DOI:
10.1039/C5CE00221D
(Paper)
CrystEngComm, 2015,
17, 3460-3467
Dinuclear organogold(I) complexes bearing uracil moieties: chirality of Au(I)–Au(I) axis and self-assembly†
Received
30th January 2015
, Accepted 18th March 2015
First published on 26th March 2015
Abstract
The conjugation of dinuclear organogold(I) complexes with a bridging diphosphine ligand as an organometallic compound and the uracil derivative as a nucleobase was demonstrated to afford bioorganometallic conjugates. Single-crystal X-ray structure determination of the dinuclear organogold(I)–uracil conjugates revealed the assembly properties of gold(I) and the uracil moieties in the solid state. The crystal structure of (U6Au)2(μ-Xantphos) (U6 = 6-ethynyl-1-octyluracil) with Xantphos as the bridging diphosphine ligand revealed the presence of an intramolecular aurophilic Au(I)–Au(I) interaction. R- and S-enantiomers based on the Au(I)–Au(I) axis exist in the unit cell, which are connected alternately to form the hydrogen-bonded assembly through intermolecular hydrogen bonds between the uracil moieties. In the case of the dinuclear organogold(I) complex (U5Au)2(μ-Xantphos) (U5 = 5-ethynyl-1-octyluracil), both enantiomers were found to form homochiral RR and SS dimers, respectively, through π–π interactions between 5-ethynyl-uracil moieties. In the crystal packing, each dimer is assembled alternately to form the hydrogen-bonded assembly through intermolecular hydrogen bonds between the uracil moieties. As expected, the utilization of (R)-BINAP as a bridging diphosphine ligand with axial chirality induced the chirality of the Au(I)–Au(I) axis. The crystal structure of the dinuclear organogold(I) complex with (R)-BINAP (U6Au)2(μ-R-BINAP) confirmed the axial chirality of the Au(I)–Au(I) axis, forming a R,R-enantiomer, wherein each molecule is arranged through intermolecular hydrogen bonds between the uracil moieties to form a helical molecular arrangement.
Introduction
Gold(I) alkynyl compounds have attracted much attention in a variety of areas such as luminescent materials and metallodrugs.1 A number of gold(I) alkynyl complexes are characterized to possess luminescence properties, which show long emission lifetimes and emissive excited states derived from alkynyl moieties.2 Several gold(I) alkynyl complexes are also reported to exhibit cytotoxicity against cancer cells.3 Gold(I) complexes are known to aggregate through a d10–d10 closed shell aurophilic bonding interaction, which plays an important role in the formation of aggregated structures and their physical properties.4 The emission properties of gold(I) complexes are influenced by the aurophilic interaction, which often expresses specific emission properties. Some gold(I) complexes show mechanochromic luminescence by switching of the aurophilic interaction through mechanical stimuli such as grinding in the solid state.5 The design of aggregation of gold(I) complexes is important for control of the aurophilic interaction. Construction of a dinuclear gold(I) complex with a bridging ligand is considered to be a convenient approach for the rearrangement of Au(I) centers. In particular, a semirigid bridging diphosphine ligand is expected to arrange the Au(I) centers on the same side of the ligand and facilitate the induction of the aurophilic interaction.6 On the other hand, biomolecules such as nucleobases, peptides, and sugars play important roles in the formation of highly-organized structures like DNA, proteins, and enzymes. The utilization of non-covalent bonds is a convenient strategy for construction of the designed assembly structure. The complementary hydrogen bond of nucleobases is regarded as a powerful tool for building various self-assembly systems based on its directionality and specificity.7 The reversibility and tunability of hydrogen bonding also plays an important factor in the chemical and/or physical properties of molecular assemblies.8 The combination of functional organometallic compounds with biomolecules such as nucleobases and peptides is envisioned to afford bioorganometallic compounds.9 We have already reported the emergence of emission based on the metallophilic interaction of the aggregated structures of organoplatinum(II)–uracil and organogold(I)–guanine conjugates.10 From these points of view, we herein designed and synthesized dinuclear organogold(I)–uracil conjugates with bridging diphosphine ligands in order to control the arrangement of the Au(I) centers and the self-assembly properties of the uracil moieties in the crystal structure.
Results and discussion
Xantphos and (R)-BINAP were used as bridging diphosphine ligands. The advantage in the use of Xantphos and (R)-BINAP depends on the ability of their semirigid backbones to arrange the phosphorus atoms on the same side. Dinuclear organogold(I)–uracil conjugates with bridging diphosphine ligands were designed by the introduction of Xantphos and (R)-BINAP to induce an intramolecular aurophilic Au(I)–Au(I) interaction. The dinuclear organogold(I)–uracil conjugates (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP) were prepared by the reaction of 6-ethynyl-1-octyluracil (1) or 5-ethynyl-1-octyluracil (2) with (ClAu)2(μ-diphosphine) (diphosphine = Xantphos or (R)-BINAP), which were obtained by the treatment of chloro(tetrahydrothiophene)gold(I) [ClAu(tht)] with the corresponding diphosphine in situ, in the presence of sodium bis(trimethylsilyl)amide (NaN(SiMe3)2) (Scheme 1). The thus-obtained dinuclear organogold(I)–uracil conjugates were fully characterized by 1H NMR, 13C NMR, 31P NMR, IR, HRMS and elemental analysis. In the 1H NMR spectra, the signal for the ethynyl proton disappeared and the upfield shift of the uracil protons was observed after the introduction of the Au(I) center. The 31P NMR spectra of (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP) showed only one kind of resonance at around 30 ppm in CD2Cl2, indicating that two phosphorus atoms of the dinuclear organogold(I)–uracil conjugates are equivalent on the NMR time scale.
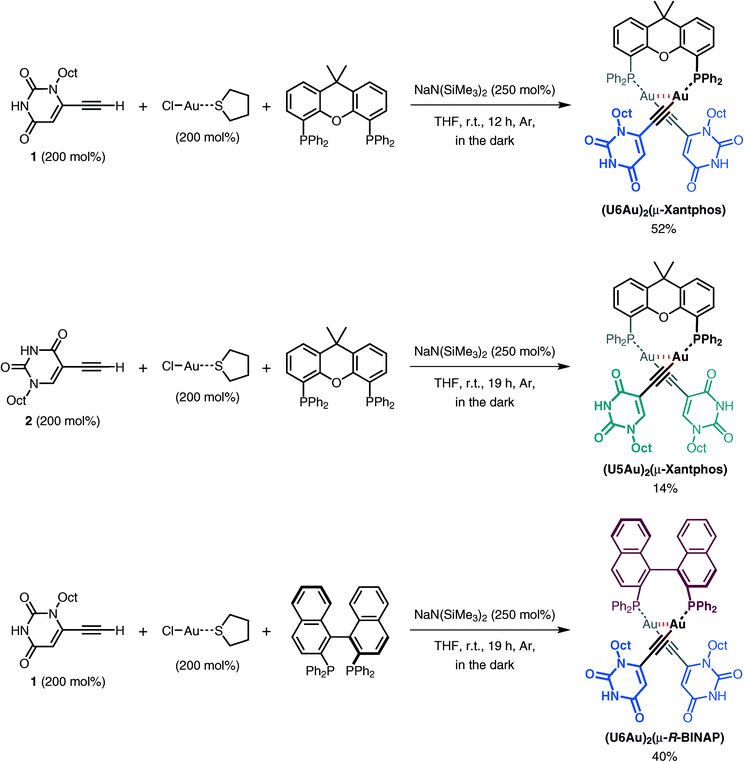 |
| | Scheme 1 Synthesis of the dinuclear organogold(I)–uracil conjugates (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP). | |
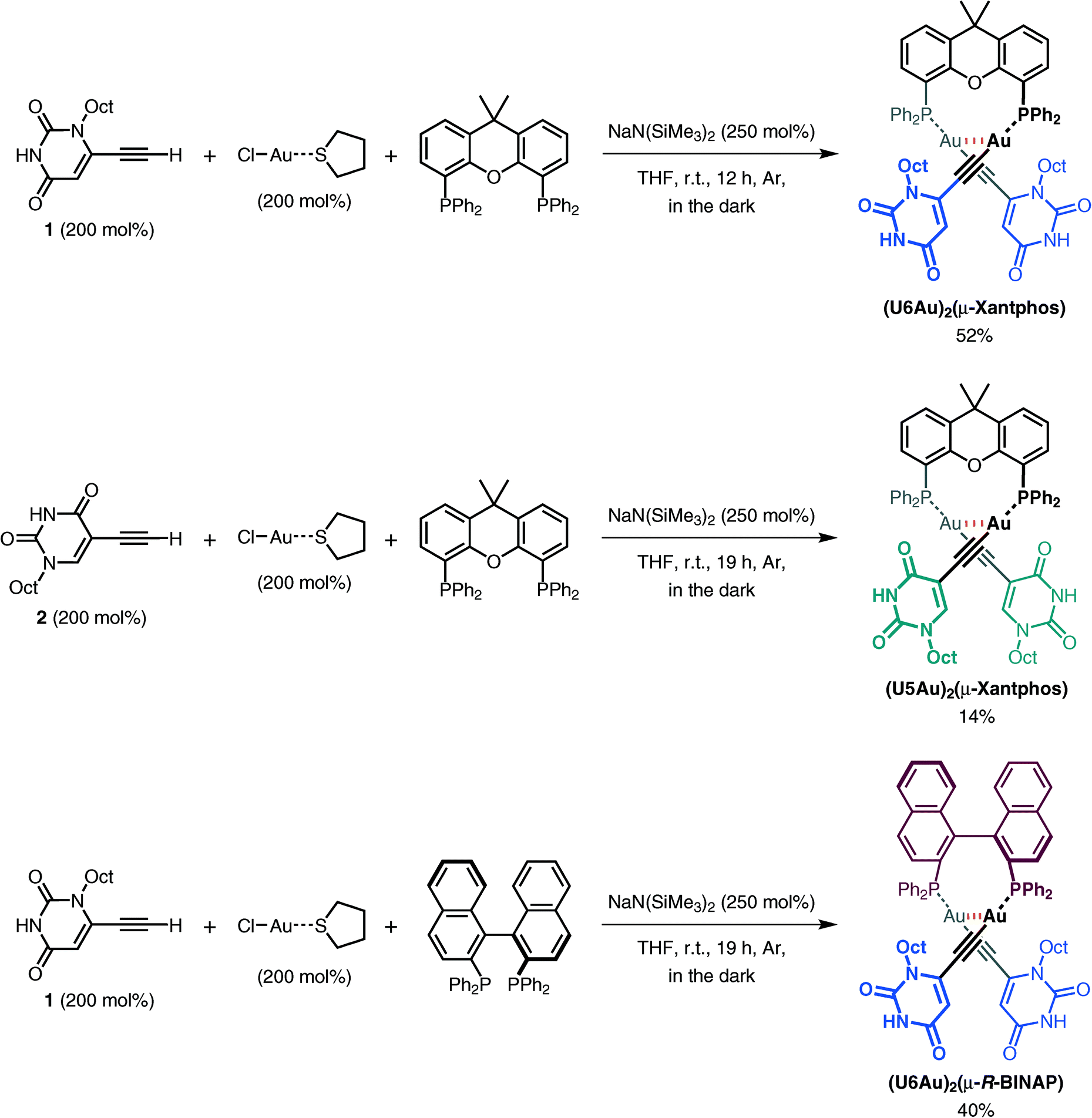
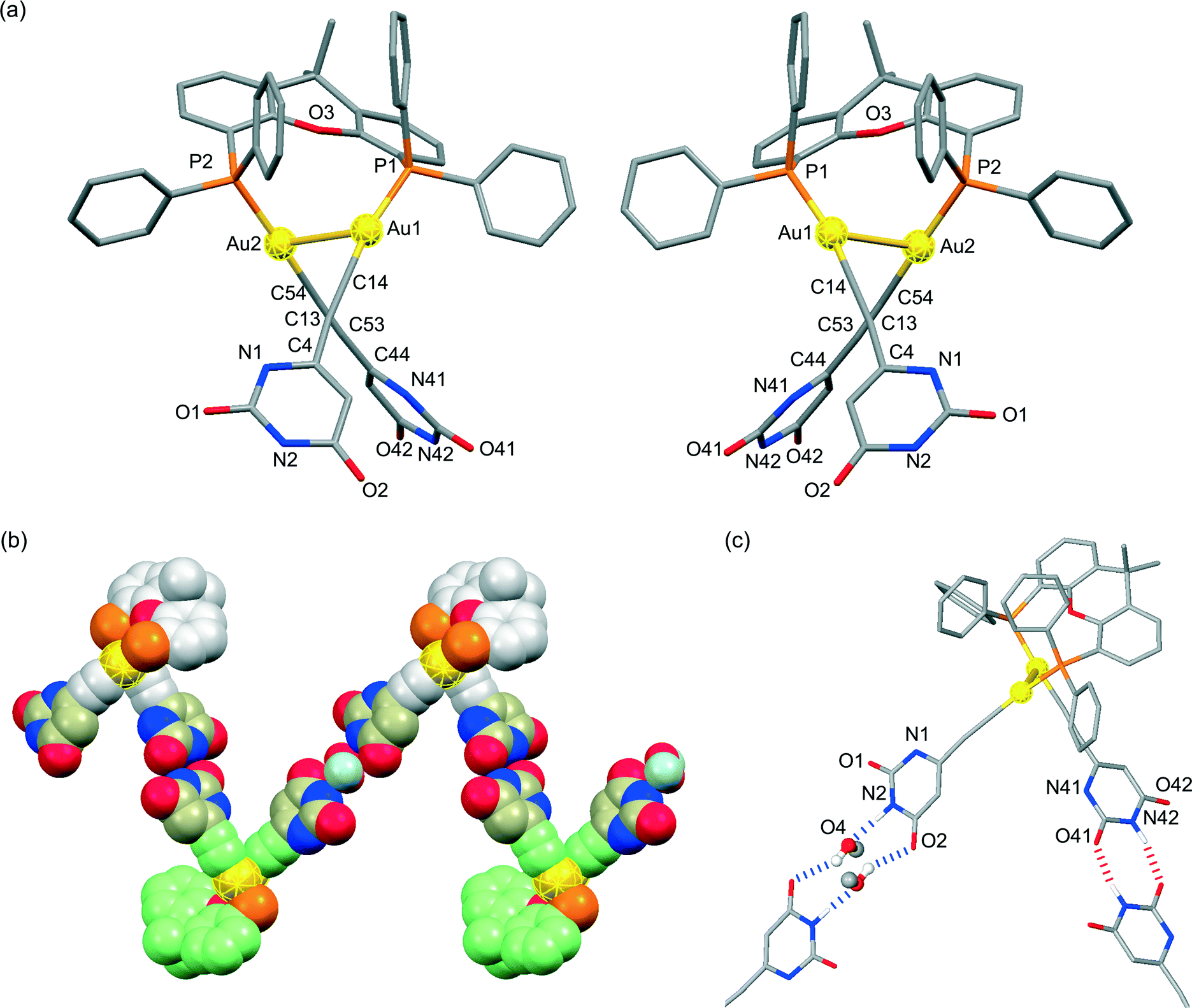
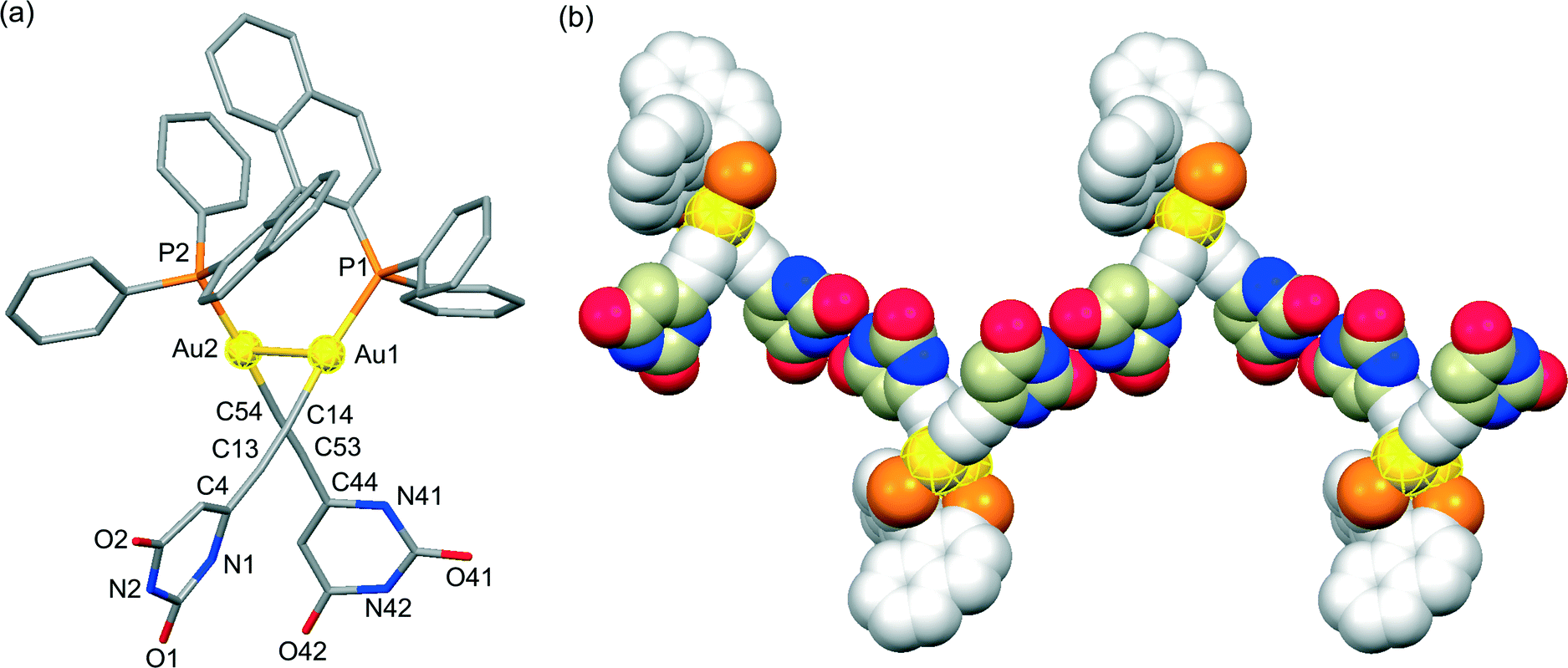
X-ray crystallographic analyses were performed in order to clarify the coordination environment of the Au(I) centers and the self-assembly properties of the dinuclear organogold(I)–uracil conjugates (Table 1). Diffraction-quality single crystals of (U6Au)2(μ-Xantphos) and (U5Au)2(μ-Xantphos) were grown by diffusion of methanol into a dichloromethane solution of (U6Au)2(μ-Xantphos) and diffusion of hexane into a dichloromethane solution of (U5Au)2(μ-Xantphos). The dinuclear structure of (U6Au)2(μ-Xantphos) composed of 6-ethynyl-1-octyluracil was confirmed by single-crystal X-ray structure determination (Fig. 1). Selected bond distances and angles are listed in Table 2. The crystal structure revealed a linear coordination geometry of the Au(I) centers bridged by the Xantphos ligand. It should be noted that an intramolecular aurophilic Au(I)–Au(I) interaction was observed with a Au(1)–Au(2) distance of 2.9994(8) Å. The semirigid xanthene backbone was found to play an important role in the arrangement of the phosphorus atoms on the same side to induce the intramolecular Au(I)–Au(I) interaction. The existence of the conformational enantiomers based on the torsional twist about the Au(I)–Au(I) axis are possible in the dinuclear gold(I)–uracil conjugates as depicted in Fig. 2. The dinuclear organogold(I) complex (U6Au)2(μ-Xantphos) crystallized in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with R- and S-enantiomers based on the Au(I)–Au(I) axis in the unit cell (Fig. 1a). The Au(I)–Au(I) interaction was found to induce the deviation from the linearity of the coordination structures of the Au centers with P–Au–C angles of 168.1(4)° and 174.5(5)°. The torsion angle of P(1)–Au(1)–Au(2)–P(2) of 82° indicates that the P–Au–C moieties are almost perpendicular to each other. Furthermore, each enantiomer is connected alternately to form the heterochiral hydrogen-bonded assembly through intermolecular hydrogen bonds between the uracil moieties (Fig. 1b and Table 3). There are two types of hydrogen bonding patterns in the heterochiral hydrogen-bonded assembly: one uracil moiety is connected by the hydrogen-bonded bridges of methanol solvent molecules and another uracil moiety is hydrogen-bonded to the uracil moiety of another molecule directly (Fig. 1c).
space group with R- and S-enantiomers based on the Au(I)–Au(I) axis in the unit cell (Fig. 1a). The Au(I)–Au(I) interaction was found to induce the deviation from the linearity of the coordination structures of the Au centers with P–Au–C angles of 168.1(4)° and 174.5(5)°. The torsion angle of P(1)–Au(1)–Au(2)–P(2) of 82° indicates that the P–Au–C moieties are almost perpendicular to each other. Furthermore, each enantiomer is connected alternately to form the heterochiral hydrogen-bonded assembly through intermolecular hydrogen bonds between the uracil moieties (Fig. 1b and Table 3). There are two types of hydrogen bonding patterns in the heterochiral hydrogen-bonded assembly: one uracil moiety is connected by the hydrogen-bonded bridges of methanol solvent molecules and another uracil moiety is hydrogen-bonded to the uracil moiety of another molecule directly (Fig. 1c).
Table 1 Crystallographic data for (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP)
|
|
(U6Au)2(μ-Xantphos)
|
(U5Au)2(μ-Xantphos)
|
(U6Au)2(μ-R-BINAP)
|
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo|.
wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
|
| Empirical formula |
C67H70N4O5P2Au2·CH2Cl2·CH3OH |
C67H70N4O5P2Au2·CH2Cl2 |
C72H70N4O4P2Au2·CHCl3 |
| Formula weight |
1584.17 |
1552.13 |
1630.63 |
| Crystal system |
Triclinic |
Monoclinic |
Orthorhombic |
| Space group |
P (no. 2) |
C2/c (no. 15) |
P212121 (no. 19) |
|
a (Å) |
10.6798(6) |
30.3688(18) |
11.0678(16) |
|
b (Å) |
14.2920(7) |
26.6704(15) |
23.228(4) |
|
c (Å) |
22.3083(13) |
21.7986(12) |
27.246(4) |
|
α (°) |
97.4663(16) |
|
|
|
β (°) |
90.7906(16) |
109.0564(16) |
|
|
γ (°) |
91.1556(14) |
|
|
|
V (Å3) |
3375.0(3) |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 688.2(16) 688.2(16) |
7004.5(18) |
|
Z
|
2 |
8 |
4 |
|
D
calcd (g cm−3) |
1.559 |
1.235 |
1.546 |
|
μ (Mo Kα) (cm−1) |
45.360 |
36.672 |
44.086 |
|
T (°C) |
4.0 |
4.0 |
−150 |
|
λ (Mo Kα) (Å) |
0.71075 |
0.71075 |
0.71075 |
|
R
1
|
0.083 |
0.054 |
0.078 |
| wR2b |
0.232 |
0.179 |
0.211 |
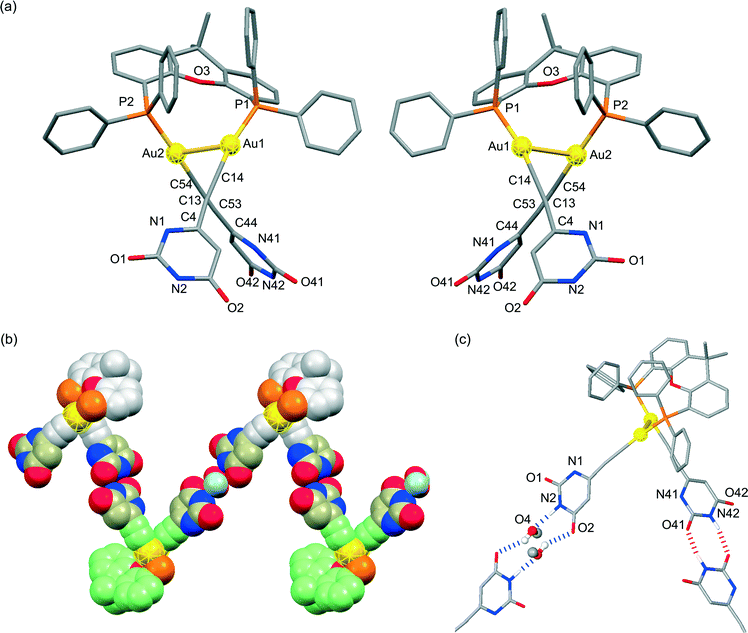 |
| | Fig. 1 (a) Molecular structures of the R- and S-enantiomers; (b) the heterochiral hydrogen-bonded assembly formed through intermolecular hydrogen bonds between the uracil moieties of the dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-Xantphos); and (c) a portion of the crystal structure showing two types of hydrogen bonding patterns in the heterochiral hydrogen-bonded assembly (hydrogen atoms and octyl moieties are omitted for clarity). | |
Table 2 Selected bond distances (Å) and angles (°) for (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP)
|
|
(U6Au)2(μ-Xantphos)
|
(U5Au)2(μ-Xantphos)
|
(U6Au)2(μ-R-BINAP)
|
| Bond distances |
| Au(1)–Au(2) |
2.9994(8) |
2.9286(5) |
3.0021(9) |
| Au(1)–P(1) |
2.283(4) |
2.281(3) |
2.281(4) |
| Au(2)–P(2) |
2.275(3) |
2.253(4) |
2.290(4) |
| Au(1)–C(14) |
2.009(14) |
1.976(10) |
2.066(14) |
| Au(2)–C(54) |
2.002(17) |
1.926(11) |
2.003(17) |
| C(13)–C(14) |
1.16(2) |
1.230(13) |
1.14(2) |
| C(53)–C(54) |
1.18(2) |
1.188(17) |
1.16(2) |
| Bond angles |
| P(1)–Au(1)–C(14) |
168.1(4) |
166.9(3) |
172.2(5) |
| P(2)–Au(2)–C(54) |
174.5(5) |
176.1(3) |
176.5(4) |
| Au(1)–C(14)–C(13) |
180.0(13) |
175.1(11) |
175.8(14) |
| Au(2)–C(54)–C(53) |
173.1(16) |
170.5(8) |
175.6(14) |
| C(14)–C(13)–C(4) |
173.3(17) |
|
168.7(18) |
| C(54)–C(53)–C(44) |
173(2) |
|
175.3(18) |
| C(14)–C(13)–C(3) |
|
171.6(13) |
|
| C(54)–C(53)–C(43) |
|
175.9(10) |
|
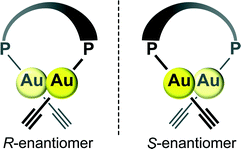 |
| | Fig. 2 Enantiomorphous conformations of the dinuclear organogold(I)–uracil conjugates with bridging diphosphine ligands. The enantiomorphs are related by the mirror plane. | |
Table 3 Intermolecular hydrogen bonds for (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP)
| Compound |
Donor |
Acceptor |
D⋯A (Å) |
D–H⋯A (°) |
|
Oxygen atom of methanol.
−X + 2, −Y + 1, −Z + 2.
−X + 2, −Y + 1, −Z + 1.
X, −Y, Z + 1/2.
X, −Y, Z + 1/2 − 1.
−X + 2, Y + 1/2 − 1, −Z + 1/2.
−X + 2, Y + 1/2, −Z + 1/2.
|
|
(U6Au)2(μ-Xantphos)
|
O(4)a |
O(2)b |
2.73(2) |
147(10) |
|
|
N(2) |
O(4)a |
2.78(2) |
172(5) |
|
|
N(42) |
O(41)c |
2.798(15) |
167(4) |
|
(U5Au)2(μ-Xantphos)
|
N(2) |
O(42)d |
2.816(13) |
166(4) |
|
|
N(42) |
O(1)e |
2.924(13) |
169(3) |
|
(U6Au)2(μ-R-BINAP)
|
N(2) |
O(41)f |
2.816(17) |
158(4) |
|
|
N(42) |
O(2)g |
2.789(18) |
176(4) |
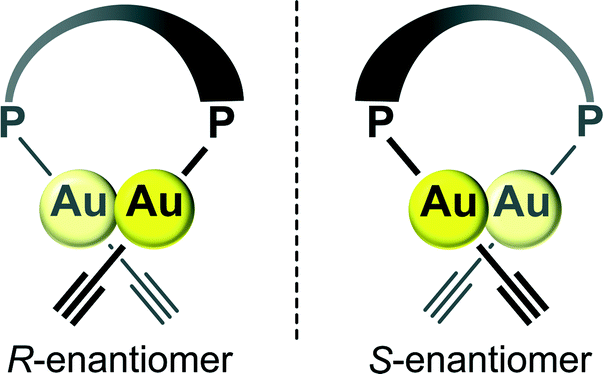
The presence of an intramolecular aurophilic Au(I)–Au(I) interaction with a Au(1)–Au(2) distance of 2.9286(5) Å in the crystal structure of (U5Au)2(μ-Xantphos) composed of 5-ethynyl-1-octyluracil, wherein the direction of the hydrogen bonding sites of the uracil moieties is different from that in (U6Au)2(μ-Xantphos), was also confirmed by single-crystal X-ray structure determination (Fig. 3). R- and S-enantiomers based on the Au(I)–Au(I) axis observed in (U5Au)2(μ-Xantphos) are present as depicted in Fig. 3a. The distortion of the linear coordination geometry of the Au centers based on the Au(I)–Au(I) interaction was also observed, resulting in P–Au–C angles of 166.9(3)° and 176.1(3)° (Table 2). Compared with (U6Au)2(μ-Xantphos), the P(1)–Au(1)–Au(2)–P(2) torsion angle of 78.96(10)° was slightly small. The Au–C bond of (U5Au)2(μ-Xantphos) was a little shorter than that of (U6Au)2(μ-Xantphos). The position of the introduced ethynyl moiety of uracil is likely to influence the electronic environment of the Au centers. Interestingly, both enantiomers form homochiral RR and SS dimers, respectively, through π–π interactions between the uracil moieties (Fig. 3b). Furthermore, each homochiral π stacked dimer is connected alternately to form the hydrogen-bonded assembly through intermolecular hydrogen bonds between the uracil moieties (Fig. 3c and Table 3). Self-assembly patterns were found to depend on the direction of the hydrogen bonding sites.
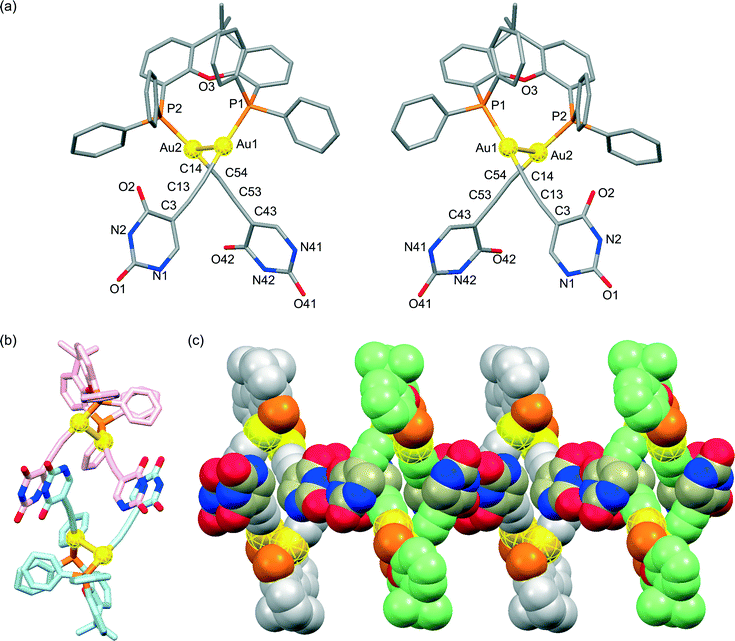 |
| | Fig. 3 (a) Molecular structures of the R- and S-enantiomers; (b) the homochiral π stacked dimer; and (c) the heterochiral hydrogen-bonded assembly formed through intermolecular hydrogen bonds between the uracil moieties of the dinuclear organogold(I)–uracil conjugate (U5Au)2(μ-Xantphos) (hydrogen atoms and octyl moieties are omitted for clarity). | |
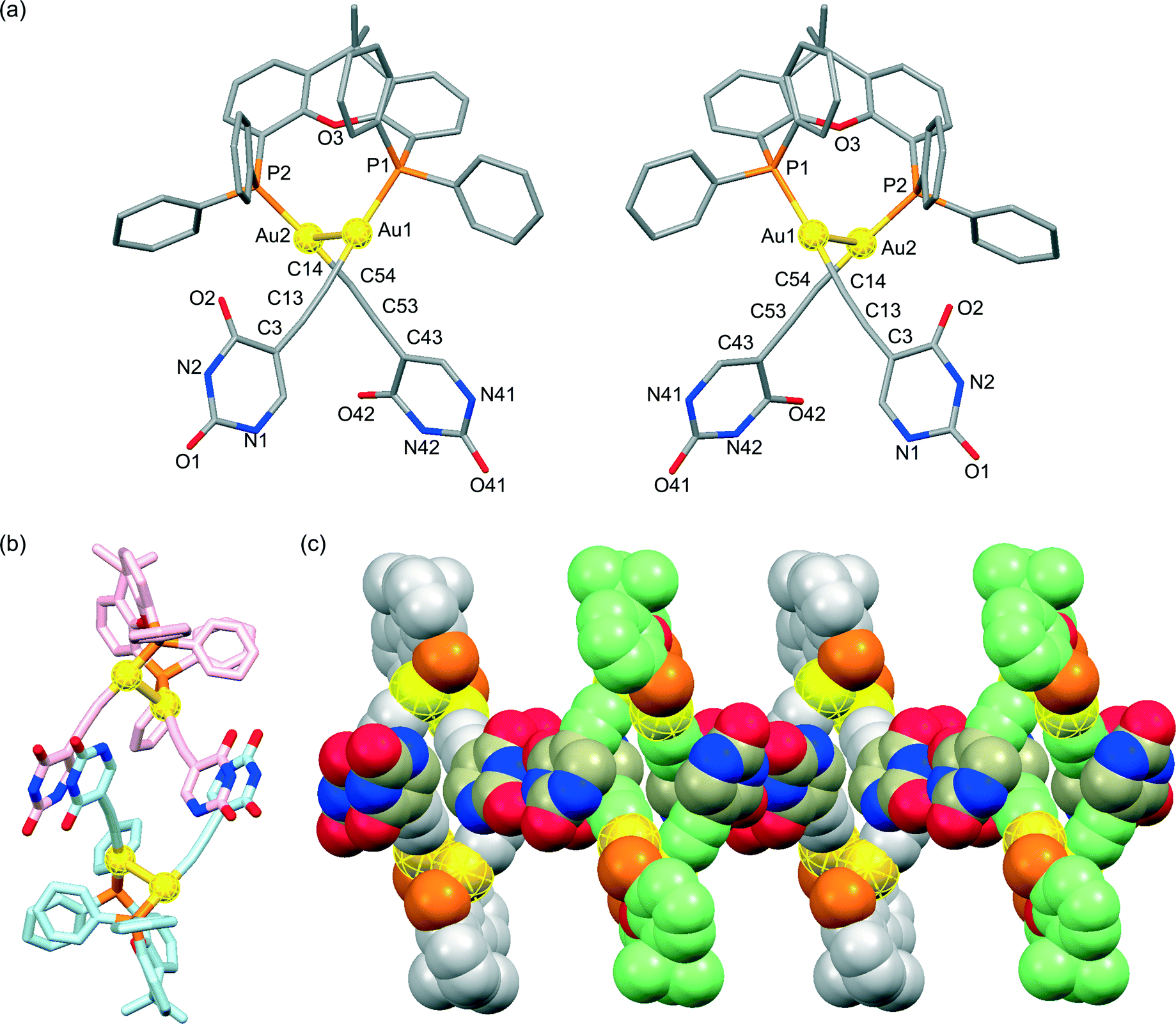
Based on the above-mentioned intriguing results, we embarked upon the chirality induction of the Au(I)–Au(I) axis by using a bridging diphosphine ligand with axial chirality. A diffraction-quality single crystal of (U6Au)2(μ-R-BINAP) was grown by diffusion of hexane into a chloroform solution of (U6Au)2(μ-R-BINAP). The dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-R-BINAP) crystallized in the P212121 space group; the molecular structure shows the presence of an intramolecular Au(I)–Au(I) interaction based on the aurophilic interaction (Fig. 4). The deviation from the linear coordination structure of the Au centers with P–Au–C angles of 172.2(5)° and 176.5(4)° based on the Au(I)–Au(I) interaction was also observed (Table 2). The crystal structure of (U6Au)2(μ-R-BINAP) showed a P(1)–Au(1)–Au(2)–P(2) torsion angle of 71.97(8)°, which is smaller than that of the gold(I)–uracil conjugates with Xantphos probably due to the difference in rigidity of the diphosphine frameworks. Gratifyingly, (U6Au)2(μ-R-BINAP) adopts a R,R-configuration through the chirality induction of the Au(I)–Au(I) axis by the axial chirality of the BINAP moiety as shown in Fig. 4a. Although Au(I)–Au(I) aurophilic interactions have been studied by using bridging diphosphine ligands,6 to the best of our knowledge, the chirality induction of the Au(I)–Au(I) axis has not been reported so far. In the crystal packing, each molecule is assembled through intermolecular hydrogen bonds between the uracil moieties to form a helical molecular arrangement (Fig. 4b and Table 3).
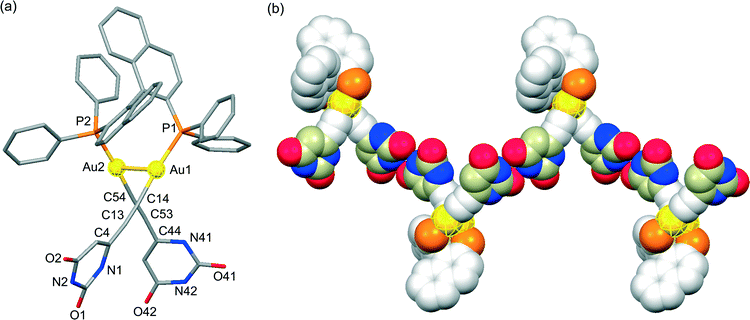 |
| | Fig. 4 (a) Molecular structure of the R-enantiomer and (b) the homochiral hydrogen-bonded assembly formed through intermolecular hydrogen bonds between the uracil moieties of the dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-R-BINAP) (hydrogen atoms and octyl moieties are omitted for clarity). | |
Conclusions
In conclusion, bioorganometallic compounds were designed and synthesized by the conjugation of dinuclear organogold(I) complexes with a bridging diphosphine ligand as an organometallic compound and the uracil derivative as a nucleobase. Single-crystal X-ray structure determination of the dinuclear organogold(I)–uracil conjugates was performed to reveal the assembly properties of gold(I) and the uracil moieties in the solid state. The semirigid bridging diphosphine ligand was found to play an important role in the arrangement of the phosphorus atoms on the same side to induce the intramolecular aurophilic Au(I)–Au(I) interaction, wherein R- and S-enantiomers based on the Au(I)–Au(I) axis exist. It is worth noting that the chirality of the Au(I)–Au(I) axis was induced by the utilization of (R)-BINAP as a bridging diphosphine ligand with axial chirality. An interesting feature of the dinuclear organogold(I)–uracil conjugates is their strong tendency to self-assemble through intermolecular hydrogen bonds between the uracil moieties, wherein hydrogen bonding patterns were found to depend on the direction of the hydrogen bonding sites. Further investigation of the application and dynamic control of the assembly of the bioconjugates including functional materials and catalysts is now in progress.
Experimental
General methods
All reagents and solvents were purchased from commercial sources and were further purified by standard methods, if necessary. All manipulations were carried out under Ar. Infrared spectra were obtained using a JASCO FT/IR-6200 spectrometer. 1H, 13C and 31P NMR spectra were recorded on a JNM-ECS 400 (400, 100 and 160 MHz, respectively) spectrometer. For 1H and 31P NMR spectra, chemical shifts were determined by using tetramethylsilane and 85% aq. H3PO4 as standard samples, respectively. Chemical shifts of 13C NMR spectra were determined relative to the solvent residual peaks. Mass spectra were run on a JEOL JMS-700 mass spectrometer.
6-Ethynyl-1-octyluracil,11 5-ethynyl-1-octyluracil,12 and chloro(tetrahydrothiophene)gold(I) [ClAu(tht)]13 were prepared by literature methods.
Synthesis of the dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-Xantphos)
A mixture of Xantphos (0.12 g, 0.21 mmol), chloro(tetrahydrothiophene)gold(I) (0.13 g, 0.41 mmol) and 6-ethynyl-1-octyluracil (1) (0.10 g, 0.40 mmol) was stirred in THF (20 mL) at room temperature for 10 minutes under Ar. Sodium bis(trimethylsilyl)amide (93 mg, 0.51 mmol) was added to the solution and the resulting solution was stirred at room temperature under Ar for 12 h. The mixture was diluted with dichloromethane, washed with water and then brine, and dried over Na2SO4. The solvent was evaporated and the residue was washed with ethyl acetate. The crude product was purified by recrystallization from dichloromethane and methanol to afford the desired dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-Xantphos) (0.15 g, 0.10 mmol) as a colorless crystal.
(U6Au)2(μ-Xantphos)
.
Yield 50%; IR (KBr) 3172, 3047, 2925, 2854, 2119, 1677, 1579, 1435, 1403, 1363, 1227 cm−1; 1H NMR (400 MHz, CD2Cl2, 5.0 × 10−3 M): δ 8.22 (br, 2H), 7.69 (dd, 2H, J = 7.8, 1.4 Hz), 7.48–7.44 (m, 4H), 7.34–7.23 (m, 16H), 7.11 (td, 2H, J = 7.8 Hz, 4JH-P = 1.2 Hz), 6.48 (ddd, 2H, J = 7.8, 1.4 Hz, 3JH-P = 12.1 Hz), 5.67 (s, 2H), 4.01 (t, 4H, J = 7.2 Hz), 1.69 (s, 6H), 1.67–1.60 (m, 4H), 1.29–1.12 (m, 20H), 0.81 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CD2Cl2, 5.0 × 10−3 M): 163, 153 (d, 2JC-P = 4.0 Hz), 151.3, 150.2 (d, 2JC-P = 140.9 Hz),140.8, 134.6 (d, 2JC-P = 14.9 Hz), 133.4, 132, 130, 129.8 (d, 1JC-P = 51.8 Hz), 129.5 (d, 3JC-P = 12 Hz), 124.9 (d, 3JC-P = 9.1 Hz), 117 (d, 1JC-P = 52.2 Hz), 104.8, 95.5 (d, 3JC-P = 26.8 Hz), 46.6, 35.1, 32.2, 31.6, 29.8, 29.6, 29.2, 27.1, 23, 14.2 ppm; 31P NMR (160 MHz, CD2Cl2, 5.0 × 10−3 M): 31.2 ppm; HRMS (FAB) m/z calcd for C67H71N4O5P2Au2 (M + H+), 1467.4225; found, 1467.4215; anal. calcd. for C67H70N4O5P2Au2: C, 54.85; H, 4.81; N, 3.82; found: C, 54.85; H, 4.94; N, 3.82.
Synthesis of the dinuclear organogold(I)–uracil conjugate (U5Au)2(μ-Xantphos)
A mixture of Xantphos (58 mg, 0.10 mmol), chloro(tetrahydrothiophene)gold(I) (64 mg, 0.20 mmol) and 5-ethynyl-1-octyluracil (2) (50 mg, 0.20 mmol) was stirred in THF (10 mL) at room temperature for 10 minutes under Ar. Sodium bis(trimethylsilyl)amide (47 mg, 0.26 mmol) was added to the solution and the resulting solution was stirred at room temperature under Ar for 19 h. The mixture was diluted with dichloromethane, washed with water and then brine, and dried over Na2SO4. The solvent was evaporated and purification of the crude product by preparative thin-layer chromatography using dichloromethane/methanol (93:7 v/v) as the mobile phase gave the desired dinuclear organogold(I)–uracil conjugate (U5Au)2(μ-Xantphos) (20 mg, 0.014 mmol). Recrystallization from dichloromethane and hexane produced a pale yellow crystal.
(U5Au)2(μ-Xantphos)
.
Yield 14%; IR (KBr) 3178, 3053, 2925, 2854, 2116, 1682, 1435, 1403, 1343, 1221 cm−1; 1H NMR (400 MHz, CD2Cl2, 5.0 × 10−3 M): δ 8.15 (br, 2H), 7.66 (d, 2H, J = 7.7 Hz), 7.45–7.41 (m, 6H), 7.36–7.27 (m, 16H), 7.09 (t, 2H, J = 7.7 Hz), 6.49 (dd, 2H, J = 7.7 Hz, 2JH-P = 11.4 Hz), 3.66 (t, 4H, J = 7.3 Hz), 1.69–1.60 (m, 10H), 1.33–1.22 (m, 20H), 0.87 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CD2Cl2, 5.0 × 10−3 M): 162.8, 153.1 (d, 2JC-P = 2.0 Hz), 151.1, 146.1, 137.5 (d, 2JC-P = 141.6 Hz), 134.8 (d, 2JC-P = 14.5 Hz), 133.3, 132, 131.5, 130.7(d, 1JC-P = 55.8 Hz), 129.6, 129.3 (d, 3JC-P =11.4 Hz), 124.6 (d, 3JC-P = 8.1 Hz) 117.8 (d, 1JC-P = 50.5 Hz), 102.3, 94.6 (d, 3JC-P = 28.6 Hz), 49.1, 35, 32.1, 31.4, 29.5, 29.4, 26.7, 23, 14.2 ppm; 31P NMR (160 MHz, CD2Cl2, 5.0 × 10−3 M): 31.8 ppm; HRMS (FAB) m/z calcd for C67H71N4O5P2Au2 (M + H+), 1467.4225; found, 1467.4171.
Synthesis of the dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-R-BINAP)
A mixture of (R)-BINAP (0.13 g, 0.21 mmol), chloro(tetrahydrothiophene)gold(I) (0.13 g, 0.41 mmol) and 6-ethynyl-1-octyluracil (1) (99 mg, 0.40 mmol) was stirred in THF (20 mL) at room temperature for 20 minutes under Ar. Sodium bis(trimethylsilyl)amide (93 mg, 0.51 mmol) was added to the solution and the resulting solution was stirred at room temperature under Ar for 19 h. The mixture was diluted with dichloromethane, washed with water and then brine, and dried over Na2SO4. The solvent was evaporated and purification of the crude product by preparative thin-layer chromatography using dichloromethane/methanol (93:7 v/v) as the mobile phase gave the desired dinuclear organogold(I)–uracil conjugate (U6Au)2(μ-R-BINAP) (0.12 g, 0.079 mmol). Recrystallization from chloroform, diethyl ether and hexane produced a pale yellow crystal.
(U6Au)2(μ-R-BINAP)
.
Yield 40%; IR (KBr) 3165, 3049, 2925, 2853, 2118, 1683, 1582, 1456, 1407, 1367 cm−1; 1H NMR (400 MHz, CD2Cl2, 1.0 × 10−2 M): δ 8.69 (br, 2H), 8.11 (d, 2H, J = 8.8 Hz), 7.93 (d, 2H, J = 8.2 Hz), 7.76–7.71 (m, 4H), 7.59 (t, 2H, J = 8.8 Hz, 3JH-P = 8.8 Hz), 7.49–7.36 (m, 10H), 7.25–7.17 (m, 8H), 6.87–6.83 (m, 2H), 6.60 (d, 2H, J = 8.5 Hz), 5.65 (s, 2H), 4.01–3.90 (m, 4H), 1.68–1.60 (m, 4H), 1.34–1.14 (m, 20H), 0.83 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CD2Cl2, 1.0 × 10−2 M): 163.3, 152.1 (d, 2JC-P = 141.9 Hz), 151.4, 143.3 (dd, 2JC-P = 16 Hz, 3JC-P = 7.2 Hz), 140.7 (d, 4JC-P = 2.6 Hz), 135.3 (d, 2JC-P = 14 Hz), 135, 134.8 (d, 2JC-P = 14.6 Hz), 134.1 (d, 3JC-P = 10 Hz), 131.8, 130.5 (d, 3JC-P = 4.8 Hz), 130.4 (d, 1JC-P = 56.6 Hz), 130.2 (d, 2JC-P = 8.4 Hz), 129.5 (d, 3JC-P = 11.5 Hz), 129.3 (d, 3JC-P = 11.7 Hz), 128.9 (d, 1JC-P = 56.6 Hz), 128.8, 128.6, 128.2 (d, 1JC-P = 56.2 Hz), 127.4, 127.3, 105.1, 93.2 (d, 3JC-P = 25.9 Hz), 46.6, 32.2, 29.8, 29.7, 29.1, 27.1, 23, 14.2 ppm; 31P NMR (160 MHz, CD2Cl2, 1.0 × 10−2 M): 33.5 ppm; HRMS (FAB) m/z calcd for C72H71N4O4P2Au2 (M + H+), 1511.4276; found, 1511.4257; anal. calcd. for C72H70Au2N4O4P2·CHCl3: C, 53.77; H, 4.39; N, 3.44; found: C, 53.78; H, 4.45; N, 3.42.
X-ray structure analysis
All measurements for (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP) were made on a Rigaku R-AXIS RAPID diffractometer using graphite monochromated Mo Kα radiation. The structures of (U6Au)2(μ-Xantphos), (U5Au)2(μ-Xantphos) and (U6Au)2(μ-R-BINAP) were solved by direct methods14 and expanded using Fourier techniques. All calculations were performed using the CrystalStructure crystallographic software package15 except for the refinement, which was performed using SHELXL-97.16 The non-hydrogen atoms were refined anisotropically. The H atoms involved in hydrogen bonding were located in electron density maps. The remainder of the H atoms were placed in idealized positions and allowed to ride on the C atoms to which each was bonded. Crystallographic details are given in Table 1. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC-1033826 for (U6Au)2(μ-Xantphos), CCDC-1033825 for (U5Au)2(μ-Xantphos) and CCDC-1033824 for (U6Au)2(μ-R-BINAP).
Acknowledgements
This work was supported partly by a Grant-in-Aid for Scientific Research on Innovative Areas (“Coordination Programming” Area 2107, no. 24108722) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the ACT-C program of Japan Science and Technology Agency (JST). Y. S. acknowledges a JSPS Fellowship for Young Scientists. Thanks are due to the Analytical Center, Graduate School of Engineering, Osaka University.
Notes and references
- J. C. Lima and L. Rodríguez, Chem. Soc. Rev., 2011, 40, 5442 RSC.
-
(a) D. Li, X. Hong, C. M. Che, W. C. Lo and S. M. Peng, J. Chem. Soc., Dalton Trans., 1993, 2929 RSC;
(b) H. Xiao, Y. X. Weng, S. M. Peng and C. M. Che, J. Chem. Soc., Dalton Trans., 1996, 3155 RSC;
(c) M. J. Irwin, J. J. Vittal and R. J. Puddephatt, Organometallics, 1997, 16, 3541 CrossRef CAS;
(d) C. M. Che, H. Y. Chan, V. M. Miskowski, Y. Li and K. K. Cheung, J. Am. Chem. Soc., 2001, 123, 4985 CrossRef CAS PubMed.
-
(a) E. Schuh, S. M. Valiahdi, M. A. Jakupec, B. K. Keppler, P. Chiba and F. Mohr, Dalton Trans., 2009, 10841 RSC;
(b) E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596 CrossRef CAS;
(c) A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895 CrossRef CAS PubMed;
(d) A. Meyer, A. Gutiérrez, I. Ott and L. Rodríguez, Inorg. Chim. Acta, 2013, 398, 72 CrossRef CAS PubMed.
-
(a) P. Pyykkő, Chem. Rev., 1997, 97, 597 CrossRef PubMed;
(b) R. J. Puddephatt, Coord. Chem. Rev., 2001, 216–217, 313 CrossRef CAS;
(c) V. W. W. Yam and E. C. C. Cheng, Chem. Soc. Rev., 2008, 37, 1806 RSC;
(d) M. J. Katz, K. Sakai and D. B. Leznoff, Chem. Soc. Rev., 2008, 37, 1884 RSC;
(e) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
-
(a) Z. Assefa, M. A. Omary, B. G. McBurnett, A. A. Mohamed, H. H. Patterson, R. J. Staples and J. P. Fackler, Inorg. Chem., 2002, 41, 6274 CrossRef CAS PubMed;
(b) H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044 CrossRef CAS PubMed;
(c) M. Osawa, I. Kawata, S. Igawa, M. Hoshino, T. Fukunaga and D. Hashizume, Chem. – Eur. J., 2010, 16, 12114 CrossRef CAS PubMed;
(d) H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 Search PubMed;
(e) T. Seki, K. Sakurada and H. Ito, Angew. Chem., Int. Ed., 2013, 52, 12828 CrossRef CAS PubMed.
-
(a) A. Pintado-Alba, H. de la Riva, M. Nieuhuyzen, D. Bautista, P. R. Raithby, H. A. Sparkes, S. J. Teat, J. M. Lopez-de-Luzuriaga and M. C. Lagunas, Dalton Trans., 2004, 3459 RSC;
(b) A. Deák, T. Megyes, G. Tárkány, P. Király, L. Biscók, G. Pálinkás and P. J. Stang, J. Am. Chem. Soc., 2006, 128, 12668 CrossRef PubMed;
(c) D. V. Partiyka, J. B. Undegraff III, M. Zeller, A. D. Hunter and T. G. Gray, Dalton Trans., 2010, 5388 RSC;
(d) D. V. Partiyka, T. S. Teets, M. Zeller, J. B. Undegraff III, A. D. Hunter and T. G. Gray, Chem. – Eur. J., 2012, 18, 2100 CrossRef PubMed.
-
(a) S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC;
(b) J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296 RSC;
(c) J. L. Sessler, C. M. Lawrence and J. Jayawickramarajah, Chem. Soc. Rev., 2007, 36, 314 RSC;
(d) K. Tanaka and M. Shionoya, Coord. Chem. Rev., 2007, 251, 2732 CrossRef CAS PubMed;
(e) J. Müller, Eur. J. Inorg. Chem., 2008, 3749 CrossRef;
(f) S. Lena, S. Masiero, S. Pieraccini and G. P. Spada, Chem. – Eur. J., 2009, 15, 7792 CrossRef CAS PubMed;
(g) G. H. Clever and M. Shionoya, Coord. Chem. Rev., 2010, 254, 2391 CrossRef CAS PubMed.
-
(a) M. M. Conn and J. Rebek Jr., Chem. Rev., 1997, 97, 1647 CrossRef CAS PubMed;
(b) E. A. Archer, H. Gong and M. J. Krische, Tetrahedron, 2001, 57, 1139 CrossRef CAS;
(c) L. J. Prins, D. N. Reinhoudt and P. Timmerman, Angew. Chem., Int. Ed., 2001, 40, 2382 CrossRef CAS.
-
(a) G. Jaouen, A. Vessiéres and I. S. Butler, Acc. Chem. Res., 1993, 26, 361 CrossRef CAS;
(b) R. Severin, R. Bergs and W. Beck, Angew. Chem., Int. Ed., 1998, 37, 1634 CrossRef;
(c) R. H. Fish and G. Jaouen, Organometallics, 2003, 22, 2166 CrossRef CAS;
(d) T. Moriuchi and T. Hirao, Chem. Soc. Rev., 2004, 33, 294 RSC;
(e) D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS PubMed;
(f) H. Song, X. Li, Y. Long, G. Schatte and H.-B. Kraatz, Dalton Trans., 2006, 4696 RSC;
(g) W. Beck, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1221 CAS;
(h) A. Lataifeh, S. Beheshti and H.-B. Kraatz, Eur. J. Inorg. Chem., 2009, 3205 CrossRef CAS;
(i) T. Moriuchi and T. Hirao, Acc. Chem. Res., 2010, 43, 1040 CrossRef CAS PubMed;
(j) G. Gasser, A. M. Sosniak and N. Metzler-Nolte, Dalton Trans., 2011, 40, 7061 RSC;
(k) B. Adhikari, R. Afrasiabi and H.-B. Kraatz, Organometallics, 2013, 32, 5899 CrossRef CAS.
-
(a) X. Meng, T. Moriuchi, M. Kawahata, K. Yamaguchi and T. Hirao, Chem. Commun., 2011, 47, 4682 RSC;
(b) X. Meng, T. Moriuchi, Y. Sakamotoa, M. Kawahata, K. Yamaguchi and T. Hirao, RSC Adv., 2012, 4349 Search PubMed;
(c) T. Moriuchi, Y. Sakamoto, S. Noguchi, T. Fujiwara, S. Akine, T. Nabeshima and T. Hirao, Dalton Trans., 2012, 41, 8524 RSC.
- T. Moriuchi, S. Noguchi, Y. Sakamoto and T. Hirao, J. Organomet. Chem., 2011, 696, 1089 CrossRef CAS PubMed.
- M. Takase and M. Inoueye, J. Org. Chem., 2003, 68, 1134 CrossRef CAS PubMed.
- A. S. K. Hashmi, T. Hengst, C. Lothschütz and F. Rominger, Adv. Synth. Catal., 2010, 352, 1315 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
-
CrystalStructure 4.0: Crystal Structure Analysis Package, Rigaku Corporation ( 2000–2010). Tokyo 196–8666, Japan Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † CCDC reference number 1033826 for (U6Au)2(μ-Xantphos), 1033825 for (U5Au)2(μ-Xantphos) and 1033824 for (U6Au)2(μ-R-BINAP). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ce00221d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence