxygen. Reflects COOH dimer formation in α and NH2 donor and acceptor capabilities in β. | ||
The atomistic contributions to the lattice energy from the asymmetric unit shown in Fig. 4 for both polymorphs reflect the intermolecular packing of both structures. The β form was found to show a significantly increased contribution from the amino nitrogen and hydrogens when compared to that for the α form, as the amino functional group acts as both a H-bonding donor and acceptor to form the primary H-bonding synthons of the β structure. Conversely, it is interesting to note the significant increase in contribution from the hydroxyl hydrogen in the α form compared to that of the β structure. This reflects the much greater strength of the OH⋯O H-bonds compared to the OH⋯N H-bonds in β, and how important they are in formation of the α crystal structure.
4.3 Bulk intrinsic synthons
To understand which interactions need to be saturated for lattice energy convergence, the strongest interactions in each polymorph were evaluated.Fig. 5 and Table 3 shows that the strongest interactions in the α form were found to be the H-bonding dimers between the carboxylic acid groups, contributing approximately 23% of the calculated lattice energy. Interestingly bond Cα, which involves the more isotropic vdW forces due to π–π interactions between close packed molecules of pABA stacking along the b-axis, was found to contribute approximately 22% of the total calculated lattice energy. Fig. 6 and Table 4 shows the contributions from the strongest interactions in the β-form is much more evenly spread in 3-dimensions with respect to the α-form. The top four interactions all contribute above 10% of the lattice energy. Of these, the two most important interactions (Aβ and Cβ) which each were found to contribute around 22% to the lattice energy, are the OH⋯N H-bond and the polar interactions between the two COOH head groups.
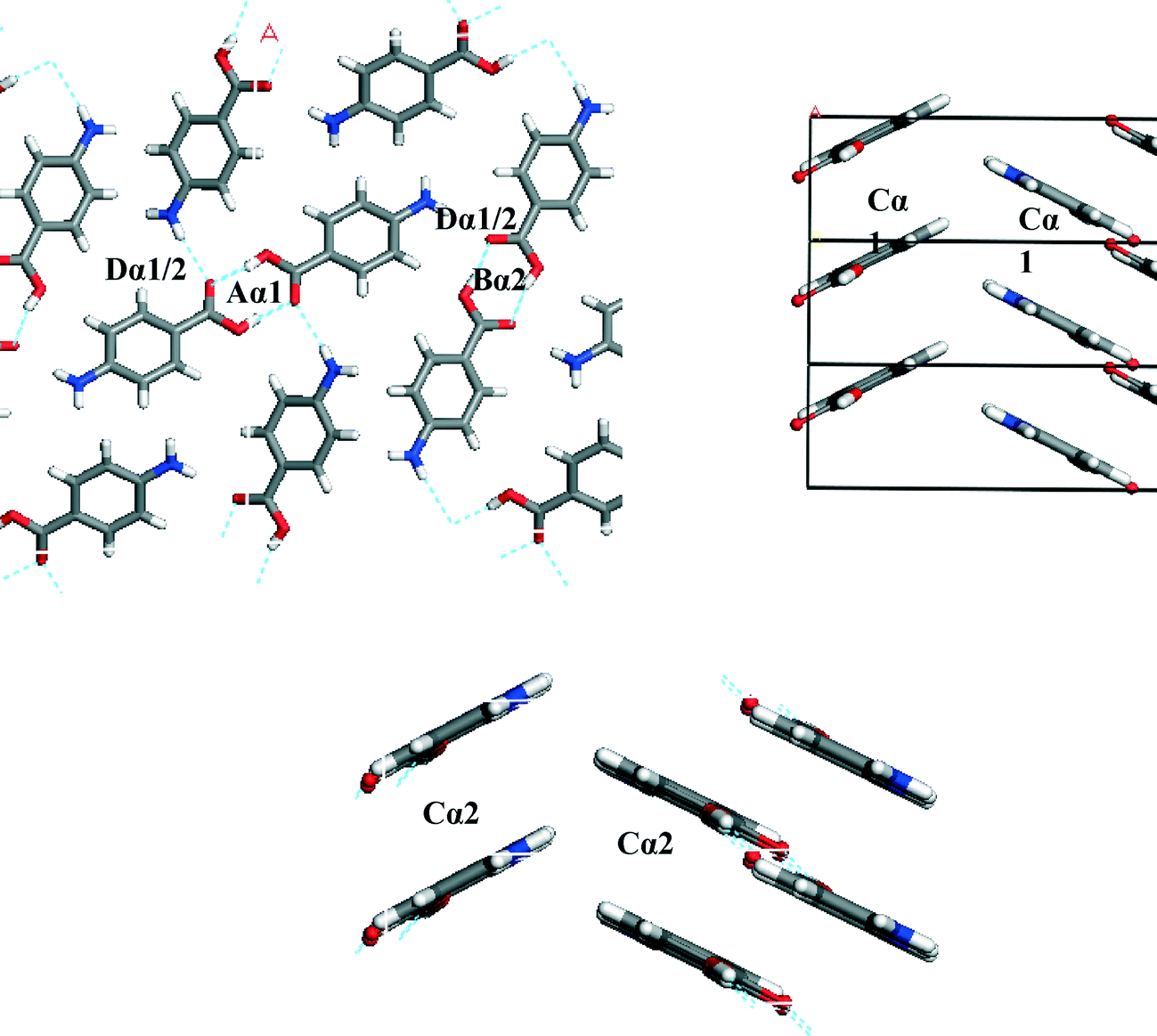 | ||
| Fig. 5 Strongest interactions of α-pABA labelled on the α packing diagram. Combination of H-bonding interactions (A, B and D) and π–π stacking (C) indicating that both types of interactions are important in the formation of α. Interactions tabulated Table 3a and b. | ||
| a | ||||||||
|---|---|---|---|---|---|---|---|---|
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) | Percentage contribution to lattice energy | Dominating interatomic interaction type | COOH % contribution to interaction | C6H4 % contribution to interaction | NH2 % contribution to interaction |
| Aα1 | 1 | 8.2 | −5.7 | 23.1 | H-bond | 96.4 | 4.0 | −0.4 |
| Cα1 | 2 | 3.9 | −5.4 | 21.8 | π–π stacking | 14.5 | 72.6 | 13.0 |
| Dα1 | 1 | 7.9 | −2.3 | 9.3 | H-bond | 41.7 | 20.7 | 37.6 |
| Eα1 | 1 | 7.8 | −2.0 | 8.2 | H-bond | 38.8 | 26.1 | 35.1 |
| Fα1 | 2 | 8.0 | −2.3 | 9.2 | vdW | 79.90 | 21.01 | −0.92 |
| Total | 18.7 | 71.6 | ||||||
| b | ||||||||
|---|---|---|---|---|---|---|---|---|
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) | Percentage contribution to lattice energy | Dominating interatomic interaction type | COOH % contribution to interaction | C6H4 % contribution to interaction | NH2 % contribution to interaction |
| Bα2 | 1 | 8.3 | −5.6 | 22.9 | H-bond | 96.7 | 3.6 | −0.4 |
| Cα2 | 2 | 3.9 | −5.3 | 21.7 | π–π stacking | 14.5 | 72.6 | 13.0 |
| Dα2 | 1 | 7.9 | −2.3 | 9.3 | H-bond | 41.7 | 20.7 | 37.6 |
| Eα2 | 1 | 7.8 | −1.2 | 4.9 | H-bond | 38.8 | 26.1 | 35.1 |
| Fα2 | 2 | 6.9 | −1.9 | 7.7 | vdW | 80.8 | 20.0 | −0.9 |
| Total | −16.3 | 66.5 | ||||||
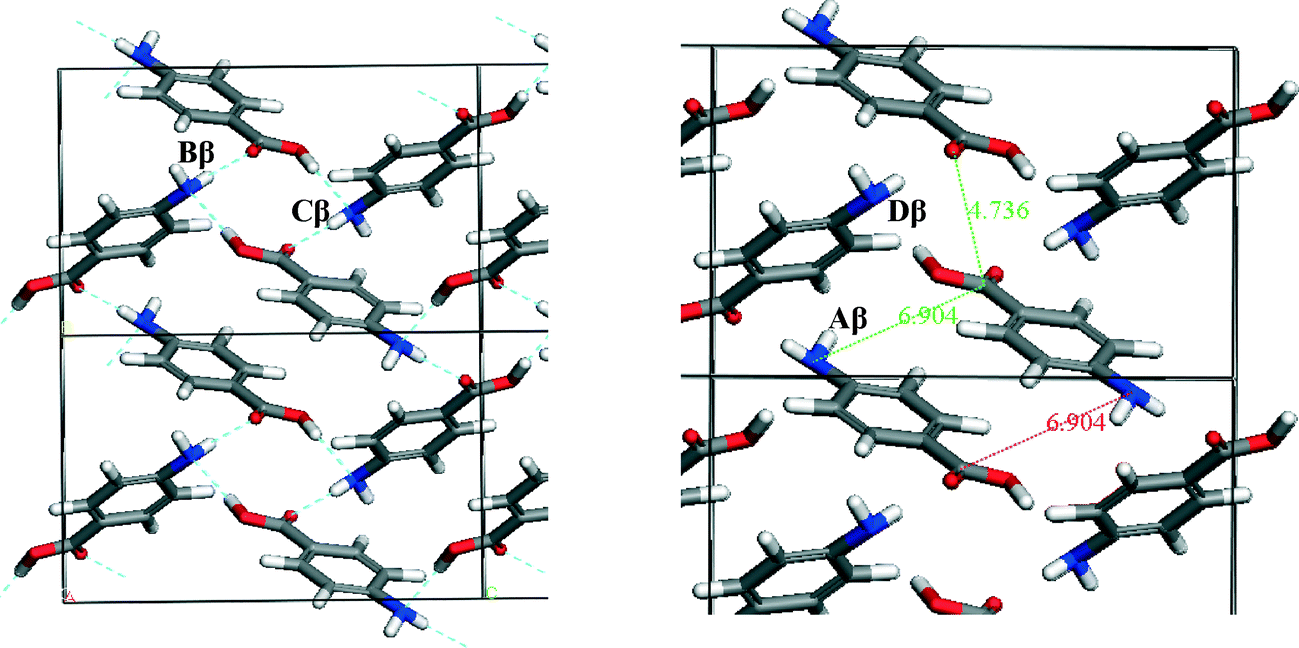 | ||
| Fig. 6 Strongest interactions of β-pABA labelled on the β packing diagram. Combination of H-bonding ring interactions (B and D) and offset stacking with interactions between the NH2 and COOH groups (A and C) indicating that both types of interactions are important in the formation of β. Interactions tabulated in Table 4. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) | % Contribution to latt eng | Dominating interatomic interaction type | COOH % contribution to interaction | C6H4 % contribution to interaction | NH2 % contribution to interaction |
|---|---|---|---|---|---|---|---|---|
| Aβ | 1 | 4.17 | −2.57 | 11.9 | π–π stacking | 33.3 | 65.2 | 1.5 |
| Bβ | 2 | 8.11 | −2.45 | 22.7 | H-bond | 46.5 | 15.7 | 37.7 |
| Cβ | 2 | 5.73 | −2.39 | 22.2 | vdW | 37.8 | 34.0 | 28.2 |
| Dβ | 2 | 6.74 | −1.46 | 13.6 | vdW | 9.1 | 44.5 | 46.4 |
| Eβ | 1 | 6.53 | −1.01 | 4.4 | vdW | 15.7 | 80.21 | 4.1 |
| Total | −16.18 | 74.8 |
The functional group contribution analysis with respect to the lattice energy as highlighted in Table 1 is further expanded in columns 7–9 in Tables 3 and 4 by considering the difference in their % contributions to the intermolecular interaction strengths, both within the ranked lists for each polymorph, as well as between the polymorphic forms. For example, the carboxylic H-bonded dimers (Aα) were found to have over 96% of its interaction centred on the COOH group, while the π–π stacking interaction (Cα) was found to be more centred on the phenyl ring, with over 72% of the interaction contributed by the phenyl ring.
These pair-wise synthonic interactions are shown in Fig. 7 and highlight the important bulk synthons for each structure.
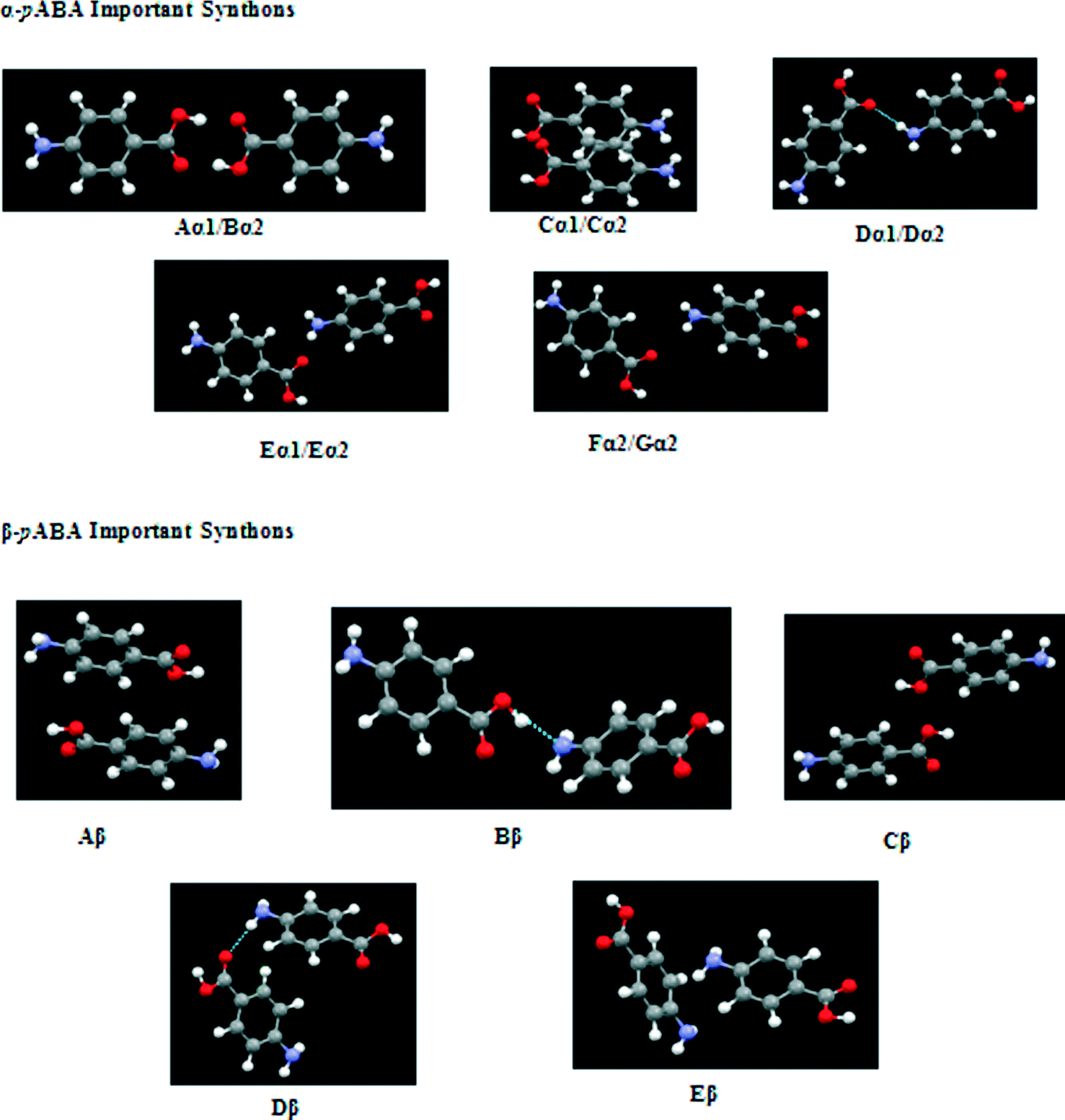 | ||
| Fig. 7 Important bulk synthons as specified in Tables 3 and 4 for both forms of pABA that are required to be satisfied to converge the lattice energy. Pairwise interactions visualised for clarity. Combination of H-bonding and vdW interacting synthons important for both structures. | ||
It is interesting to observe that both structures were found to contain a π–π stacking motif, with the α structure containing a head to head stack and the β structure containing a head to tail stack.
Fig. 8a shows the head to head α stacking motif. The stacking motifs Cα1 and Cα2 associated with the two different molecules in the asymmetric unit are almost identical and form a strong intermolecular interaction. The stacking is slightly offset so that the negative nitrogen and positive hydrogen atoms can form stronger atom–atom electrostatic interactions at one end, whilst the negative oxygen and positive carbon can interact in the same way at the other end.
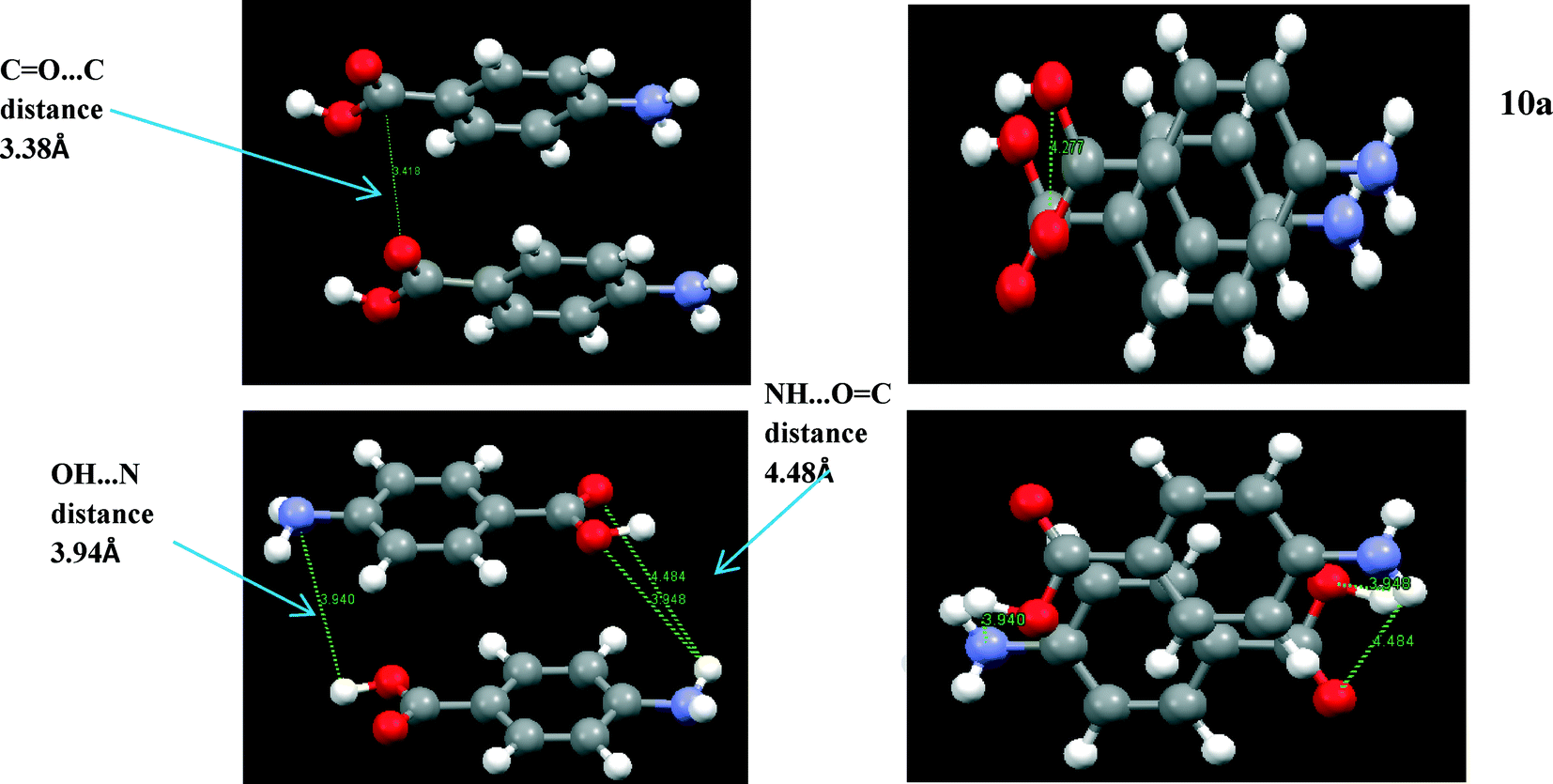 | ||
| Fig. 8 (a) (Above left and right) α π–π stacking dimer, head to head stacking 3.8 Å intermolecular distance between the corresponding NH2 and COOH groups. Molecules slightly offset creating stronger interactions between the functional groups. (b) (Below left and right) β π–π stacking. Head to tail stack around 4 Å distance between functional groups. Molecules more offset than α to maximise strength of interactions between NH2 and COOH groups. | ||
Fig. 8b shows the β motif to be a head to tail stacking dimer that is even more offset than that in the α stacking motif. This suggests that the electrostatic interactions between the more polar atoms in the NH2 and COOH functional groups are the dominating atom–atom interactions in this dimer motif. This is despite the interatomic distances of the strongly interacting atoms being slightly longer in the β stack compared to that present in the α stack. This particular stacking motif was predicted to be the strongest synthon in the β structure, although the energies of this interaction and the OH⋯N and NH⋯O H-bonding interactions are very similar.
4.4 CSD analysis of important H-bonding interactions
Fig. 9 shows the density of hits in the CSD of the OH⋯O, OH⋯N and NH⋯O interactions as examined as a function of distance and angle.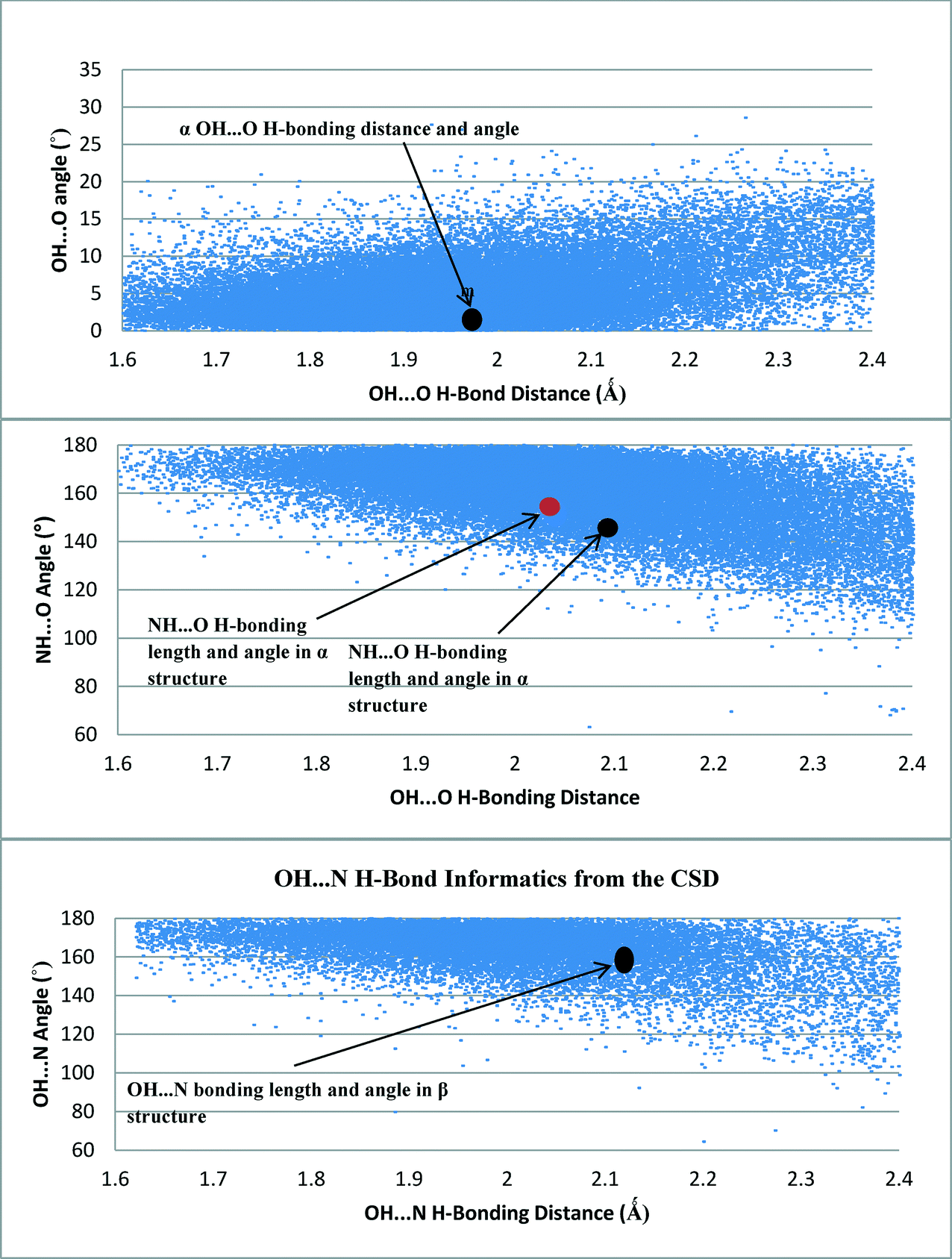 | ||
| Fig. 9 H-bonding data from the CSD of H-bonding angles and distances of OH⋯O (top), NH⋯O (middle) and OH⋯N (bottom) interacting groups. All interactions from the pABA crystal structures were found to be in a dense area of hits suggesting these are stable common interactions. | ||
Fig. 9 revealed that the hit density of structures with OH⋯O H-bond lengths between 1.8 Å and 2.1 Å was very high. It also showed that the more linear the bond angle between the molecules, then the higher the amount of hits. The H-bond length and orientation of the carboxylic acid H-bonding dimer interactions in the α structure were found to be close to the centre of this dense area of structures, consistent with this being a common and stable interaction.
The majority of the OH⋯N H-bonding interactions found in the search of the CSD were between 160° and 180° and had bond lengths of 1.8–2.1 Å. The OH⋯N H-bonding length of 2.15 Å in the β structure was also found to be within a dense area of structures containing a similar bond length, once again suggesting that this is a common stable interaction that is a key synthon in the molecular self-assembly and formation of the β structure.
The spread of hits for the NH⋯O interactions in the CSD was found to be a little wider in terms of bond length compared to the OH⋯O and OH⋯N interactions, though the highest density of hits was found to be around 2 Å. The shorter interactions tended to have more linear interactions, but as the NH⋯O bond length increased, the bond angle was found to move away from a linear conformation, suggesting that these structures could be more amenable to a change in geometry as the NH⋯O bond length increases, mindful that these interactions would be expected to be weaker and possibly not the major interactions that stabilise the crystal structure. This appears to be the case for the NH⋯O interactions present in the α and β forms of pABA.
4.5 Morphological simulations and surface chemistry analysis
| a | ||||
|---|---|---|---|---|
| Face (hkl) | d hkl (Å) | Slice energy (kcal mol−1) | Attachment energy (kcal mol−1) | % Saturation of surface molecule (anisotropy factor) |
| 1 0 1 | 12.7 | −24.5 | −1.7 | 93.6 |
| 1 0 −1 | 13.6 | −14.1 | −10.4 | 66.2 |
| 0 1 −1 | 3.8 | −9.2 | −15.4 | 35.9 |
| 1 1 −1 | 3.7 | −8.2 | −16.3 | 34.6 |
| 1 −1 0 | 3.8 | −9.1 | −15.5 | 39.5 |
| 0 0 2 | 9.3 | −14.7 | −9.6 | 59.8 |
| 2 0 0 | 9.3 | −15.0 | −9.8 | 60.5 |
| b | |||||
|---|---|---|---|---|---|
| Face (hkl) | d hkl (Å) | d hkl (Å) | Slice energy (kcal mol−1) | Attachment energy (kcal mol−1) | % Saturation of surface molecule (anisotropy factor) |
| 1 0 1 | 12.7 | 5.2 | −26.6 | −8.3 | 76.1 |
| 1 0 −1 | 13.6 | 6.0 | −20.7 | −14.3 | 59.2 |
| 0 1 −1 | 3.8 | 7.0 | −9.4 | −25.4 | 27.0 |
| 1 1 −1 | 3.7 | 4.9 | −7.6 | −27.3 | 21.8 |
| 1 −1 0 | 3.8 | 5.0 | −8.5 | −26.4 | 24.4 |
| 0 0 2 | 9.3 | 6.1 | −20.7 | −14.2 | 59.3 |
| 2 0 0 | 9.3 | 3.1 | −21.5 | −13.4 | 61.6 |
| c | ||||
|---|---|---|---|---|
| Face (hkl) | d hkl (Å) | Slice energy (kcal mol−1) | Attachment energy (kcal mol−1) | % Saturation of surface molecule (anisotropy factor) |
| 0 1 1 | 5.2 | −12.2 | −10.5 | 53.8 |
| 0 0 2 | 6.0 | −8.9 | −13.8 | 39.2 |
| 1 0 −1 | 7.0 | −10.6 | −12.2 | 46.5 |
| 1 0 1 | 4.9 | −12.0 | −10.7 | 53.0 |
| 1 1 1 | 5.0 | −10.5 | −12.2 | 46.2 |
| 1 1 0 | 6.1 | −11.5 | −11.2 | 50.8 |
| 1 1 −1 | 3.09 | −8.34 | −14.39 | 36.69 |
The degree of satisfaction of the intermolecular interactions of a molecule at a surface compared to a molecule in the bulk can be related to how labile a surface is to accepting molecules from solution and therefore how fast a given surface will grow. Table 5a shows the degree of satisfaction of a molecule within the α-structure for the different faces from the monomer binding calculation is markedly diverse, with the slow growing (1 0 1) surface having approximately 93% of possible interactions satisfied. Compared to the capping (0 1 −1), (1 −1 0) and (1 1 −1) surfaces, which have approximately 35–39% of interactions satisfied. Hence, it can be expected that the capping faces would grow significantly faster than the (1 0 1) surface.
4.5.2.1 α-form. The attachment energies for the monomeric growth unit model in Table 5a resulted in the flat lathe-like morphological prediction shown in Fig. 10a and c. A comparison of the attachment energies calculated for α-pABA using the dimer growth unit, revealed that the attachment energy of the (1 0 1) surface is increased when compared to that calculated for the monomer form, with the attachment energies of the (1 0 −1) and the capping surfaces being relatively reduced. This reduction of the attachment energy of the capping surfaces resulted in the prediction of a less plate like morphology shown in Fig. 10b and d, with the predicted inclusion of the (0 0 2) and (2 0 0) surfaces.
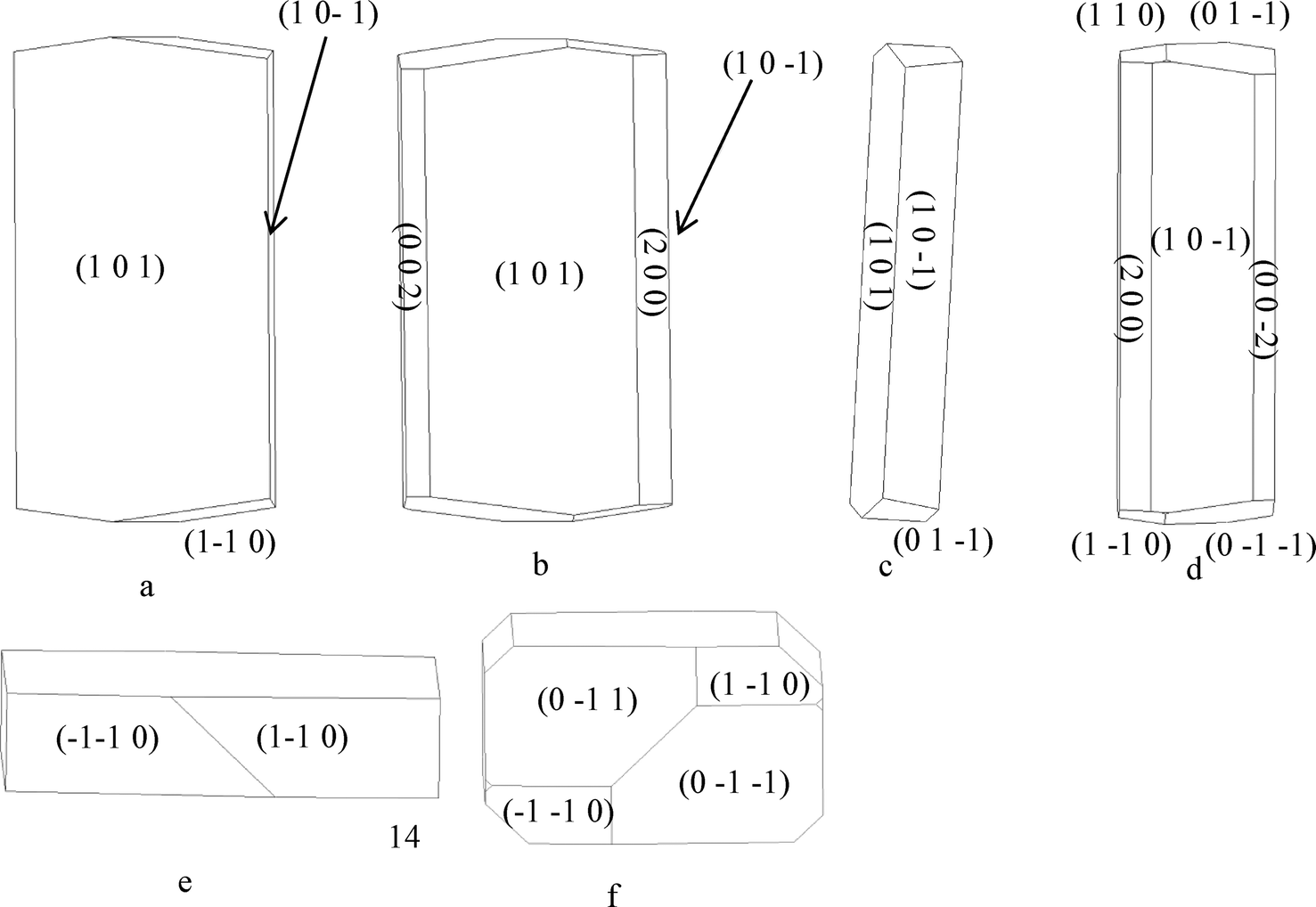 | ||
| Fig. 10 (a, c and e) Attachment energy morphological prediction of α-pABA, assuming the attaching growth units are monomers, showing the major faces that are predicted in the final morphology. (b, d and f) Attachment energy morphological prediction of α-pABA, assuming the attaching growth units are carboxylic acid H-bonding dimers, showing the major faces that are predicted in the final morphology. | ||
Both monomer and dimer based morphological simulations have lower aspect ratios with respect to those observed from the experimentally grown crystals shown in Fig. 11. Studies of the crystal growth rates for the capping faces of α-pABA is consistent with their growth by a linear dependence of the growth rate as a function of supersaturation, suggesting a rough interfacial growth mechanism (RIG). This reflects the strong intermolecular solute/surface recognition from the solution phase to crystal habit surface due to the strong π–π stacking interactions.56 In contrast, the side (1 0 −1) surfaces were found to grow by a B&S mechanism.56 The attachment energy morphology is essentially a prediction of the growth morphology under equilibrium conditions, i.e. at zero supersaturation, and this provides a good prediction for crystals that grow by a BCF14 or B & S15 mechanism. This higher growth rate for the capping face, with respect to that of the side faces, probably explains why the growth (kinetic) morphology is less consistent with the predicted equilibrium morphology.
 | ||
| Fig. 11 (a) α-pABA grown in EtOH at σ = 0.03 for ten min. (b) α-pABA grown in EtOH at σ = 0.07 for ten min. | ||
Fig. 11a shows that at very low supersaturations α-pABA appears to present a more flat and lathe-like morphology. Though still longer than the monomer morphology prediction, the general flat shape appears to correlate to the low supersaturation crystal featuring a dominant flat face being the (1 0 1) surface. Fig. 11b shows at σ = 0.07, the shape of the crystal appears thicker and seems to include more faces in the b-axis zone of the crystal, probably the (0 0 2) and (2 0 0) surfaces that appear in the dimer morphological prediction. Recent work by Sullivan and Davey suggests that the (1 0 1) and (1 0 −1) surfaces do not completely dominate the b-axis zone of facets (Fig. 12b and d), and that the morphology is 6 or even 8 sided, with increased importance of the (0 0 2) face.64
 | ||
| Fig. 12 (a) 6-sided morphological sketch of α-pABA adapted to match Fig. 12b. (b) α-pABA crystal grown from slow solvent evaporation of EtOH. (c) 8-sided morphological sketch adapted to match Fig. 12d. (d) α-pABA crystal grown from slow solvent evaporation of EtOAc. (b and d) Reproduced with permission from Sullivan and Davey, CrystEngComm, 2014.49 | ||
The morphological sketch in Fig. 12a suggests that in the 6 sided shape in Fig. 12b, the extra face is indeed the (0 0 2) surface. However, the morphological sketch in Fig. 12c suggests that both the (0 0 2) and (2 0 0) faces can be present in the growth morphology of α-pABA. Table 5a and b show that the predicted attachment energy for these minor habit surfaces were found to be very similar to each other for both the monomer- and dimer-based calculation. The latter would suggest that the growth rates for these faces would be very similar and, hence, there would be a competition between these two faces in terms of them appearing in the final growth morphology. Table 6 shows experimental interplanar angles in the (0 1 0) zone for an α-pABA crystal with respect to those calculated based on the unit cell parameters.
| Plane angle measured | Calculated angle (°) | Measured angle (°) |
|---|---|---|
| (0 0 2) → (1 0 1) | 43.3 | 43 |
| (1 0 1) → (2 0 0) | 43.13 | 45 |
| (2 0 0) → (1 0 −1) | 46.69 | 46 |
The interplanar angles were found to match reasonably well to the calculated interplanar angles, suggesting the appearance of the (0 0 2) and (2 0 0) faces in the experimental crystal morphology that are shown in Fig. 12c.
As with the b-axis zone of the crystal facets, the end capping faces also appeared to show variations in the final experimental growth morphology with respect to predictions. The monomer attachment energy prediction (Fig. 10e) showed the (1 −1 0) and (−1 −1 0) faces at the end of the crystal. However, comparison of calculated and measured interplanar angles between the edge (1 0 −1) surfaces and with those of the capping faces suggested that the capping face is more likely to be the (0 1 −1) face. That said, the attachment energies of the (0 1 −1) and (1 −1 0) faces were seen to be very similar (Table 5a), suggesting that the appearance of these faces at the capping end of the crystal is very competitive.
4.5.2.2 β-form. The attachment energy, and hence the anisotropy factor, for the morphologically important surfaces of the β-form were found to be relatively similar, and thus, more isotropic growth would be expected in 3D. Fig. 12a and b show the attachment energy morphological prediction of β-pABA to have a diamond-shaped morphology, with more equal growth in the different crystallographic directions. This is consistent with the attachment energies calculations given in Table 5c.
The attachment energy prediction for the β polymorph compared to experimentally grown crystals is shown in Fig. 13a and b and shows that the morphological prediction this gives a good match to the shape of the experimentally grown β-pABA crystal shown in Fig. 13c.
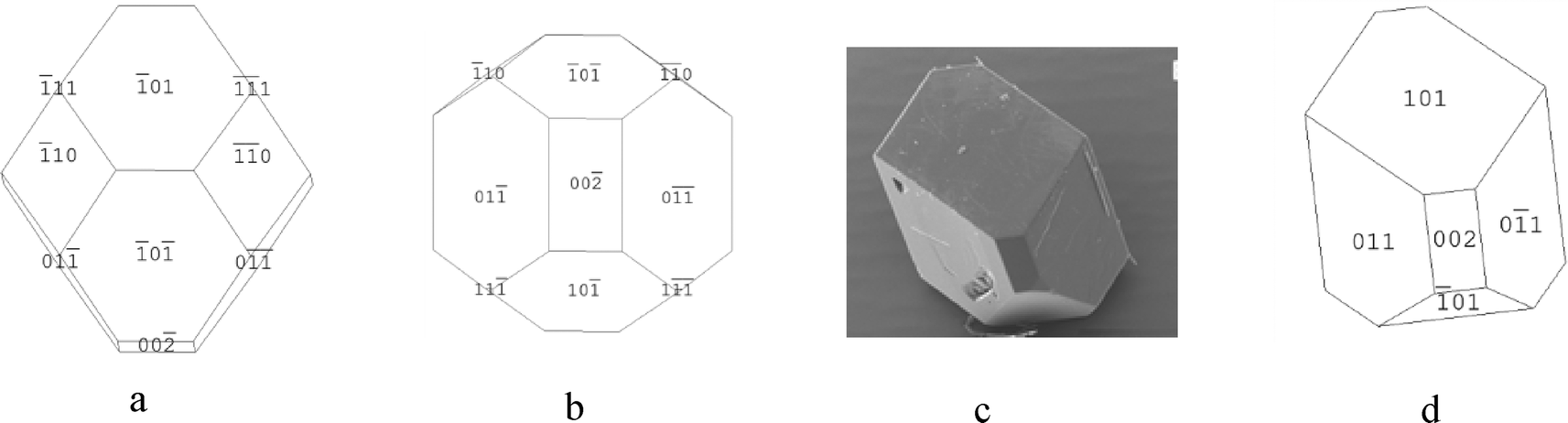 | ||
| Fig. 13 (a and b) Attachment energy morphological predictions of the crystal structure of β-pABA. (c) SEM of β-pABA grown from water showing flat top face, no evidence of multi faceting. (d) Morphological sketch of β-pABA made to resemble the experimental crystal in Fig. 13c. (c) Reproduced with permission of Sullivan and Davey, CrystEngComm, 2015.49 | ||
However, the simulation shown in Fig. 13a shows a multifaceted top surface, whereas Fig. 13c shows a flat top surface. From the morphological sketch in Fig. 13d, it would appear likely that the dominating top face is the (1 0 1) surface. Table 5c shows that the attachment energies of the individual faces of β-pABA were found to be similar, suggesting that the growth rates are quite similar to each other and hence the associated crystal growth mechanisms are probably the same. This is in contrast to α-pABA and is probably a factor as to why the attachment energy morphological prediction of β-pABA gave a greater resemblance to experimental crystals when compared to α-pABA.
4.5.3.1 α-form. The previous analysis of the intermolecular interactions was from the bulk crystal structure (Fig. 6, Table 4). In this case, the specific unsaturated interactions that contribute to the attachment energy at each of the present surfaces were characterised for the monomer attachment energy prediction of both forms.
Fig. 14a shows that the carboxylic acid H-bonding dimers were found to run in-plane at the (1 0 1) surface. Fig. 14b shows the π–π stacking in the b direction was found to be perpendicular to the growth direction of the (1 0 1) surface, and therefore not contributing to the attachment energy. Table 7 shows the extrinsic synthons were found to be made up of vdW interactions between the polar atoms of the COOH and NH2 functional groups. These interactions were found to be quite weak with all of them being less than 1 kcal mol−1, hence the very low attachment energy predicted at this surface. Compared to the strongest bulk interactions, e.g. A, B and C representing the H-bonding carboxylic acid dimers and π–π stacking interactions, the strongest interaction for this face was found to be comparatively weak and is 10th (Jth) in terms of morphological importance. Such a low attachment energy and concomitantly weak interactions at this surface would be consistent with a slow growth rate for this surface (Fig. 15, Table 8).
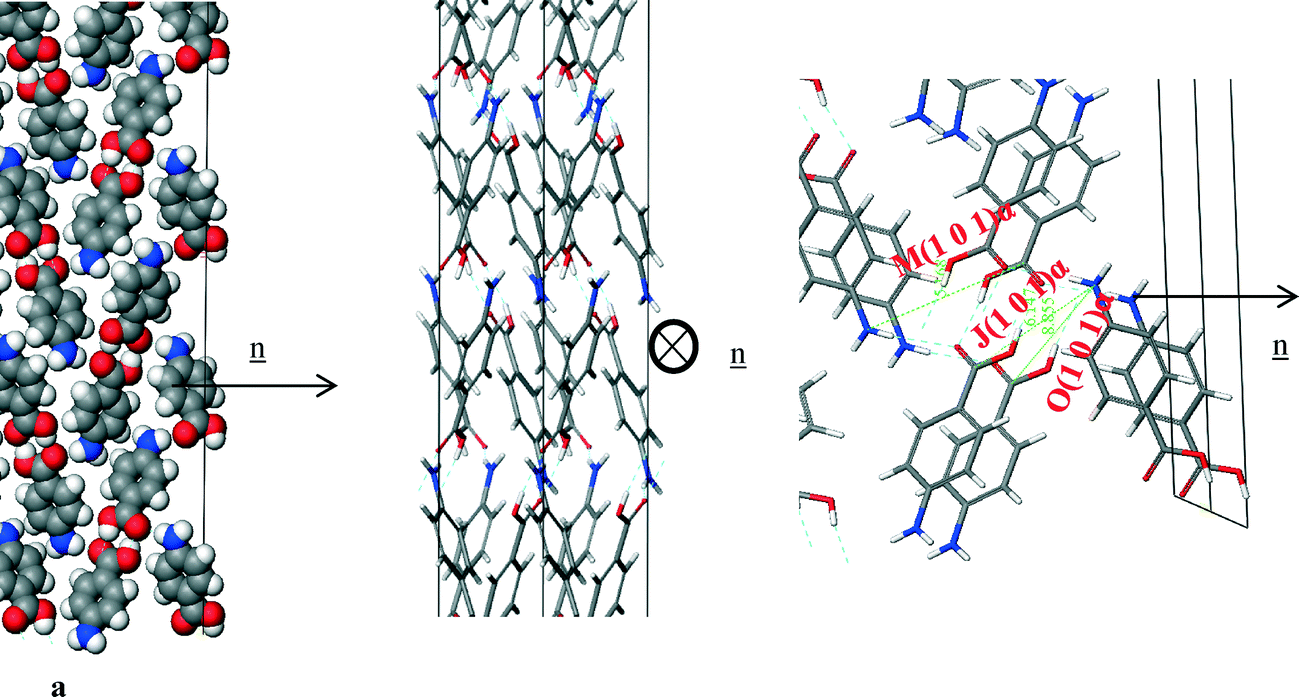 | ||
| Fig. 14 Crystal chemistry of the (1 0 1) surface of α-pABA: (a) space fill model of side view; (b) stick model of plan view; (c) stick model of side view. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| J(1 0 1)α | 2 | 6.9 | −0.7 |
| M(1 0 1)α | 2 | 6.7 | −0.4 |
| O(1 0 1)α | 2 | 8.9 | −0.2 |
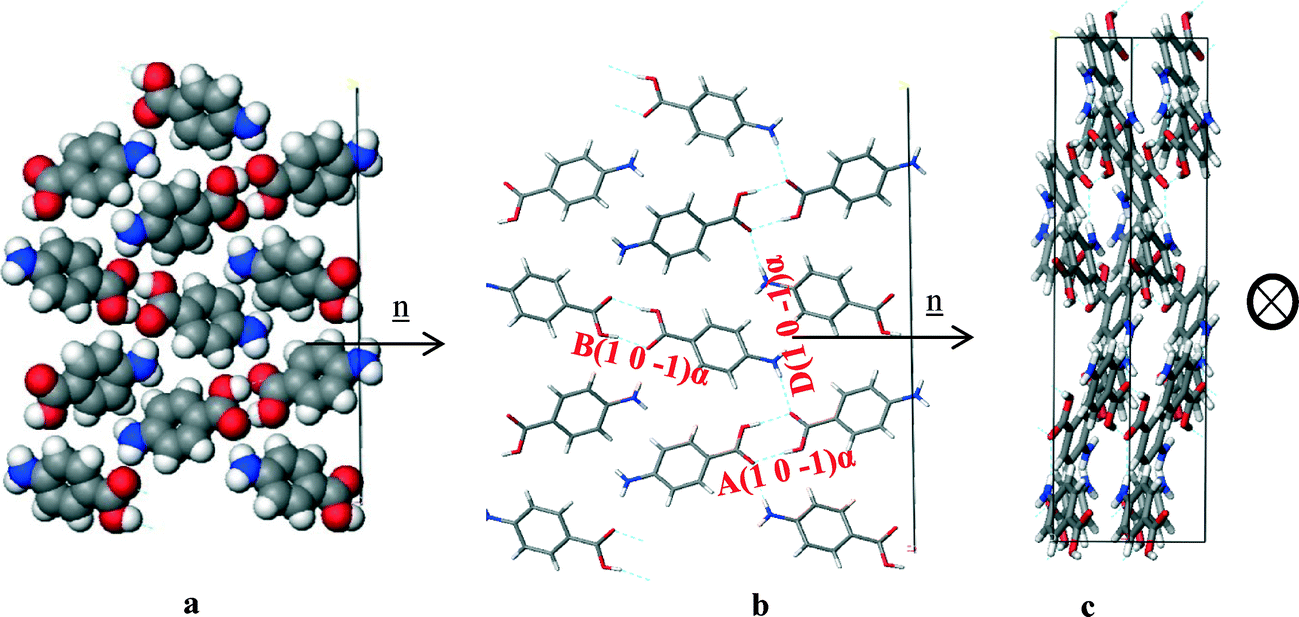 | ||
| Fig. 15 Crystal chemistry (1 0 −1) surface of α-pABA: (a) space fill model of side view; (b) stick model of side view; (c) stick model of plan view. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| A(1 0 −1)α | 1 | 8.2 | −5.7 |
| B(1 0 −1)α | 1 | 8.3 | −5.6 |
| D(1 0 −1)α | 2 | 7.9 | −2.3 |
Fig. 12a and b shows the COOH and NH2 functional groups were found to be orientated almost parallel to the direction of growth of the α-pABA (1 0 −1) surface. The H-bonds between the COOH groups were found to form almost parallel to the growth direction of this surface, hence promoting much faster growth in this direction when compared to that of the (1 0 1) surface. Fig. 12c shows reactive H-bonding functional groups exposed at this surface, while the π–π stacking were found to form almost perpendicular to this surface and are therefore would not be involved with the growth of the (1 0 −1) surface. Table 7 shows that the interactions contributing to the attachment energy for this surface were found to be some of the strongest bulk interactions with the attachment energy predicted to be more than five times higher than that for the (1 0 1) surface.
The capping face (0 1 −1) was predicted to be the fastest growing of the morphologically important crystal surfaces.
Fig. 16a of the space fill model shows how the molecules were found to close pack in zig-zag chains stacking along the b direction of the structure. The molecules were found pack more closely along this growth direction than the (1 0 1) or (1 0 −1) directions, and hence it is no coincidence that the (0 1 −1) surface was found to grow much faster than the (1 0 1) or (1 0 −1) habit surfaces. Fig. 16b shows the C intermolecular interaction that represent the intermolecular interactions created by the close π–π packing, and this interaction was found to be close to parallel with respect to the direction of growth. Table 9 shows the three strongest interactions contributing to the attachment energy at this surface, which were found to be the same as the three strongest interactions measured for the bulk interactions.
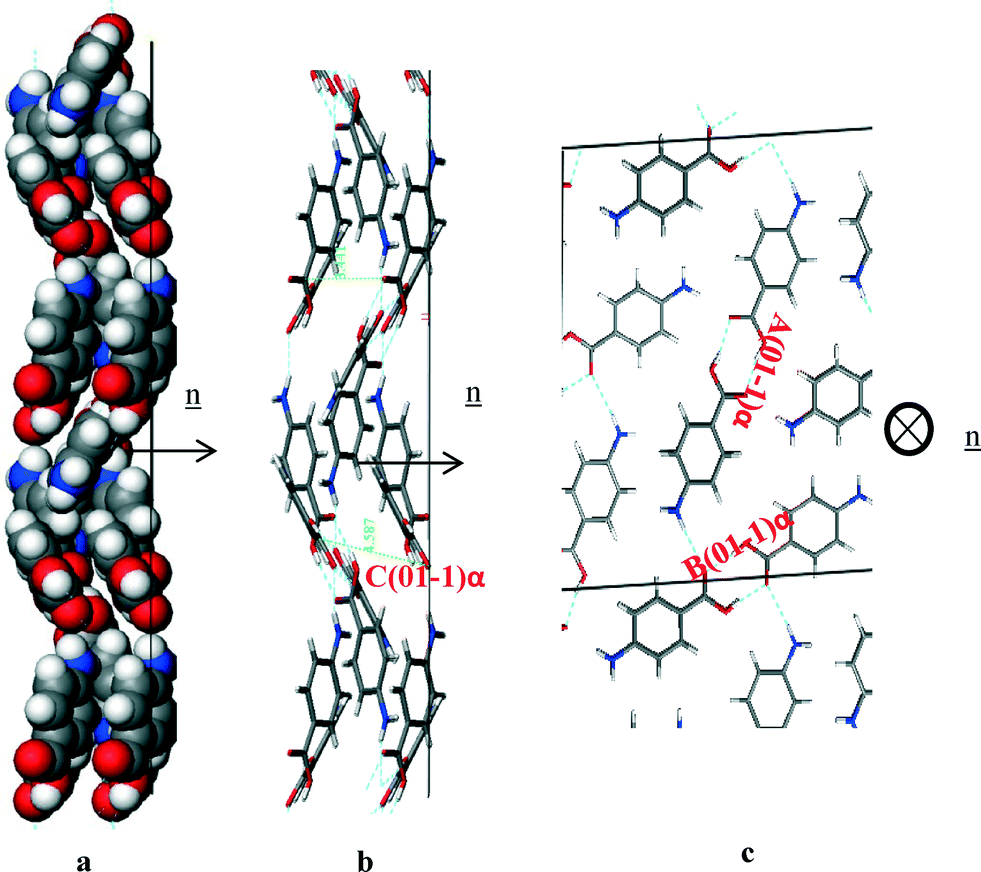 | ||
| Fig. 16 Crystal chemistry of (0 1 −1) surface of α-pABA: (a) space fill model of side view; (b) stick model of side view; (c) stick model of plan view. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| A(0 1 −1)α | 1 | 8.2 | −5.7 |
| B(0 1 −1)α | 1 | 8.3 | −5.67 |
| C(0 1 −1)α | 2 | 3.9 | −2.7 |
The attachment energy model also predicted contribution from the OH⋯O intermolecular interactions between the H-bonding dimers to the growth of this surface. However, examining the in-plane molecular packing of the (0 1 −1) surface revealed that these interactions are not orientated significantly along the growth direction of this surface, which would be consistent with their reduced role in the growth of the (0 1 −1) surface.
These orientation effects then suggest that the dominant interactions promoting the fast growth of the (0 1 −1) surface are the π–π stacking interactions. The close packing is very favourable and coupled with the fact that the solvents used for crystallisation have strongly contrasting molecular structures, i.e. without any aromatics, would suggest that the surface/solvent interaction would be unlikely to disrupt this interaction and hence the growth process of the (0 1 −1) surface.
In this respect, it is also important to consider that α-pABA crystallises from polar protic solvents such as EtOH, MeOH etc. which can form strong H-bonds and thus each growing surface must de-solvate before incorporation of solute and growth can occur. The (1 0 −1) surface was found to have exposed H-bonding sites orientated directly out at the surface, not only having potential to form strong interactions with pABA, but also with H-bonding solvents. Such binding would have the effect of slowing down the de-solvation process, and hence through this the growth rate of the surface. In comparison to the capping (0 1 −1) surface, where the growth process was found to be dominated by the π–π solute binding interactions, the solvent binding strength for polar protic solvents would be expected to be much lower and hence the solvent effect on the growth process would be expected to be relatively low. Such a solvent binding effect on the (1 0 −1) surface might be a further factor explaining the discrepancy between the predicted and observed morphology i.e. reflecting the fact that the actual solvent-mediated growth rate could be much lower than that predicted.
4.5.3.2 β-form. Fig. 13 reveals the (0 1 −1) face is the largest face visible at the surface, but it does not dominate to the same extent as the (1 0 1) face in the α form. The (1 1 0), (1 0 1) and (1 0 −1) faces also contribute significantly to the surface area of the crystal habit. The analysis of the unsaturated synthons at the β faces present at the crystal surface was performed with the approach used for the α form (Fig. 17).
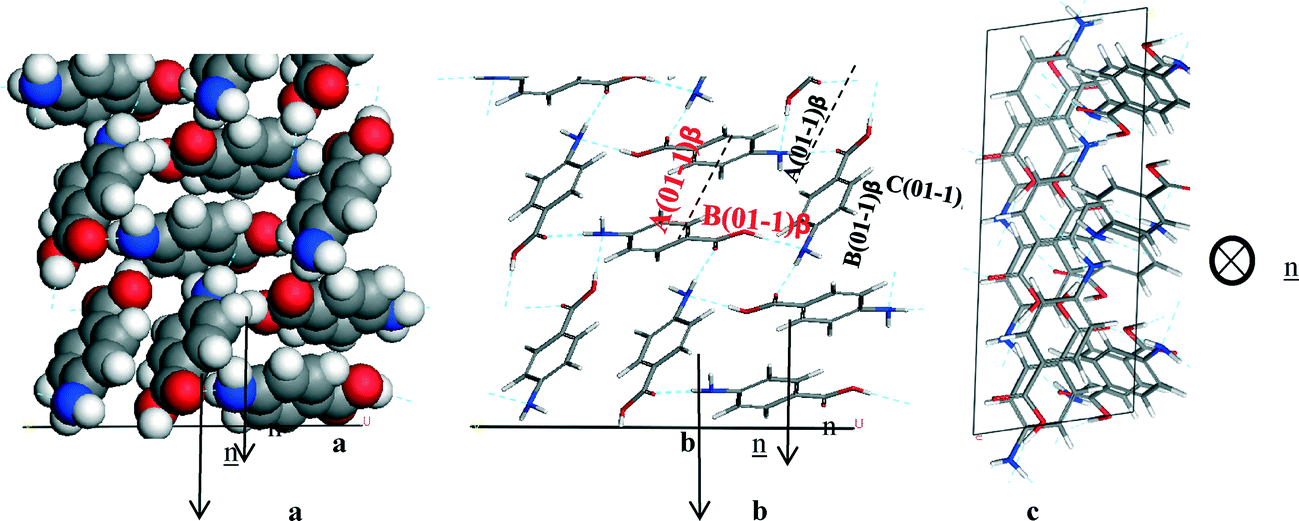 | ||
| Fig. 17 Crystal chemistry of (0 1 −1) surface of β-pABA: (a) space fill model of side view; (b) stick model of side view; (c) stick model of plan view. | ||
Analysis of the (0 1 −1) surface revealed that it has exposed NH2 and COOH groups that form the 4-membered H-bonding ring. The molecules were also found to stack out of the plane of this face to form the head to tail π–π stacking, hence Table 10 shows that the strongest interactions contributing to the attachment energy of this face were found to be in fact the same as the strongest bulk interactions.
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| A(0 1 −1)β | 1 | 4.2 | −2.6 |
| B(0 1 −1)β | 2 | 8.1 | −2.5 |
| C(0 1 −1)β | 1 | 5.7 | −2.4 |
Fig. 14a of the space fill model shows how the molecules stack almost perpendicular to each other, resulting in more equal growth in different directions, consistent with the isotropic nature of the morphology observed for the β-form. The same can be observed for the NH⋯O and OH⋯N H-bonding interactions (Fig. 18, Table 11).
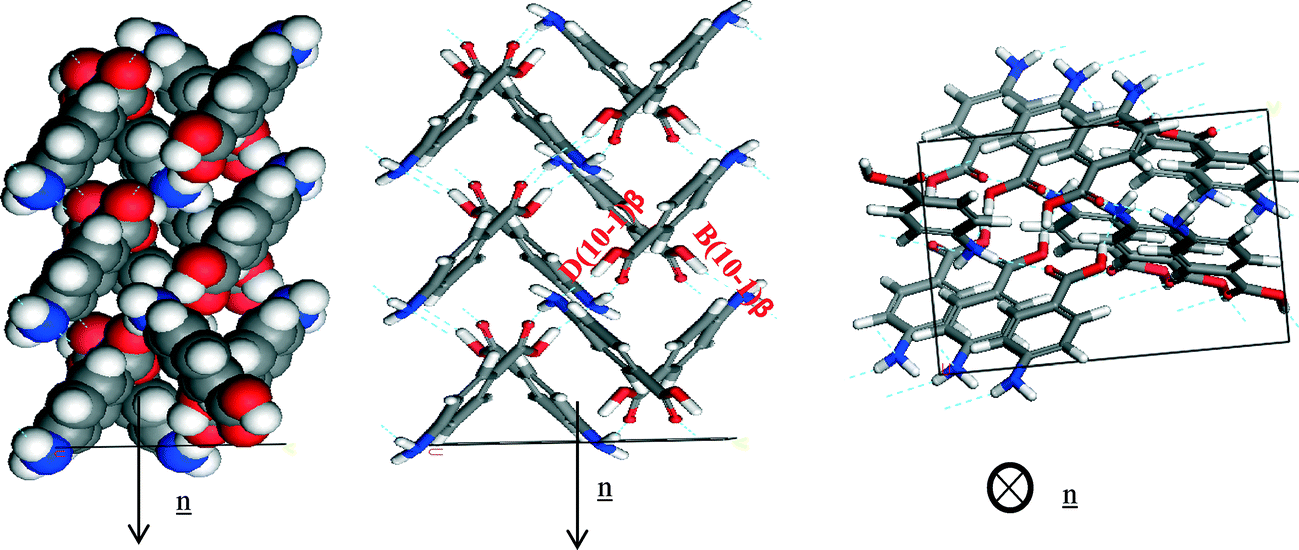 | ||
| Fig. 18 Crystal chemistry of (1 0 −1) surface of β-pABA: (a) space fill model of side view; (b) stick model of side view; (c) stick model of plan view. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| B(1 0 −1)β | 2 | 8.1 | −2.5 |
| D(1 0 −1)β | 2 | 6.7 | −1.5 |
The pABA molecules were found to stack close to perpendicular to the β-(1 0 −1) surface growth direction and hence the stacking interactions were found not to contribute to the attachment energy of this surface. The OH group and the nitrogen were found to be orientated almost parallel to the growth direction of the (1 0 −1) surface, and hence these interactions were found to dominate the growth of this surface
The smaller faster growing (0 0 2) surface was found to have contributions from the H-bonding and π–π stacking interactions to the growth of this face. The zig-zag chains of OH⋯N and NH⋯O hydrogen bonds making up the 4-membered ring structure were observed to run closer to the growth direction of the (0 0 2) surface compared to the other important surfaces present in the β morphology.
Fig. 19c reveals that the phenyl rings are found to be at about a 45° tilt away from parallel to the growth direction showing that there is some contribution to growth from the π–π stacking interactions as well as the H-bonds formed to the exposed NH2 group. Table 12 shows that all three of the strongest bulk interactions were found to contribute to the attachment energy of this surface.
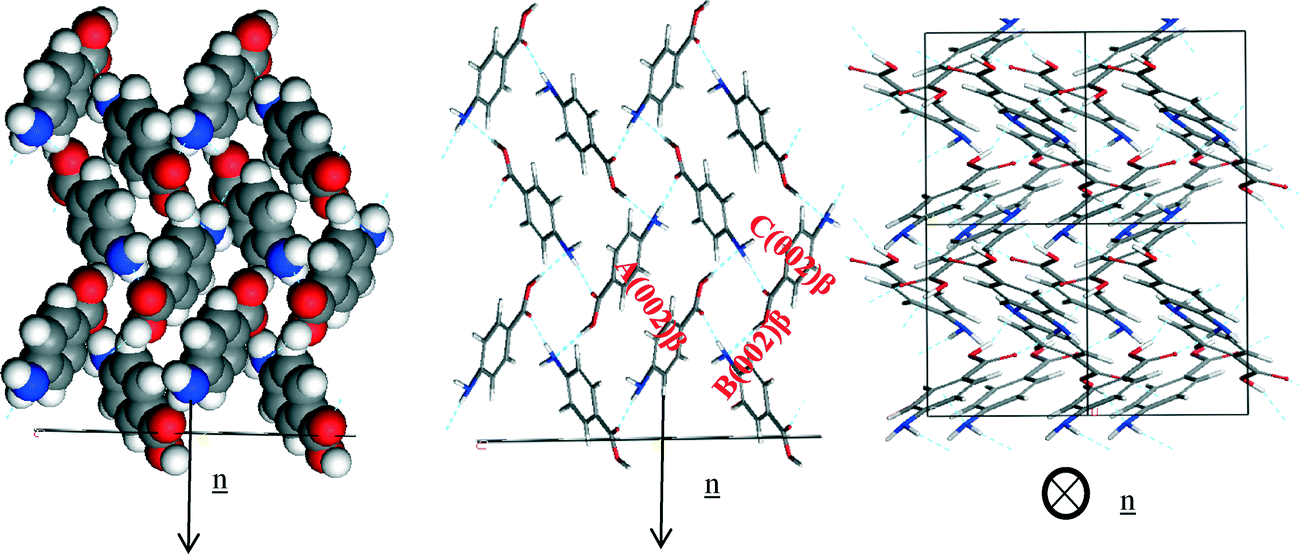 | ||
| Fig. 19 Crystal chemistry of (1 0 −1) surface of α-pABA: (a) space fill model of side view; (b) stick model of side view; (c) stick model of plan view. | ||
| Bond | Multiplicity | Distance (Å) | Intermolecular energy (kcal mol−1) |
|---|---|---|---|
| A(0 0 2)β | 1 | 4.2 | −2.6 |
| B(0 0 2)β | 2 | 8.1 | −2.5 |
| C(0 0 2)β | 1 | 5.7 | −2.4 |
The strength and character of the extrinsic synthons associated with the major faces of the β form were found to be not dissimilar to the synthons found at the surface of the smaller faster growing faces. The more isotropic nature of the β packing which is dominated by the H-bonding ring means that the major interactions were found to be orientated in more than one crystallographic direction; hence they affect growth in different directions in 3D. The large, slow growing, (0 1 −1) and faster growing (0 0 2) surface were also found to have significant contributions from all of the 4 strongest intermolecular interactions. However, the amount of these interactions outside of the slice is found to be less in the (0 1 −1) surface than the (0 0 2) surface, hence the larger predicted area of this face at the surface. The (1 0 −1) surface has only two strong intermolecular interactions outside the slice, but there was found to be a large contribution from both interactions, hence it has a smaller area than the (0 1 −1) surface but a larger area than the (0 0 2).
Experimentally it appears that the growth via π–π stacking in the α form was found to be more dominant than observed in the β form. Interestingly the amino hydrogens also showed a similar contribution for both polymorphs. This could suggest the NH⋯O interaction, that seems relatively unimportant for the α form, could facilitate the transition pathway between the α and β form as it is the main interaction that is found to be shared by both forms.
5. Conclusions
In this paper, the strength of the intermolecular interactions of pABA were calculated and their contribution to the lattice energy and morphology predictions of each polymorph was rigorously analysed for the first time.The NH2 group of the β form was found to contribute more to the lattice energy than the NH2 of the α form, reflecting the H-bonding donor and acceptor role that the NH2 plays in the β crystal structure, compared to the NH2 acting solely as a H-bonding donor in the α structure. The COOH group was found to contribute significantly more to the lattice energy of the α form than the β form due to the formation of the strong carboxylic acid H-bonding dimers. In addition, the formation of these H-bonding dimers appears to hold the carboxylic acid groups rigidly planar with respect to the phenyl ring in the α structure, while the β carboxylic acid group was found to have a slight torsion angle of around 10°.
The morphological prediction of α-pABA with a monomer growth unit gave a flat, lathe like morphology, while prediction with a carboxylic acid dimer growth unit gave a less plate like morphology. Both of these morphologies were predicted to have a large (1 0 1) surface, and it is observed that the (1 0 1) surface interactions consist of weak vdW forces. Compared to the surface interactions of the side (1 0 −1) which were found to contain H-bonding interactions, and the capping faces which were found to contain π–π stacking interactions, these much weaker interactions result in a much slower growth rate for the (1 0 1) surface. The experimental morphology often appears much more needle like than the prediction. However, such morphological predictions reflect an equilibrium situation, i.e. zero supersaturation, while the experimental morphologies are, by definition, grown under supersaturated conditions. As seen from a previous study,56 the capping faces were found to grow by a linear growth rate dependence with respect to supersaturation consistent with an RIG mechanism. Hence, the equilibrium morphological predictions would not be expected to predict the relative growth rates of a 3D set of surfaces crystallised under different interface kinetic mechanisms.
The dimer morphological prediction included the (2 0 0) and (0 0 2) surfaces, and comparison of some of the images from the publication by Sullivan and Davey49 to morphological sketches suggest that these faces can indeed appear in the α morphology. This assertion was reinforced by interplanar angle measurements from optical goniometry. However, optical microscopy at differing supersaturation suggests that the morphology can vary in different conditions, and that it is not as simple as one set morphology for the α form.
Comparatively, the β morphological prediction gave a reasonable match to the general shape of the experimental crystal. There seemed to be an underestimation of the morphological importance of the (1 0 1) surface. The higher resemblance of the β morphological prediction to the experimental crystal compared to α is probably due to the more isotropic ring like crystal structure giving similar growth in all directions, and hence the growth mechanisms and growth rates are probably similar. This postulation was reinforced by the fact that the nature and strength of the intermolecular interactions at the morphologically important faces of the β structure were found to be relatively similar compared to the interactions at the morphologically important faces of the α form.
Overall this paper presents the results of a thorough, holistic analysis and methodology for understanding the interrelationship between the molecular and solid-state polymorphic structures with the morphology and surface chemistry of a crystalline system at the molecular level.
Nomenclature
| BFDH | Bravais, Friedel, Donnay, Harker |
| PBC | Periodic bond chains |
| vdW | van der Waals |
| H-bonding | Hydrogen bonding |
| CSD | Cambridge Structural Database |
| BCF | Burton, Carbera and Frank |
| B & S | Birth and spread |
| RIG | Rough interfacial growth |
| 3D | 3 dimensions |
List of symbols
| E cr | Lattice energy |
| E sl | Slice energy |
| E att | Attachment energy |
| ΔHs | Sublimation enthalpy |
| R | Gas constant |
| T | Temperature |
| ξ | Anisotropy factor |
| n | Growth direction |
| ⊗ n | Growth direction perpendicular to plane of the page |
Acknowledgements
The authors gratefully acknowledge the UK's EPSRC for the funding of this research through a joint collaborative Critical Mass project between the Universities of Leeds and Manchester (grant references EP/IO14446/1 and EP/IO13563/1) which forms part of the doctoral studies of one of us (IR). We also gratefully acknowledge the Critical Mass team at the University of Manchester lead by Professors Davey and Schroeder for stimulating discussions that inspired and contributed to the research and ideas presented in this paper. In addition, we gratefully acknowledge Dr. D. Toroz (University of Leeds) for calculating the atomistic charges and Dr. R. B. Hammond (University of Leeds) for his assistance in creating the carboxylic acid H-bonding unit for the dimer attachment energy calculation.References
- P. York, Int. J. Pharm., 1983, 14, 1–28 CrossRef CAS.
- R. B. Hammond, K. Pencheva and K. J. Roberts, Cryst. Growth Des., 2007, 7, 875–884 CAS.
- R. B. Hammond, S. Jeck, C. Y. Ma, K. Pencheva, K. J. Roberts and T. Auffret, J. Pharm. Sci., 2009, 98, 4589–4602 CrossRef CAS PubMed.
- V. Ramachandran, D. Murnane, R. B. Hammond, J. Pickering, K. J. Roberts, M. Soufian, B. Forbes, S. Jaffari, G. P. Martin, E. Collins and K. Pencheva, Mol. Pharmaceutics, 2014, 12, 18–33 CrossRef PubMed.
- R. Docherty, G. Clydesdale, K. J. Roberts and P. Bennema, J. Phys. D: Appl. Phys., 1991, 24, 89–99 CrossRef CAS.
- A. Bravais, Etudes Crystallographiques, Gauthiers Villars, Paris, 1886 Search PubMed.
- J. D. H. Donnay and D. Harker, Am. Mineral., 1937, 22, 446–467 CAS.
- G. Friedel, Bull. Soc. Fr. Mineral. Crist., 1907, 30, 326 Search PubMed.
- R. Docherty, K. J. Roberts and E. Dowty, Comput. Phys. Commun., 1988, 51, 423–430 CrossRef.
- R. Docherty and K. J. Roberts, J. Cryst. Growth, 1988, 88, 159–168 CrossRef CAS.
- P. Hartman and W. G. Perdok, Acta Crystallogr., 1955, 8, 49–52 CrossRef.
- M. Born, Atom Theorie des Feste Zlrsfande 2nd Edn, 1923 Search PubMed.
- E. Dowty, Am. Mineral., 1976, 61, 448–459 CAS.
- W. K. Burton, N. Cabrera and F. C. Frank, Philos. Trans. R. Soc., A, 1951, 243, 299–358 CrossRef.
- M. Ohara and R. C. Reid, Modelling Crystal Growth Rates from Solution, 1973, vol. 225 Search PubMed.
- P. Hartman and P. Bennema, J. Cryst. Growth, 1980, 49, 145–156 CrossRef CAS.
- Z. Berkovitch-Yellin, J. Am. Chem. Soc., 1985, 107, 8239–8253 CrossRef CAS.
- G. Clydesdale, R. Docherty and K. J. Roberts, Comput. Phys. Commun., 1991, 64, 311–328 CrossRef CAS.
- G. Clydesdale, K. J. Roberts and E. M. Walker, The crystal habit of molecular materials: A structural perspective, In Molecular Solid State: Syntheses, Structure, Reactions, Applications, 1996 Search PubMed.
- M. C. Etter, J. C. Macdonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, USA, 2002 Search PubMed.
- G. Desiraju, J. J. Vittal and A. Ramanan, Crystal Engineering: The Design of Organic Solids, Elsevier, 1989 Search PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS PubMed.
- Q. Wang, D. B. Sheen, E. E. A. Shepherd, J. N. Sherwood, G. S. Simpson and R. B. Hammond, J. Cryst. Growth, 1997, 181, 418–426 CrossRef CAS.
- G. Clydesdale, K. J. Roberts, G. B. Telfer, V. R. Saunders, D. Pugh, R. A. Jackson and P. Meenan, J. Phys. Chem. B, 1998, 102, 7044–7049 CrossRef CAS.
- G. Clydesdale, K. J. Roberts, G. B. Telfer and D. J. W. Grant, J. Pharm. Sci., 1997, 86, 135–141 CrossRef CAS PubMed.
- D. E. Williams, J. Chem. Phys., 1966, 45, 3770–& CrossRef CAS PubMed.
- F. A. Momany, L. M. Carruthers, R. F. McGuire and H. A. Scheraga, J. Phys. Chem., 1974, 78, 1595–1620 CrossRef CAS.
- G. Nemethy, M. S. Pottle and H. A. Scheraga, J. Phys. Chem., 1983, 87, 1883–1887 CrossRef CAS.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- A. T. Hagler, S. Lifson and P. Dauber, J. Am. Chem. Soc., 1979, 101, 5122–5130 CrossRef CAS.
- S. Lifson, A. T. Hagler and P. Dauber, J. Am. Chem. Soc., 1979, 101, 5111–5121 CrossRef CAS.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- J. D. Gale, J. Chem. Soc., Faraday Trans., 1997, 93, 629–637 RSC.
- J. D. Gale and N. J. Henson, J. Chem. Soc., Faraday Trans., 1994, 90, 3175–3179 RSC.
- R. B. Hammond, K. Pencheva, V. Ramachandran and K. J. Roberts, Cryst. Growth Des., 2007, 7, 1571–1574 CAS.
- M. K. Singh and A. Banerjee, Cryst. Growth Des., 2013, 13, 2413–2425 CAS.
- J. Chen and B. L. Trout, Cryst. Growth Des., 2010, 10, 4379–4388 CAS.
- D. Winn and M. F. Doherty, AIChE J., 1998, 44, 2501–2514 CrossRef CAS PubMed.
- S. Gnanasambandam and R. Rajagopalan, CrystEngComm, 2010, 12, 1740–1749 RSC.
- C. Schmidt and J. Ulrich, Chem. Eng. Technol., 2012, 35, 1009–1012 CrossRef CAS PubMed.
- C. Schmidt and J. Ulrich, Chem. Eng. Technol., 2011, 34, 563–570 CrossRef CAS PubMed.
- C. Cambridge Crystallographic Data, Cambridge Structural Database, 1965 Search PubMed.
- W. R. Busing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 340–347 CrossRef.
- S. Athimoolam and S. Natarajan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2007, 63, O514–O517 CAS.
- S. Gracin and A. Fischer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, O1242–O1244 CAS.
- R. Benali-Cherif, R. Takouachet, E.-E. Bendeif and N. Benali-Cherif, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 323–325 CAS.
- R. A. Sullivan and R. J. Davey, CrystEngComm, 2015, 17, 1015–1023 RSC.
- M. A. Lovette and M. F. Doherty, Cryst. Growth Des., 2013, 13, 3341–3352 CAS.
- N. Panina, R. van de Ven, F. F. B. J. Janssen, H. Meekes, E. Vlieg and G. Deroover, Cryst. Growth Des., 2009, 9, 840–847 CAS.
- A. S. Inc., San Diego, 5.5 edn., 2011.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- G. Clydesdale, K. J. Roberts and R. Docherty, J. Cryst. Growth, 1996, 166, 78–83 CrossRef CAS.
- R. B. Hammond, K. J. Roberts, E. D. L. Smith and R. Docherty, J. Phys. Chem. B, 1999, 103, 7762–7770 CrossRef CAS.
- D. Toroz, I. Rosbottom, T. Turner, D. M. C. Corzo, R. B. Hammond, X. Lai and K. J. Roberts, Faraday Discuss., 2015, 179, 79–114 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- E. Dowty, Am. Mineral., 1980, 65, 465–472 Search PubMed.
- H. J. Human, J. P. Van Der Eerden, L. A. M. J. Jetten and J. G. M. Odekerken, J. Cryst. Growth, 1981, 51, 589–600 CrossRef CAS.
- L. A. M. J. Jetten, H. J. Human, P. Bennema and J. P. Van Der Eerden, J. Cryst. Growth, 1984, 68, 503–516 CrossRef CAS.
- J. S. Stevens, C. R. Seabourne, C. Jaye, D. A. Fischer, A. J. Scott and S. L. M. Schroeder, J. Phys. Chem. B, 2014, 118(42), 12121–12129 CrossRef CAS PubMed.
- M. Nabavian, R. Sabbah, R. Chastel and M. Laffitte, J. Chim. Phys. Phys.-Chim. Biol., 1977, 74, 115–126 CAS.
- C. G. de Kruif, J. Voogd and J. C. A. Offringa, J. Chem. Thermodyn., 1979, 11, 651–656 CrossRef CAS.
- R. A. Sullivan and R. J. Davey, CrystEngComm, 2015, 17, 1015–1023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ce00302d |
| This journal is © The Royal Society of Chemistry 2015 |